

Abstract
Small molecule inhibitors (SMIs) of MDM2 are known to restore the apoptotic and cell cycle regulatory functions of p53 by disrupting the MDM2-p53 interaction. In principle, these SMIs are not effective against tumours with mut-p53, which is known to be present in approximately 50% of all cancers. In this study we are reporting, for the first time, that MI-319 in combination with cisplatin induced cell growth inhibition and apoptosis in pancreatic cancer (PC) cells irrespective of their p53 mutational status. MI-319-cisplatin combination synergistically suppressed cell growth (MTT CI<1) and colony formation (clonogenic assay) and induced apoