

Abstract
Mutations in BRCA1/2 increase the risk of developing breast and ovarian cancer. Germline BRCA1/2 mutations occur in 8.6–13.7% of unselected epithelial ovarian cancers, somatic mutations are also frequent. BRCA1/2 mutated or dysfunctional cells may be sensitive to PARP inhibition by synthetic lethality. The aim of this study is to comprehensively characterise the BRCA1/2 status of a large panel of ovarian cancer cell lines available to the research community to assist in biomarker studies of novel drugs and in particular of PARP inhibitors. The BRCA1/2 genes were sequenced in 41 ovarian cell lines, mRNA expression of BRCA1/2 and gene methylation status of BRCA1 was also examined. The cytotoxicity of PARP inhibitors olaparib and veliparib was examined in 20 cell lines.
The cell line SNU‐251 has a deleterious BRCA1 mutation at 5564G > A, and is the only deleterious BRCA1/2 mutant in the panel. Two cell lines (UPN‐251 and PEO1) had deleterious mutations as well as additional reversion mutations that restored the protein functionality. Heterozygous mutations in BRCA1/2 were relatively common, found in 14.6% of cell lines. BRCA1 was methylated in two cell lines (OVCAR8, A1847) and there was a corresponding decrease in gene expression. The BRCA1 methylated cell lines were more sensitive to PARP inhibition than wild‐type cells. The SNU‐251 deleterious mutant was more sensitive to PARP inhibition, but only in a long‐term exposure to correct for its slow growth rate. Cell lines derived from metastatic disease are significantly more resistant to veliparib (2.0 fold p = 0.03) compared to those derived from primary tumours. Resistance to olaparib and veliparib was correlated Pearsons‐R 0.5393, p = 0.0311.
The incidence of BRCA1/2 deleterious mutations 1/41 cell lines derived from 33 different patients (3.0%) is much lower than the population incidence. The reversion mutations and high frequency of heterozygous mutations suggest that there is a selective pressure against BRCA1/2 in cell culture similar to the selective pressure seen in the clinic after treatment with chemotherapy. PARP inhibitors may be useful in patients with BRCA1 deleterious mutations or gene methylation.
Keywords: BRCA1/2, Ovarian, Mutation, Methylation, Parp inhibitor, Olaparib, Veliparib
Highlights
-
►
First study of BRCA1/2 mutation and methylation in ovarian cancer cell lines.
-
►
BRCA1/2 mutations are much less common in cell lines than in the clinic.
-
►
There is a selective pressure against BRCA1/2 mutations in cell culture.
-
►
BRCA1/2 methylated and mutated cell lines are more sensitive to parp inhibitors.
1. Introduction
The BRCA1 and BRCA2 proteins play a critical role in DNA damage repair and mutations in these genes increase the risk of developing breast and ovarian cancer. BRCA1/2 germline mutations have been shown occur in 8.6–13.7% of unselected epithelial ovarian cancer patients (Pal et al., 2005; Risch et al., 2001; Rubin et al., 1998). The rates of BRCA1 or combined BRCA1/2 mutation are much higher in ovarian cancer than in unselected invasive breast cancer 1.42–1.96% (Anonymous, 2000; Newman et al., 1998; Papelard et al., 2000).
Synthetic lethality is the phenomenon whereby cell death results from the loss of function of two different gene products, when loss of either gene product in isolation does not. This concept has led to the idea that a new class of agents, PARP inhibitors, could be effective at treating BRCA1/2 dysfunctional cancers. PARP‐1 is thought to be a regulator of base excision repair (BER) (Helleday, 2011). One model suggests that PARP‐1 inhibition traps one of the single strand break intermediates in the BER process and prevents the final ligation step (Helleday, 2011). PARP also plays a role at stalled replication forks and is often hyper‐activated in cells defective in homologous recombination repair such as BRCA1/2 mutant or defective cells (Helleday, 2011). Inhibition of PARP in cells with dysfunctional homologous recombination repair therefore results in the inhibition of two major DNA repair pathways. Studies have demonstrated this synthetic lethality by inhibiting PARP in cells deficient in BRCA1/2 (Bryant et al., 2005; Farmer et al., 2005). The results of proof of concept clinical trials of the PARP inhibitor olaparib in breast and ovarian cancer patients with germline BRCA1/2 mutations have also been encouraging (Audeh et al., 2010; Tutt et al., 2010).
Recently it has been shown that somatic BRCA1/2 mutations are also frequent in ovarian cancer. BRCA1/2 mutations were detected in 18.3% of tumour samples (n = 235). Of the 42 patients found to have a BRCA1/2 mutation in their tumor, germline DNA was available from 28. Eleven of these patients (39%) had somatic BRCA1/2 mutations not present in the germline (Hennessy et al., 2010). These somatic BRCA1/2 mutations may expand the cohort of ovarian cancer patients which may benefit from treatment with PARP inhibitors. PARP inhibitors may also be useful in sporadic ovarian cancer patients which have other non‐mutation defects in the BRCA1/2 pathways in their tumours, such as loss of gene or protein expression which can occur in up to 90% of patients (Russell et al., 2000).
To understand the role of BRCA1/2 mutations in ovarian cancer and study potential therapeutic strategies including the use of PARP inhibitors, suitable cell lines are needed. A previous study has characterised BRCA1/2 status in a large panel of breast cancer cell lines (Elstrodt et al., 2006). Our study characterises the mutation, expression and methylation status of BRCA1/2 in 41 ovarian cancer cell lines. The responsiveness to PARP inhibitors olaparib and veliparib was examined in a subset of 20 cell lines.
2. Results
2.1. BRCA1 mutation screening
The clinical characteristics of the ovarian tumors from which the cell lines were established are presented in Table 1. Sequencing of BRCA1 revealed 18 different alterations in the gene sequence among 41 human ovarian cell lines (Table 2). The majority of alterations are nonpathogenic polymorphisms as described by the Breast Cancer Information Core (BIC) mutation database (http://research.nhgri.nih.gov/bic/). Three novel mutations in BRCA1 were identified in this study 1199del29, 2921G > A, and 2925G > A (Table 3). These variants have not been previously identified and are not present in the BIC or Myriad Genetics databases. 1199del29 is deleterious as it results in a frameshift and a premature stop codon; 2132G > A is a polymorphism and 2136G > A is a variant of unknown significance.
Table 1.
Clinical characteristics of ovarian tumors from which cell lines were established.
| Cell line | Original tumor histology | Isolated from | Pre‐isolation | Post‐isolation | References | ||
|---|---|---|---|---|---|---|---|
| Treatment received | Response | Treatment received | Response | ||||
| 59M | Endometriod/clear cell | Ascites | None | N/A | IFO, MEL | PR, died | (Hills et al., 1989) |
| A1847 | Undifferentiated carcinoma | Metastasis | Unknown | Unknown | Unknown | Unknown | (Aaronson, 2012) |
| A2780 | Unknown | Primary tumour | None | N/A | Unknown | Unknown | (2012a) |
| CAOV3 | Adenocarcinoma | Unknown | Unknown | Unknown | Unknown | Unknown | (2012c) |
| DOV13 | Adenocarcinoma | Unknown | Unknown | Unknown | Unknown | Unknown | (Young et al., 1995) |
| EFO27 | Mucinous | Solid metastasis | None | N/A | Unknown | Unknown | (Kunzmann and Hozel, 1987) |
| ES2 | Serous/clear cell | Primary tumour | None | N/A | Chemotherapy | Refractory | (2012b) (Sikic, 2012) |
| FUOV1 | Serous | Primary tumour | None | N/A | CIS, ASR, CYC | Responded | (Emoto et al., 1999) |
| HEY | Serous | Peritoneal deposit and xenograft | Radiotherapy, radium | CR | CIS, ADR, CHL | Died | (Buick et al., 1985; Hills et al., 1989) |
| HI0180 | Normal OSE | Normal OSE | N/A | N/A | N/A | N/A | (Sood et al., 2002) |
| HOC1 | Serous | Ascites | MEL, CIS, ADR, CYC | PR, PR | None | N/A | (Buick et al., 1985; Mackillop et al., 1983) |
| HOC7a | Serous | Ascites | MEL, CIS, ADR, CYC | PR, PR | Unknown | Unknown | (Buick et al., 1985; Mackillop et al., 1983) |
| HOC8 | Serous | Ascites | MEL | PR | (Filmus et al., 1986; Filmus and Buick, 1985) | ||
| IGROV1 | Endometriod/clear cell | Primary tumour | None | N/A | Unknown | Unknown | (Benard et al., 1985) |
| IOSE29 | Normal OSE | Normal OSE + SV40 | N/A | N/A | N/A | N/A | (Wong et al., 2004) |
| IOSE80 | Normal OSE | Normal OSE + SV40 | N/A | N/A | N/A | N/A | (Choi et al., 2005) |
| ML46 | Serous | Primary Tumour + SV40 | Unknown | Unknown | Unknown | Unknown | (Yu et al., 2007) |
| MPSCI | Serous | Unknown | Unknown | Unknown | Unknown | Unknown | (Pohl et al., 2005) |
| OAW28 | Adenocarcinoma | Ascites | CIS, MEL | NR, NR | Unknown | Died | (Hills et al., 1989) |
| OAW42 | Serous | Ascites | CIS | CR | Unknown | PD, died | (Hills et al., 1989) |
| OC316 | Serous | Ascites | CIS, ETO, CYC, TAX | PD, SD | Epirubicin | PD | (Alama et al., 1996) |
| OCC1 | Clear cell | Ascites | Unknown | Unknown | Unknown | Unknown | (Wong et al., 1990) |
| OVCA420 | Serous | Unknown | Unknown | Unknown | Unknown | Unknown | (Syed et al., 2001) |
| OVCA432 | Serous | Unknown | Unknown | Unknown | Unknown | Unknown | (Syed et al., 2001) |
| OVCA433 | Serous | Unknown | Unknown | Unknown | Unknown | Unknown | (Syed et al., 2001) |
| OVCAR5 | Adenocarcinoma | Unknown | None | N/A | Unknown | Unknown | (Schilder et al., 1990) |
| OVCAR3 | Serous | Ascites | CYC, CIS, DOX | Unknown | Unknown | Unknown | (Hamilton et al., 1983; Hills et al., 1989; Schilder et al., 1990) |
| OVCAR8 | Adenocarcinoma | Unknown | CAR | PD | Unknown | Unknown | (Schilder et al., 1990) |
| OVCAR10 | Adenocarcinoma | Unknown | CIS, CAR | PD | Unknown | Unknown | (Schilder et al., 1990) |
| PA1 | Germ cell tumor | Ascites | Chemotherapy | NR | Unknown | Died | (Hills et al., 1989) |
| PEA1 | Adenocarcinoma | Pleural effusion | None | N/A | CIS, PDM | Relapsed | (Langdon et al., 1988) |
| PEA2b | Adenocarcinoma | Ascites | CIS, PDM | Relapsed | Unknown | Unknown | (Langdon et al., 1988) |
| PEO1 | Serous | Ascites | CIS, CHL, 5‐FU | CR | CIS, CHL, 5‐FU | CR | (Langdon et al., 1988; Sakai et al., 2009) |
| PEO4c | Serous | Ascites | CIS, CHL, 5‐FU | CR | Higher dose CIS | PD | (Langdon et al., 1988; Sakai et al., 2009) |
| PEO6c | Serous | Ascites | CIS, CHL, 5‐FU, CIS | CR, PD | None | Died | (Langdon et al., 1988; Sakai et al., 2009) |
| PEO14 | Serous | Ascites | None | N/A | CIS, CHL | Relapsed | (Langdon et al., 1988) |
| PEO23d | Serous | Ascites | CIS, CHL | Relapsed | Unknown | Unknown | (Langdon et al., 1988) |
| SKOV3 | Adenocarcinoma | Ascites | THI | Unknown | Unknown | Unknown | (Hills et al., 1989) |
| SNU251 | Endometriod | Ascites | CYC, ADR, CIS | Unknown | Unknown | Unknown | (Yuan et al., 1997) |
| SW626 | Adenocarcinoma | Primary tumor or ovarian metastasis | Unknown | Unknown | Unknown | Unknown | (2012d; Furlong et al., 1999) |
| UPN251 | Unknown | Unknown | Pt/TAX, TAX | NR | Unknown | Unknown | (Hamilton, 2012) |
ADR – Adriamycin, CAR – Carboplatin, CIS – Cisplatin, CHL – Chlorambucil, CR – Complete Response, CYC – Cyclophosphamide, DOX – Doxurubicin, ETO – Etoposide, MEL – Melphalan, N/A – Not Applicable, NR – No Response, OSE – Ovarian Surface Epithelium, PD – Progressive Disese, PR – Partial Response, PDM – Prednimustine, Pt – Platinum, TAX – Taxol, THI – Thiotepa.
Same patient as HOC‐1.
Same patient as PEA1.
Same patient as PEO1.
Same patient as PEO14.
Table 2.
BRCA1 sequence variants among ovarian cell lines.
| Variant | Nucleotide change | Exon | Predicted protein effect | Type of variant | Number mutations in ovarian cell lines | No. in BIC |
|---|---|---|---|---|---|---|
| 1 | 1186A > G | 11 | Q356R | Poly | 1 | 82 |
| 2 | 1199del29 (ctcagagaatcctagagatactgaagatg) | 11 | Frameshift and then premature stop at base 1218. | Deleterious | 1 | 0 |
| 3 | 1246delA | 11 | Stop 393 | Deleterious | 1 | 4 |
| 4 | 2080delA | 11 | Stop 700 | Deleterious | 1 | 31 |
| 5 | 2196G > A | 11 | D693N | Poly | 7 | 16 |
| 6 | 2201C > T | 11 | S694S | Poly | 22 | 14 |
| 7 | 2430T > C | 11 | L771L | Poly | 21 | 25 |
| 8 | 2577A > G | 11 | K820E | Poly | 1 | 28 |
| 9 | 2731C > T | 11 | P871L | Poly | 24 | 30 |
| 10 | 2921G > A | 11 | Q934Q | Poly | 1 | 0 |
| 11 | 2925G > A | 11 | D936N | Unclassified | 1 | 0 |
| 12 | 3232A > G | 11 | E1038G | Poly | 20 | 37 |
| 13 | 3238G > A | 11 | S1040N | Poly | 1 | 42 |
| 14 | 3667A > G | 11 | K1183R | Poly | 22 | 32 |
| 15 | 4427T > C | 13 | S1436S | Poly | 21 | 35 |
| 16 | 4956A > G | 16 | S1613G | Poly | 22 | 40 |
| 17 | IVS21 + 1G > C (first base after exon 21, ie intron 22) | 21 | Splice variant | Suspected deleterious | 1 | 0 |
| 18 | 5564G > A | 23 | W1815X | Deleterious | 1 | 0 |
Table 3.
BRCA1/2 mutation, methylation and LOH analysis of ovarian cell lines.
| Cell line | BRCA1 | BRCA2 | |||||
|---|---|---|---|---|---|---|---|
| LOH | Gene variants | Mutation status | Methylated | LOH | Gene variants | Mutation status | |
| 59M | Yes | None | Wild‐type | No | No | 5,8,11,25 | Wild‐type |
| A1847 | Yes | 5,6,7,9,12,14,15,16 | Wild‐type | Yes | Yes | 5 | Wild‐type |
| A2780 | No | 9 | Wild‐type | No | No | 2,9,11,20,25 | Wild‐type |
| CAOV3 | ND | None | Wild‐type | No | ND | 1,10,23 | Wild‐type |
| DOV13 | No | 6,7,12,14,15,16 | Wild‐type | No | No | 11,25,26,33 | Wild‐type |
| EFO27 | No | 9 | Wild‐type | No | No | 1,10,14,21,23 | Wild‐type |
| ES2 | ND | 5,6,7,9,12,14,15,16 | Wild‐type | No | ND | 11,25,33 | Wild‐type |
| FUOV1 | Yes | None | Wild‐type | No | No | 11,25,33 | Wild‐type |
| HEY | No | 1,6,7,9,12,14,15,16 | Wild‐type | No | No | 1,10,23 | Wild‐type |
| HI0180 | ND | 6,7,9,14,15,16,17* | Wild‐type | No | ND | 1,10,23 | Wild‐type |
| HOC1 | No | 6,7,9,12,14,15,16 | Wild‐type | No | No | 1,5,10,23 | Wild‐type |
| HOC7 | No | 6,7,9,12,14,15,16 | Wild‐type | No | No | 1,5,10,23 | Wild‐type |
| HOC8 | ND | 6,7,9,12,14,15,16 | Wild‐type | No | ND | 10,23 | Wild‐type |
| IGROV1 | No | 4*,6,7,9,12,14,15,16 | Wild‐type | No | No | 1,10,13,23 | Wild‐type |
| IOSE29 | No | 6,7,9,12,14,15,16 | Wild‐type | No | No | 5,25 | Wild‐type |
| IOSE80 | No | 6,7,9,12,14,15,16 | Wild‐type | No | No | 1,23,31a | Wild‐type |
| ML46 | No | None | Wild‐type | No | No | None | Wild‐type |
| MPSCI | No | 6,8,9,14,16 | Wild‐type | No | No | 10,11,23,25,32 | Wild‐type |
| OAW28 | ND | 6,7,9,12,14,15,16 | Wild‐type | No | ND | 1,10,23 | Wild‐type |
| OAW42 | No | None | Wild‐type | No | No | 5,11,25 | Wild‐type |
| OC316 | No | 5,6,7,9,12,14,15,16 | Wild‐type | No | No | 5,18,19a,25 | Wild‐type |
| OCC1 | No | None | Wild‐type | No | No | 11,25 | Wild‐type |
| OVCA420 | No | 9 | Wild‐type | No | No | 10,11,22,23,25,33 | Wild‐type |
| OVCA432 | Yes | 9 | Wild‐type | No | No | 5,25,28,30a | Wild‐type |
| OVCA433 | Yes | None | Wild‐type | No | No | 5,25,28,30a | Wild‐type |
| OVCAR5 | No | 5,6,7,9,12,14,15,16 | Wild‐type | No | No | 5,11,25 | Wild‐type |
| OVCAR3 | Yes | None | Wild‐type | No | Yes | 1,10,23 | Wild‐type |
| OVCAR8 | Yes | 5,6,7,12,14,15,16, | Wild‐type | Yes | Yes | None | Wild‐type |
| OVCAR10 | ND | 9,10,11 | Wild‐type | No | ND | 3,9,11,20,24,25,27 | Wild‐type |
| PA1 | No | 13 | Wild‐type | No | No | 5,12,25 | Wild‐type |
| PEA1 | Yes | None | Wild‐type | No | Yes | 1,10,23 | Wild‐type |
| PEA2 | Yes | None | Wild‐type | No | Yes | 1,10,23 | Wild‐type |
| PEO1 | Yes | 6,7,9,12,14,15,16 | Wild‐type | No | Yes | 11,15c,16b,25 | Wild‐type |
| PEO4 | Yes | 6,7,9,12,14,15,16 | Wild‐type | No | Yes | 11,17c,25 | Wild‐type |
| PEO6 | Yes | 6,7,9,12,14,15,16 | Wild‐type | No | Yes | 11,17c,25 | Wild‐type |
| PEO14 | ND | 5,6,7,9,12,14,15,16 | Wild‐type | No | ND | 1,5,10,23,25 | Wild‐type |
| PEO23 | Yes | 5,6,7,9,12,14,15,16 | Wild‐type | No | No | 1,5,10,23,25 | Wild‐type |
| SKOV3 | No | None | Wild‐type | No | No | 1,10,21,23 | Wild‐type |
| SNU251 | ND | 18 | Mutant | No | ND | 5,25,29 | Wild‐type |
| SW626 | Yes | None | Wild‐type | No | Yes | 4,6,7,9 | Wild‐type |
| UPN251 | Yes | 2b,3b | Wild‐type | No | Yes | 5,25 | Wild‐type |
Numbered gene variants for BRCA1 and BRCA2 refer to Tables 2 and 4 respectively.
Heterozygous deleterious mutation.
Homozygous Deleterious mutation compensated for by another reversion mutation.
Reversion mutation.
Deleterious BRCA1 mutations were identified in 4 ovarian cell lines IGROV‐1, UPN‐251, HI0180 and SNU‐251 (Tables 2 and 3). The deletion of an adenine at position 2080 in the IGROV‐1 cell line, leads to a premature stop of protein translation at amino acid 700. However, IGROV‐1 is heterozygous not homozygous for the 2080delA BRCA1 mutation (Figure 1A) as previously reported (Foray et al., 1999). The germline genotype of BRCA1 in the patient who IGROV‐1 was derived from is unknown, so we cannot determine if this mutation is germline or somatic (Benard et al., 1985; Fajac et al., 1996). The immortalised ovarian epithelial cell line HI0180 (Sood et al., 2002) has a suspected deleterious mutation at IVS21+1G > C, the first base after exon 21. However, this mutation is also heterozygous.
Figure 1.

Sequence diagrams for BRCA1/2 deleterious mutations. (A) Top: SNU‐251 showing homozygous mutation W1815X (5564G > A) Bottom: BRCA1 wild‐type sequence. (B) Top: IGROV‐1 showing heterozygous deletion – 2080delA Bottom: BRCA1 wild‐type sequence. (C) Top: UPN‐251 showing homozygous deletions – 1199del29 + 1246delA reversion mutation. Bottom: BRCA1 wild‐type sequence.
The 1199del29 mutation in BRCA1 is defined as deleterious as it results in a frameshift causing a premature stop codon. UPN‐251 has the 1199del29 mutation however there is an additional mutation (1246delA), which restores the reading frame of the protein (Figure 1C). Therefore, despite having two mutations that would independently be deleterious, the UPN‐251 cell line likely has a functional BRCA1 due to the combination of the two mutations. We believe this is an example of a deleterious mutation, followed by a reversion mutation.
SNU‐251 has a homozygous deleterious mutation at 5564G > A which converts a tryptophan to a stop codon truncating the protein in exon 23. SNU‐251 was developed from malignant ascites fluid from a Korean patient with endometrioid ovarian carcinoma (Yuan et al., 1997). The germline BRCA1 status of this patient is unknown.
2.2. BRCA2 mutation screening
Sequencing of BRCA2 revealed 33 different alterations in the gene sequence among 43 human ovarian cell lines (Tables 3 and 4). Similar to BRCA1 the majority of alterations are nonpathogenic polymorphisms. Five novel mutations in BRCA2 were identified in this study (Table 3). 3624A > G, 4542C > T, 4680C > T are polymorphisms. 5192A > T and 5193C > T are reversion mutations correcting a deleterious mutation, explained below.
Table 4.
BRCA2 sequence variants among ovarian cell lines.
| Variant | Nucleotide change | Exon | Predicted protein effect | Type of variant | Number mutations in ovarian cell lines | No. in BIC |
|---|---|---|---|---|---|---|
| 1 | 5′UTR203G > A | n/a | No effect | Poly | 16 | 12 |
| 2 | 602A > G | 4 | D125G | Uncertain | 1 | 0 |
| 3 | IVS5‐9insT (in intron 5 upstream of exon 6) | 6 | No effect | Favour Poly | 1 | 2 |
| 4 | 1093A > C | 10 | N289H | Poly | 1 | 9 |
| 5 | 1342C > A | 10 | H372N | Poly | 16 | 9 |
| 6 | 1593A > G | 10 | S455S | Poly | 1 | 7 |
| 7 | 2457T > C | 10 | H743H | Poly | 1 | 3 |
| 8 | 3031G > A | 11 | D935N | Poly | 1 | 114 |
| 9 | 3199A > G | 11 | N991D | Poly | 3 | 3 |
| 10 | 3624A > G | 11 | K1132K | Poly | 18 | 0 |
| 11 | 4035T > C | 11 | V1269V | Poly | 14 | 3 |
| 12 | 4097G > A | 11 | C1290Y | Poly | 1 | 47 |
| 13 | 4542C > T | 11 | V1438V | Poly | 1 | 0 |
| 14 | 4680C > T | 11 | D1484D | Poly | 1 | 0 |
| 15 | 5192A > T | 11 | Y1655L | Poly | 1 | 0 |
| 16 | 5193C > G | 11 | Stop 1655 | Deleterious | 1 | 21 |
| 17 | 5193C > T | 11 | Y1655Y | Poly | 2 | 0 |
| 18 | 5427C > T | 11 | S1733S | Poly | 1 | 2 |
| 19 | 5579delA | 11 | Stop 1790 | Deleterious | 1 | 7 |
| 20 | 5932G > A | 11 | D1902N | Poly | 2 | 87 |
| 21 | 5972C > T | 11 | T1915M | Poly | 2 | 0 |
| 22 | 6575A > G | 11 | H2116R | Poly | 1 | 88 |
| 23 | 7470A > G | 14 | S2414S | Poly | 18 | 10 |
| 24 | 7829C > T | 15 | A2534V | Favour Poly | 1 | 5 |
| 25 | IVS16‐14C > T (in intron 16 upstream of exon 17) | 17 | None | Poly | 25 | 15 |
| 26 | 8688A > C | 19 | V2820V | Poly | 1 | 0 |
| 27 | 9145C > T | 22 | R2973C | Favour Poly | 1 | 14 |
| 28 | IVS24‐16T > C (intron 24 upstream of exon 25) | 25 | None | Poly | 3 | 6 |
| 29 | IVS26‐19G > A (intron 26 upstream of exon 27) | 27 | None | Poly | 1 | 0 |
| 30 | 10204A > T | 27 | Stop 3326 | Poly | 3 | 293 |
| 31 | 10323delCins11 | 27 | Stop 3369 | Poly | 1 | 11 |
| 32 | 10349C > T | 27 | T3374I | Poly | 1 | 8 |
| 33 | 10462A > G | 27 | I3412V | Poly | 4 | 110 |
Deleterious BRCA2 mutations were identified in 5 ovarian cell lines IOSE80, OC316, OVCA432, OVCA433 and PEO1 (Tables 3 and 4). The deletion of an adenine at position 5579 in the OC316 cell line, leads to a premature stop of protein translation at amino acid 1790. However, OC316 is heterozygous, not homozygous for the 5579delA BRCA2 mutation. The germline genotype of BRCA2 in the patient who OC316 was derived from is unknown (Alama et al., 1996). The deleterious mutations in IOSE80, OVCA432, and OVCA433 are also all heterozygous.
The PEO1 cell line has a BRCA2 homozygous mutation 5193C > G, which would normally result in a stop codon at amino acid 1655. However, the PEO1 cell line has another mutation 5192A > T which prevents the stop codon. We believe that this is an example of a reversion mutation. The cell lines PEO4 and PEO6 were established from the ascites fluid of the same patient as PEO1 after further treatment with chemotherapy (Langdon et al., 1988). The PEO4 and PEO6 cells have a different mutation at 5193, it is now a thymine and the 5192A > T mutation seen in PEO1 is gone. This appears to be a different reversion mutation at the same site.
2.3. LOH, methylation and QPCR
Loss of heterozygosity (LOH) was common at both the BRCA1 and BRCA2 locus (Table 3). Out of the 34 cell lines examined 15 had an LOH at BRCA1 and 10 had an LOH at BRCA2 (44% and 29% respectively). Of the 6 cell lines with heterozygous deleterious mutations, 5 were examined for LOH. All of these cell lines (IGROV‐1, IOSE80, OC316, OVCA432, and OVCA433) did not show LOH at the respective BRCA1/2 locus. Methylation was examined on the BRCA1 gene in all 41 cell lines and was found in 2 cell lines A1847 and OVCAR8 (Table 3). The methylated cell lines had a corresponding decrease in gene expression of BRCA1 (Figure 2). The deleterious mutated cell line SNU‐251 had similar BRCA1 gene expression to the wild‐type cell lines. The premature stop codon results from a single base change and this does not have an impact on the length of the mRNA produced.
Figure 2.
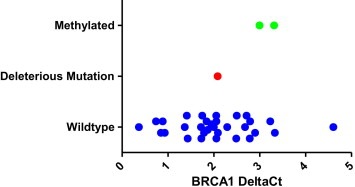
Quantitative PCR of BRCA1 in ovarian cell line panel. Wild‐type cells are shown with blue circles, methylated cells with green circles and the deleterious mutation with a red circle.
2.4. Cytotoxicity and growth assays
A panel of 20 cell lines was chosen to examine the relationship between BRCA1 mutation, methylation and resistance to PARP inhibition. The following cell lines were examined, deleterious mutation (SNU‐251), methylated (OVCAR8 and A1847) and 17 wild‐type cell lines. The IC50s of olaparib in the cell line panel is shown in Figure 3A. The methylated cell lines OVCAR8 and A1847 were more sensitive than the wild‐type cells on average to olaparib supporting the synthetic lethality theory between BRCA1 dysfunction and PARP inhibition (Figure 3B). Unexpectedly, SNU‐251, the mutated cell line, was the second most resistant to olaparib. The SNU‐251 cells were the slowest growing cell line in the panel tested with a doubling time of 3.32 ± 0.85 days (Figure 3C). In the 7‐day cytotoxicity assay the SNU‐251 cells only divide twice, the other cell lines divide 3–4 times. The methylated cell lines are on average slower growing than wild‐type cells but this difference was not significant (Figure 3D). To compensate for the slower growth rate the SNU‐251 cells were examined with a 2 week cytotoxicity assay. This longer exposure time gives an equivalent number of cell doublings to the rest of the cell panel in a 1 week assay. SNU‐251 with a 2 week exposure is relatively sensitive to olaparib (Figure 3A and B).
Figure 3.
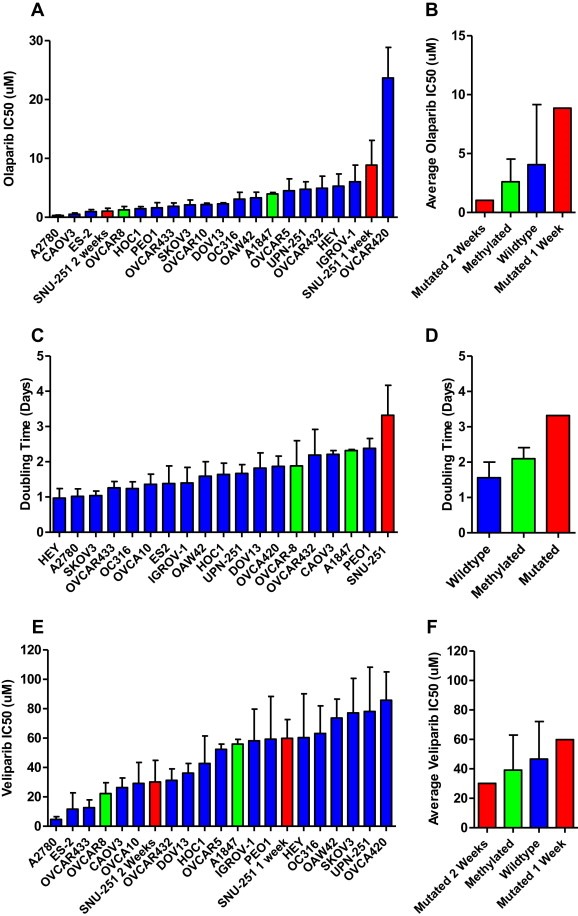
Cytotoxicity and doubling time in ovarian cell line panel. Wild‐type cells are shown with blue bars, methylated cells with green bars and the deleterious mutation with a red bar. A) Cytotoxicity of olaparib in 20 ovarian cancer cell lines. B) Average cytotoxicity of olaparib by cell and assay type. C) Doubling time of 19 ovarian cancer cell lines. D) Average doubling time by cell type. E) Cytotoxicity of veliparib in 20 ovarian cancer cell lines. F) Average cytotoxicity of veliparib by cell and assay type.
The results for veliparib were similar (Figure 3E); however a higher dose of veliparib was needed than olaparib to achieve an IC50 across the cell line panel. The IC50s of olaparib and veliparib are also correlated in the panel, Pearsons‐R 0.5393, p = 0.0311. The methylated cell lines OVCAR8 and A1847 were also on average more sensitive to veliparib than wild‐type cells (Figure 3F). SNU‐251 was also relatively resistant to veliparib in a 1 week cytotoxicity assay and more sensitive in a 2 week assay. Two cell lines were relatively sensitive to both olaparib and veliparib (A2780 and ES‐2), and OVCAR420 was highly resistant to both agents. However, the next most resistant cell lines were different for olaparib (OVCAR432, HEY, IGROV‐1) compared to veliparib (SKOV3, UPN‐251).
T‐tests were used to analyse if the clinical characteristics of the patients the cell lines were derived from were related to the response of the cell lines to parp inhibitors (Table 5). Cell lines derived from primary tumors were significantly more sensitive to veliparib (p = 0.03) than cells from metastases, including A2780 and ES‐2 cells. The response to olaparib was not significant as the cell line IGROV‐1 responded differently to A2780 and ES‐2 and was more resistant to Olaparib. Cell lines which were derived from patients after receiving chemotherapy tended to be more resistant to veliparib than cells derived after chemotherapy, 1.83‐fold p = 0.08. All other factors were non‐significant in this relatively small sample set.
Table 5.
Response of BRCA1/2 wild‐type, unmethylated cells to parp inhibitors based on patient characteristics of original tumors.
| Number of cell lines | Mean olaparib IC50 | Fold | T‐testp‐value | Mean veliparib IC50 | Fold | T‐test p‐value | |
|---|---|---|---|---|---|---|---|
| Primary tumour | 3 | 2.44 ± 3.14 | 24.82 ± 29.06 | ||||
| Metastatic disease | 6 | 2.92 ± 1.49 | 1.19 | 0.87 | 59.76 ± 11.12 | 2.00 | 0.03 |
| Chemotherapy naïvea | 4 | 2.96 ± 2.76 | 31.69 ± 27.42 | ||||
| Chemotherapy pre‐treateda | 7 | 2.91 ± 1.40 | 0.99 | 0.98 | 58.15 ± 17.71 | 1.83 | 0.08 |
| Responder to chemotherapy | 4 | 2.92 ± 1.79 | 59.00 ± 12.67 | ||||
| Non‐responder to chemotherapy | 4 | 2.74 ± 1.60 | 0.93 | 0.89 | 45.53 ± 30.48 | 0.77 | 0.45 |
| Serous histology | 6 | 3.50 ± 3.59 | 34.91 ± 32.84 | ||||
| Non‐serous histology | 2 | 3.03 ± 1.71 | 0.86 | 0.80 | 44.90 ± 20.04 | 1.28 | 0.61 |
| BRCA1/2+/+ | 15 | 2.29 ± 1.75 | 42.00 ± 25.99 | ||||
| BRCA1/2+/− | 4 | 3.98 ± 1.87 | 1.73 | 0.14 | 42.30 ± 23.68 | 1.01 | 0.96 |
| BRCA1/2−/− (reverted) | 2 | 3.18 ± 2.22 | 1.38 | 0.54 | 68.71 ± 13.32 | 1.63 | 0.20 |
Bold font indicates a significant p‐value, p < 0.05.
Prior to isolation of the cell line.
3. Discussion
When BRCA1/2 mutations are detected in cell lines they represent either a germline mutation or a somatic mutation in the tumour, or a combination of both. Currently in clinical practice BRCA1/2 mutations are screened for in the germline with a blood test. This test is more economical, does not require tumor tissue and addresses the issue of familial risk of breast and ovarian cancer. However, with the advent of PARP inhibitors as a potential targeted therapy for BRCA1/2 mutated cancer additional tumour testing for somatic mutations may become a new standard as a significant number of somatic mutations are present in ovarian cancers (Hennessy et al., 2010).
The results of this study show that deleterious BRCA1/2 mutations are rare in ovarian cancer cell lines, only 1/41 cell lines studied (2.4%). The clinical characteristics of the patients the ovarian cell lines were derived from is presented in Table 1. Five cell lines in the panel represent later stages of cancer progression in patients cell lines were already derived from (HOC‐7, PEA2, PEO4, PEO6, PEO14). Three cell lines are immortalised ovarian epithelial cells, HIO180, IOSE29 and IOSE80. Therefore, our panel of 41 cell lines represents 33 different ovarian cancer patients and the BRCA1/2 deleterious mutation rate per patient the cell lines were derived from is 1/33 (3.0%).
The histology of the cell line panel (Table 1) is reflective of clinical ovarian cancer; only one cell line is of non‐epithelial origin, PA‐1 which was derived from a germ‐cell tumour (Hills et al., 1989). Therefore 32/33 patients that the cell lines were derived from had epithelial ovarian cancer (97.0%), which is similar to the reported clinical rates of epithelial ovarian cancer (∼90.0%) (Colombo et al., 2009). Out of the patients where the subtype of epithelial ovarian cancer is known (20), serous dominates over other epithelial subtypes in the cell line panel 14/20 (70.0%), slightly lower than what is seen in the clinic (∼80–85%) (Colombo et al., 2010). This discrepancy is likely due to many cell lines being classified as adenocarcinoma, rather than into epithelial subgroups. Interestingly, although the majority of BRCA1 mutations occur in serous ovarian cancer (Berchuck et al., 1998; Lakhani et al., 2004) the SNU‐251 BRCA1 mutant is derived from an endometrial ovarian tumour (Yuan et al., 1997). The PEO1, PEO4 and PEO6 cell lines which have reversions of a BRCA2 mutation are of also of serous origin from the same patient (Langdon et al., 1988)
Deleterious BRCA1 mutations have also been shown to be rare in breast cancer cell lines, 3/41 (7.3%) (Elstrodt et al., 2006). However, due to the higher reported rates of deleterious BRCA1/2 mutations in unselected invasive ovarian versus unselected invasive breast cancer patients (11.7–15.3% versus 1.71%) (Pal et al., 2005; Papelard et al., 2000; Risch et al., 2001; Rubin et al., 1998), one would expect to see a higher number of cell lines with mutations in ovarian cancer when comparing unselected cell‐line panels of similar size. This is in contrast to what we have observed; this discrepancy may be in part due to random sampling, the cell models we have studied may have been derived more often from patients without BRCA1/2 mutations; or there may be factors selecting against these mutations during adaptation to cell culture.
Unusually, heterozygous mutations in BRCA1/2 were seen in 4 ovarian cancer cell lines studied (IGROV‐1, OC316, OVCAR432 and OVCAR433), representing a frequency of 4/33 (12.12%). Heterozygous mutations in BRCA1/2 were also seen in two of the immortalised ovarian cell lines IOSE80 and HI0180. LOH was studied in all of the heterozygous mutated cancer cell lines and no LOH were observed (Table 3). One explanation for why heterozygous BRCA1/2 mutations may be observed in a cancer cell line is the cancer patient the cells were derived from was a BRCA1/2 mutation carrier but a second mutation in BRCA1/2 was not part of the carcinogenesis of their tumour. Breast cancers in patients who are heterozygous for BRCA1/2 are rarely ER+. However, there is a subset of patients that are both (Foulkes et al., 2004; Tung et al., 2010b), these patients may be ER+ because their cancer occurred due to hormonal carcinogenesis rather than loss of the second BRCA1/2 allele. One study examined 77 breast cancer patients women with BRCA1 germline mutations, 12 tumours remained heterozygous (15.5%), and 8 of these were ER+ (Tung et al., 2010a). It appears that heterozygous BRCA1/2 mutations may be more common in breast cancer where the lifetime risk of sporadic disease is higher than ovarian cancer, 1 in 8 vs. 1 in 72 (National Cancer Institute. 2010). In contrast to our observations with cell lines, of the BRCA1/2 mutant ovarian tumours identified to date by Myriad Genetics, only ∼1/100 is heterozygous (Timms, 2011).
When BRCA1/2 mutated cells are treated with chemotherapeutics, one mechanism of resistance that can occur is a reversion of the deleterious mutation. This has been shown to occur in cancer cell lines (Sakai et al., 2008) as well as in cancer patients after treatment with platinum‐based chemotherapy (Swisher et al., 2008). This demonstrates that when the selective pressure of drug treatment is applied to BRCA1/2 mutated cells a common occurrence is a reversion of the mutation. The development of cell lines from tumour tissue is a process that involves multiple levels of selective pressure. Primary culture protocols involve physical disruption of the tissue, enzymatic digestion and selection of attached colonies in tissue culture. The more robust the cell, the more likely it is to survive the process. Cells without the full complement of DNA repair pathways, such as BRCA1/2 mutants may be at a disadvantage.
Loss of the second BRCA1/2 allele has also been shown to be heterogeneous within a tumour from BRCA1/2 mutation carriers (Martins et al., 2012). Mutations in BRCA1, PTEN and p53 were analysed at the single cell level in 55 BRCA1‐associated breast tumours. Based on the frequency of each mutation loss of PTEN was the most common ‘first’ carcinogenic event associated with a basal‐like subtype rather than BRCA1 (Martins et al., 2012). When a cell line is isolated from a heterogeneous tumour from a BRCA1/2 carrier cells with PTEN and p53 mutations could have been more robust in culture and BRCA1/2 mutated cells did not survive in the established cell line.
The heterozygous BRCA2 mutated cell line OC316 was derived from the malignant ascites of a cancer patient who had already received multiple rounds of chemotherapy (Alama et al., 1996) (Table 1). The metastasis and treatment with chemotherapy may have provided enough selective pressure for BRCA2 heterozygous cells to dominate the population. IGROV‐1 was derived from an untreated primary tumour (Benard et al., 1985) and its heterozygosity may be explained by an alternate path of tumorigenesis. IGROV‐1 is PTEN null (Cruet‐Hennequart et al., 2003) but p53 wild‐type (Casalini et al., 2001). It is possible that the primary tumour IGROV‐1 was derived from was a mixture of PTEN and BRCA1 mutant cells and the PTEN mutation survived the selective pressure of cell culture.
The reversion mutations in UPN‐251, PEO1, PEO4 and PEO6 are also evidence of a selective pressure against BRCA1/2 mutations. In the PEO4 and PEO6 cells, the reversions occurred after the patient received further chemotherapy (Langdon et al., 1988). In the PEO1 cells, we see an additional variation compared to that reported by Sakai et al. (2009), 5192 > T which has been previously reported by Stronach et al. and has been seen in other unselected stocks of PEO1 (Stronach et al., 2011). Our PEO1 cells were of relatively high passage number (p119) at the time of analysis for BRCA1/2 mutation. Stronach et al. hypothesise that 5192 > T exists as a sub‐dominant population within the original PEO1 line given that the same mutation has been reported to emerge independently either following selection with cisplatin (Sakai et al., 2009) or spontaneously following long term cell culture. It has also been shown that PEO4 and PEO6 are genetically diverse from PEO1, consistent with the selection of a pre‐existing resistant subclone rather than the transformation of PEO1 into PEO4/6 (Cooke et al., 2010). In the UPN‐251 cells, we don't know if these reversions occurred in vivo or in vitro. However, the UPN‐251 cells were derived from a patient who had failed frontline platinum/taxol as well as salvage taxol, which may have provided the selective pressure for the reversion (Hamilton, 2012).
The scientists who established SNU‐251 commented on the difficulty developing cell lines from ovarian cancer patients, their success rate was only 17% (Yuan et al., 1997). Another BRCA1‐mutated ovarian cell line (UWB1.289) was developed from a patient with a particularly aggressive cancer (DelloRusso et al., 2007). The authors of this study commented on the difficulty of establishing BRCA1 mutated cell lines in cell culture and speculate that the aggressiveness of this patient's tumour may have facilitated the propagation of this particular deleterious mutated cell line in vitro (DelloRusso et al., 2007).
An unselected breast cancer cell‐line panel was skewed towards the triple‐negative subgroup, 21/48 cell lines studied (43.75%) (Kao et al., 2009), higher than the incidence of triple negative breast cancer (TNBC) in unselected invasive breast cancer (12.4%) (Bauer et al., 2007). One would expect that BRCA1 mutations would be enriched in the panel as BRCA1 mutated tumours are common in TNBC (20.93%) (Merkel et al., 1989). However, BRCA1 mutations were relatively uncommon 2/14 (14.2%) of the TNBC cell lines examined (Kao et al., 2009). This suggests that the clinical aggressiveness of TNBC may make development of cell lines easier in the laboratory, but that the presence of BRCA1/2 mutations hinders the development of cell lines, resulting in a bias in the available cell lines.
The SNU‐251 BRCA1 deleterious mutated cell line was relatively resistant to PARP inhibition in a 1 week cytotoxicity assay (Figure 3A, E). This is the opposite of what was predicted by the synthetic lethality theory between PARP inhibition and BRCA1/2 dysfunction. The SNU‐251 cell line is likely to have many changes in its DNA repair mechanisms in addition to the BRCA1 mutation and more detailed study is needed to understand any DNA repair defects present. The deleterious mutation is also located near the end of the gene sequence and may have little impact on protein function. However, SNU‐251 has a very slow growth rate compared to the other cell lines in the panel (Figure 3C). One of the causes of toxicity from PARP inhibitors is the generation of DSBs at collapsed replication forks (Rouleau et al., 2010). It is possible that the slow growth rate masks the true sensitivity of the cell line as the cells do not go through as many cell divisions as their wild‐type counterparts. When SNU‐251 was examined with a 2‐week cytotoxicity assay their sensitivity to both olaparib and veliparib increases and they are more sensitive than wild‐type cells.
The rate of BRCA1 methylation in the cell line panel 2/33 different patients (6.0%) is similar to the observed rate clinically in ovarian tumours 12/98 (12.2%) (Baldwin et al., 2000). BRCA1 methylation status has been shown to be stable in recurrent ovarian cancers (Baldwin et al., 2000). The BRCA1‐methylated cell lines OVCAR8 and A1847 were more sensitive to PARP‐inhibition than wild‐type cell lines (Figure 3B, F). These cells also had a lower expression of BRCA1 mRNA (Figure 2). This data supports the synthetic lethality theory between PARP inhibition and BRCA1 dysfunction. This observation needs to be expanded upon but examining BRCA1 methylation may be relevant to predicting sensitivity to PARP inhibitors.
Two BRCA1/2 wild‐type unmethylated cell lines were relatively sensitive to both olaparib and veliparib (A2780 and ES‐2). This demonstrates that parp inhibitors may be useful for the treatment of a broader spectrum of patients with ovarian cancer. Cells lines derived from metastases were significantly more resistant to veliparib than cells derived from primary tumours (Table 5). This is consistent with the concept that pathways that govern invasion and metastasis overlap with drug resistance (Liang et al., 2002). It is interesting that this observation is only the case for veliparib and not olaparib, suggesting a divergent mechanism of resistance between the two parp inhibitors. Some of the most resistant cell lines were different for olaparib (OVCAR432, HEY, IGROV‐1) compared to veliparib (SKOV3, UPN‐251). One possible explanation for this difference in resistance mechanisms may be due to the expression of ABC transporters. There has been some evidence for olaparib being a P‐glycoprotein substrate in an animal model (Rottenberg et al., 2008) and for veliparib not being a P‐glycoprotein substrate in a transfected cell line (Li et al., 2011).
Some studies have suggested that BRCA1/2 heterozygosity has a distinct phenotype and is associated with increased sensitivity to DNA damaging agents (Warren et al., 2003). Other studies have shown no difference in DNA repair capacity in response to radiation in BRCA1/2 heterozygotes (Nieuwenhuis et al., 2002). In this study we found no difference in the response of the BRCA1/2+/− cell lines to the parp inhibitors olaparib and veliparib compared to wild‐type cells (Table 5).
4. Conclusions
We provide herein detailed BRCA1/2 status in a large panel of ovarian cancer cell lines to facilitate in vitro studies of parp inhibitors and other DNA damaging agents. The incidence of BRCA1/2 deleterious mutations in 1/41 cell lines, from 33 patients (3.0%) is much lower than the population incidence. The reversion mutations and high frequency of heterozygous mutations (14.6%) suggest that there is a selective pressure against BRCA1/2 mutations during adaption to cell culture similar to the selective pressure seen in the clinic after treatment with chemotherapy. PARP inhibitors may be useful in patients with BRCA1 deleterious mutations or gene methylation but further studies of this observation are needed. Some BRCA1/2 wild‐type cells are also sensitive to PARP inhibition, particularly those derived from primary tumors. Further research is needed to determine which patients who are BRCA1/2 wild‐type will benefit from treatment with PARP inhibitors.
5. Methods
5.1. Cell culture
Cell lines HOC1, HOC7, HOC8, and IGROV1 were grown in DMEM (Invitrogen, Grand Island, NY, USA # 11995) 10% FBS (Hyclone, Logan, Utah, USA #sv30014.03); OAW42 and CAOV3 were grown in DMEM 10% FBS with the addition of sodium pyruvate (Sigma St. Louis, MO, USA, #S8636), 20 ug/mL insulin (Invitrogen #12585‐014), l‐glutamine (Sigma #G7513) or NEAA (Sigma #M7145)/glucose respectively. FUOV1 was grown in DMEM:F12 (Invitrogen #11330) 10% FBS. OVCA420, OVCA432, OVCA433 were grown in EMEM (ATTC, Manassas, VA, USA, #30‐2003) 10% FBS; SW626 was grown in Leibovitz's L15 (ATTC #30.2008) 10% FBS with no CO2. IOSE29 and IOSE80 were grown in M199:MCDB105 (Invitrogen #11150, Sigma #M6395) 5% FBS. ES2 cells were grown in McCoy's 5a (Invitrogen #116600) 10% FBS with l‐glutamine. DOV13 were grown in MEM (Invitrogen #11095) 10% FBS with NEAA. EFO27 were grown in RPMI (ATCC #41458) 20% FBS with the addition of l‐glutamine, NEAA and Na Pyruvate; HTB121, was grown in RPMI 20% FBS with the addition of 10 μg/ml insulin. The remainder of cells were grown in RPMI‐1640 10% FBS, the following cell lines had additives 2 mM l‐glutamine (HEY, OVCA5, OVCAR3, OVCAR8, A2780, OC316); 2 mM l‐glutamine and 10 μg/ml insulin (SNU‐251and UPN‐251). All cell lines were fingerprinted in the MD Anderson CCSG supported cell line characterisation core to establish identity.
5.2. DNA extraction
DNA extractions were performed using the Qiagen QIAamp DNA mini kit “Appendix B: Protocol for Cultured cells” spin column protocol adding 0.4 mg RNaseA to each sample prior to the AL buffer step.
5.3. RNA extraction
All cell lines were grown in Advanced RPMI plus 3% FBS and 1% penicillin/streptomycin 24–48 h prior to harvesting cells. RNA was extracted using the Qiagen RNeasy mini kit “Animal cells spin” protocol using a QIAshredder for homogenisation and the optional on‐column DNase digestion.
5.4. BRCA1/2 analysis
BRCA1/2 mutation screening and qPCR was performed as previously published (Hennessy et al., 2010).
5.5. BRCA1 promoter methylation qPCR assays
The Methyl‐Profiler DNA Methylation PCR Array System (SA BioSciences) was used to quantify methylation levels following the manufacturers recommended protocol. DNA methylation‐sensitive and methylation‐dependent restriction enzymes were used to selectively digest unmethylated or methylated genomic DNA, respectively. Post‐digest DNA was quantified by real‐time PCR using primers flanking the regions of interest, BRCA1 (MePH28472‐1A). The relative concentrations of differentially methylated DNA are determined by comparing the amount of each digest with that of a mock digest.
5.6. Cytotoxicity‐proliferation assays
To determine the resistance to chemotherapy drugs, cells were plated into flat‐bottomed, 96‐well plates at the cell density shown of 1 × 103 cells/well and allowed to attach overnight. Slower growing cells (SNU‐251) were plated at 2 × 103 cells/well. Olaparib (AZD2281) and veliparib (ABT888) were purchased from Selleck Chemicals (Boston, MA, USA) and made up in DMSO. Wells were treated in triplicate with serial dilutions of drug in a final volume of 200 μL. Drug‐free controls were included in each assay. DMSO controls were also performed for each cell line. Plates were incubated for a further 5 days at 37 °C in a humidified atmosphere with 5% CO2 and cell viability was determined using an acid phosphatase assay (Martin and Clynes, 1993). For consistency, all cell lines for cytotoxicity and growth assays were growth in RPMI 10% FCS containing l‐glutamine and sodium bicarbonate (Sigma‐R8758). Insulin (10 μg/ml) was and Sodium pyruvate (1 mM) was added to SNU‐251. No antibiotics were used in the cell culture. The 2 week SNU‐251 cytotoxicity assays followed the same procedure, the media containing drug was changed after 1 week of incubation.
5.7. Growth assays
To determine the growth rate of each cell line 1 × 104 cells were plated in 2 ml of media in 6‐well plates. Duplicate wells were trypsinised and counted every day for 4 days. Data was graphed using Graphpad Prism and doubling time interpolated.
6. Conflict of interest
Authors KT, DW, JP, TT, are all employees and shareholders of Myriad Genetics. The remaining authors have no conflict of interests to declare.
Acknowledgements
This research was supported by an Irish Cancer Society Postdoctoral Fellowship (BS). It was also supported by a Science Foundation Ireland/Health Research Board Translational Research Award (BTH) and a Career Development Award from the Conquer Cancer Foundation (CCF) of the American Society of Clinical Oncology (BTH).
Stordal Britta, Timms Kirsten, Farrelly Angela, Gallagher Danielle, Busschots Steven, Renaud Mickaël, Thery Julien, Williams Deborah, Potter Jennifer, Tran Thanh, Korpanty Greg, Cremona Mattia, Carey Mark, Li Jie, Li Yang, Aslan Ozlem, O'Leary John J., Mills Gordon B., Hennessy Bryan T., (2013), BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation, Molecular Oncology, 7, doi: 10.1016/j.molonc.2012.12.007.
Contributor Information
Britta Stordal, Email: stordalb@tcd.ie.
Kirsten Timms, Email: ktimms@myriad.com.
Angela Farrelly, Email: angelafarrelly@rcsi.ie.
Danielle Gallagher, Email: daniellegallagher@rcsi.ie.
Steven Busschots, Email: busschos@tcd.ie.
Mickaël Renaud, Email: renaud.mickael@yahoo.fr.
Julien Thery, Email: julien-thery@orange.fr.
Deborah Williams, Email: dtrem@myriad.com.
Jennifer Potter, Email: jpotter@myriad.com.
Thanh Tran, Email: thanh@myriad.com.
Greg Korpanty, Email: greg.korpanty@gmail.com.
Mattia Cremona, Email: mattiacremona@rcsi.ie.
Mark Carey, Email: Mark.carey@vch.ca.
Jie Li, Email: jane.li@mdanderson.org.
Yang Li, Email: yli2@mdanderson.org.
Ozlem Aslan, Email: ozlemaslan@rcsi.ie.
John J. O'Leary, Email: olearyjj@tcd.ie
Gordon B. Mills, Email: gmills@mdanderson.org
Bryan T. Hennessy, Email: bryanhennessy74@gmail.com
References
- Anonymous, 2012. A2780 ECACC Datasheet http://www.hpacultures.org.uk [Google Scholar]
- Anonymous, 2012. CRL-1978™ (ES-2) ATCC Datasheet http://www.lgcstandards-atcc.org [Google Scholar]
- Anonymous, 2012. HTB-75™ (CAOV3) ATCC Datasheet http://www.lgcstandards-atcc.org [Google Scholar]
- Anonymous, 2012. HTB-78™ (SW626) ATCC Datasheet http://www.lgcstandards-atcc.org/ [Google Scholar]
- Anonymous, 2000. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Anglian breast cancer study group. British Journal of Cancer 83, 1301–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaronson, S.A. , 2012. Personal Communication. [Google Scholar]
- Alama, A. , Barbieri, F. , Favre, A. , 1996. Establishment and characterization of three new cell lines derived from the ascites of human ovarian carcinomas. Gynecologic Oncology 62, 82–88. [DOI] [PubMed] [Google Scholar]
- Audeh, M.W. , Carmichael, J. , Penson, R.T. , 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376, 245–251. [DOI] [PubMed] [Google Scholar]
- Baldwin, R.L. , Nemeth, E. , Tran, H. , 2000. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Research 60, 5329–5333. [PubMed] [Google Scholar]
- Bauer, K.R. , Brown, M. , Cress, R.D. , 2007. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype. Cancer 109, 1721–1728. [DOI] [PubMed] [Google Scholar]
- Benard, J. , Da Silva, J. , De Blois, M.C. , 1985. Characterization of a human ovarian adenocarcinoma line, IGROV1, in tissue culture and in nude mice. Cancer Research 45, 4970–4979. [PubMed] [Google Scholar]
- Berchuck, A. , Heron, K.A. , Carney, M.E. , 1998. Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clinical Cancer Research 4, 2433–2437. [PubMed] [Google Scholar]
- Bryant, H.E. , Schultz, N. , Thomas, H.D. , 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Buick, R.N. , Pullano, R. , Trent, J.M. , 1985. Comparative properties of five human ovarian adenocarcinoma cell lines. Cancer Research 45, 3668–3676. [PubMed] [Google Scholar]
- Casalini, P. , Botta, L. , Menard, S. , 2001. Role of p53 in HER2-induced proliferation or apoptosis. Journal of Biological Chemistry 276, 12449–12453. [DOI] [PubMed] [Google Scholar]
- Choi, J.H. , Choi, K.C. , Auersperg, N. , 2005. Gonadotropins upregulate the epidermal growth factor receptor through activation of mitogen-activated protein kinases and phosphatidyl-inositol-3-kinase in human ovarian surface epithelial cells. Endocrine-related Cancer 12, 407–421. [DOI] [PubMed] [Google Scholar]
- Colombo, N. , Peiretti, M. , Castiglione, M. , 2009. Non-epithelial ovarian cancer: ESMO Clinical Recommendations for diagnosis, treatment and follow-up. Annals of Oncology 20, iv24–iv26. [DOI] [PubMed] [Google Scholar]
- Colombo, N. , Peiretti, M. , Parma, G. , 2010. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Annals of Oncology 21, v23–v30. [DOI] [PubMed] [Google Scholar]
- Cooke, S.L. , Ng, C.K.Y. , Melnyk, N. , 2010. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene 29, 4905–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruet-Hennequart, S. , Maubant, S. , Luis, J. , 2003. [Alpha]v integrins regulate cell proliferation through integrin-linked kinase (ILK) in ovarian cancer cells. Oncogene 22, 1688–1702. [DOI] [PubMed] [Google Scholar]
- DelloRusso, C. , Welcsh, P.L. , Wang, W. , 2007. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Molecular Cancer Research: MCR 5, 35–45. [DOI] [PubMed] [Google Scholar]
- Elstrodt, F. , Hollestelle, A. , Nagel, J.H. , 2006. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Research 66, 41–45. [DOI] [PubMed] [Google Scholar]
- Emoto, M. , Oshima, K. , Ishiguro, M. , 1999. Establishment and characterization of a serous papillary adenocarcinoma cell line of the human ovary in a serum-free culture. Pathology – Research and Practice 195, 238–243. [PubMed] [Google Scholar]
- Fajac, A. , Da Silva, J. , Ahomadegbe, J.C. , 1996. Cisplatin-induced apoptosis and p53 gene status in a cisplatin-resistant human ovarian carcinoma cell line. International Journal of Cancer 68, 67–74. [DOI] [PubMed] [Google Scholar]
- Farmer, H. , McCabe, N. , Lord, C.J. , 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Filmus, J. , Trent, J.M. , Pullano, R. , 1986. A cell line from a human ovarian carcinoma with amplification of the K-ras gene. Cancer Research 46, 5179–5182. [PubMed] [Google Scholar]
- Filmus, J.E. , Buick, R.N. , 1985. Stability of c-K-ras amplification during progression in a patient with adenocarcinoma of the ovary. Cancer Research 45, 4468–4472. [PubMed] [Google Scholar]
- Foray, N. , Randrianarison, V. , Marot, D. , 1999. Gamma-rays-induced death of human cells carrying mutations of BRCA1 or BRCA2. Oncogene 18, 7334–7342. [DOI] [PubMed] [Google Scholar]
- Foulkes, W.D. , Metcalfe, K. , Sun, P. , 2004. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer. Clinical Cancer Research 10, 2029–2034. [DOI] [PubMed] [Google Scholar]
- Furlong, M.T. , Hough, C.D. , Sherman-Baust, C.A. , 1999. Evidence for the colonic origin of ovarian cancer cell line SW626. Journal of the National Cancer Institute 91, 1327–1328. [DOI] [PubMed] [Google Scholar]
- Hamilton, T.C. , 2012. Personal Communication. [Google Scholar]
- Hamilton, T.C. , Young, R.C. , McKoy, W.M. , 1983. Characterization of a human ovarian carcinoma cell line (NIH: OVCAR-3) with androgen and estrogen receptors. Cancer Research 43, 5379–5389. [PubMed] [Google Scholar]
- Helleday, T. , 2011. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Molecular Oncology 5, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy, B.T. , Timms, K.M. , Carey, M.S. , 2010. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. Journal of Clinical Oncology 28, 3570–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills, C.A. , Kelland, L.R. , Abel, G. , 1989. Biological properties of ten human ovarian carcinoma cell lines: calibration in vitro against four platinum complexes. British. Journal of Cancer 59, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, J. , Salari, K. , Bocanegra, M. , 2009. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One 4, e6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzmann, R. , Hozel, F. , 1987. Karyotype alterations in human ovarian carcinoma cells during long-term cultivation and nude mouse passage. Cancer Genetics and Cytogenetics 28, 201–212. [DOI] [PubMed] [Google Scholar]
- Lakhani, S.R. , Manek, S. , Penault-Llorca, F. , 2004. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clinical Cancer Research 10, 2473–2481. [DOI] [PubMed] [Google Scholar]
- Langdon, S.P. , Lawrie, S.S. , Hay, F.G. , 1988. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Research 48, 6166–6172. [PubMed] [Google Scholar]
- Li, X. , Delzer, J. , Voorman, R. , 2011. Disposition and drug-drug interaction potential of veliparib (ABT-888), a novel and potent inhibitor of poly(ADP-ribose) polymerase. Drug Metabolism and Disposition 39, 1161–1169. [DOI] [PubMed] [Google Scholar]
- Liang, Y. , McDonnell, S. , Clynes, M. , 2002. Examining the relationship between cancer invasion/metastasis and drug resistance. [Review] [134 refs] Current Cancer Drug Targets 2, 257–277. [DOI] [PubMed] [Google Scholar]
- Mackillop, W.J. , Trent, J.M. , Stewart, S.S. , 1983. Tumor progression studied by analysis of cellular features of serial Ascitic ovarian carcinoma tumors. Cancer Research 43, 874–878. [PubMed] [Google Scholar]
- Martin, A. , Clynes, M. , 1993. Comparison of 5 microplate colorimetric assays for in vitro cytotoxicity testing and cell proliferation assays. Cytotechnology 11, 49–58. [DOI] [PubMed] [Google Scholar]
- Martins, F.C. , De, S. , Almendro, V. , 2012. Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discovery 2, 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel, D.E. , Fuqua, S.A. , Tandon, A.K. , 1989. Electrophoretic analysis of 248 clinical breast cancer specimens for P-glycoprotein overexpression or gene amplification. Journal of Clinical Oncology 7, 1129–1136. [DOI] [PubMed] [Google Scholar]
- National Cancer Institute, 2010. EER Cancer Statistics Review 1975–2007. Lifetime Risk (Percent) of Dying from Cancer by Site and Race/Ethnicity: Males, Total US, 2005–2007 (Table 1.18) and Females, Total US, 2005–2007 (Table 1.19) http://seer.cancer.gov/csr/1975_2007/results_merged/topic_lifetime_risk_death.pdf [Google Scholar]
- Newman, B. , Mu, H. , Butler, L.M. , 1998. Frequency of breast cancer attributable to BRCA1 in a population-based series of American women. JAMA 279, 915–921. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuis, B. , Assen-Bolt, A.J.V. , Van Waarde-Verhagen, W.H. , 2002. BRCA1 and BRCA2 heterozygosity and repair of X-ray-induced DNA damage. International Journal of Radiation Oncology Biology 78, 285–295. [DOI] [PubMed] [Google Scholar]
- Pal, T. , Permuth-Wey, J. , Betts, J.A. , 2005. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104, 2807–2816. [DOI] [PubMed] [Google Scholar]
- Papelard, H. , De Bock, G.H. , van Eijk, R. , 2000. Prevalence of BRCA1 in a hospital-based population of Dutch breast cancer patients. British Journal of Cancer 83, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl, G. , Ho, C.L. , Kurman, R.J. , 2005. Inactivation of the mitogen-activated protein kinase pathway as a potential target-based therapy in ovarian serous tumors with KRAS or BRAF mutations. Cancer Research 65, 1994–2000. [DOI] [PubMed] [Google Scholar]
- Risch, H.A. , McLaughlin, J.R. , Cole, D.E. , 2001. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. American Journal of Human Genetics 68, 700–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg, S. , Jaspers, J.E. , Kersbergen, A. , 2008. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences U S A 105, 17079–17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau, M. , Patel, A. , Hendzel, M.J. , 2010. PARP inhibition: PARP1 and beyond. Nature Reviews Cancer 10, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin, S.C. , Blackwood, M.A. , Bandera, C. , 1998. BRCA1, BRCA2, and hereditary nonpolyposis colorectal cancer gene mutations in an unselected ovarian cancer population: relationship to family history and implications for genetic testing. American Journal of Obstetrics and Gynecology 178, 670–677. [DOI] [PubMed] [Google Scholar]
- Russell, P.A. , Pharoah, P.D. , De Foy, K. , 2000. Frequent loss of BRCA1 mRNA and protein expression in sporadic ovarian cancers. International Journal of Cancer 87, 317–321. [DOI] [PubMed] [Google Scholar]
- Sakai, W. , Swisher, E.M. , Karlan, B.Y. , 2008. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. [see comment] Nature 451, 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, W. , Swisher, E.M. , Jacquemont, C. , 2009. Functional Restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Research 69, 6381–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilder, R.J. , Hall, L. , Monks, A. , 1990. Metallothionein gene expression and resistance to cisplatin in human ovarian cancer. International Journal of Cancer 45, 416–422. [DOI] [PubMed] [Google Scholar]
- Sikic, B.I. , 2012. Personal Communication. [Google Scholar]
- Sood, A.K. , Fletcher, M.S. , Gruman, L.M. , 2002. The paradoxical expression of maspin in ovarian carcinoma. Clinical Cancer Research 8, 2924–2932. [PubMed] [Google Scholar]
- Stronach, E.A. , Alfraidi, A. , Rama, N. , 2011. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Research 71, 4412–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher, E.M. , Sakai, W. , Karlan, B.Y. , 2008. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Research 68, 2581–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed, V. , Ulinski, G. , Mok, S.C. , 2001. Expression of gonadotropin receptor and growth responses to key reproductive hormones in normal and malignant human ovarian surface epithelial cells. Cancer Research 61, 6768–6776. [PubMed] [Google Scholar]
- Timms, K.M. , 2011. Personal Communication. [Google Scholar]
- Tung, N. , Miron, A. , Schnitt, S. , 2010. Prevalence and predictors of loss of wild type BRCA1 in estrogen receptor positive and negative BRCA1-associated breast cancers. Breast Cancer Research 12, R95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung, N. , Wang, Y. , Collins, L. , 2010. Estrogen receptor positive breast cancers in BRCA1 mutation carriers: clinical risk factors and pathologic features. Breast Cancer Research 12, R12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutt, A. , Robson, M. , Garber, J.E. , 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. The Lancet 376, 235–244. [DOI] [PubMed] [Google Scholar]
- Warren, M. , Lord, C.J. , Masabanda, J. , 2003. Phenotypic effects of heterozygosity for a BRCA2 mutation. Human Molecular Genetics 12, 2645–2656. [DOI] [PubMed] [Google Scholar]
- Wong, A.S.T. , Roskelley, C.D. , Pelech, S. , 2004. Progressive changes in Met-dependent signaling in a human ovarian surface epithelial model of malignant transformation. Experimental Cell Research 299, 248–256. [DOI] [PubMed] [Google Scholar]
- Wong, W.S.F. , Wong, Y.F. , Ng, Y.T.A. , 1990. Establishment and characterization of a new human cell line derived from ovarian clear cell carcinoma. Gynecologic Oncology 38, 37–45. [DOI] [PubMed] [Google Scholar]
- Young, T.N. , Pizzo, S.V. , Stack, M.S. , 1995. A plasma membrane-associated component of ovarian adenocarcinoma cells enhances the catalytic efficiency of matrix metalloproteinase-2. Journal of Biological Chemistry 270, 999–1002. [DOI] [PubMed] [Google Scholar]
- Yuan, Y. , Kim, W.H. , Han, H.S. , 1997. Establishment and characterization of human ovarian carcinoma cell lines. Gynecologic. Oncology 66, 378–387. [DOI] [PubMed] [Google Scholar]
- Yu, J. , Roy, D. , Brockmeyer, A.D. , 2007. Increased chromosomal stability in cultures of ovarian tumours of low malignant potential compared to cystadenomas. British Journal of Cancer 96, 1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]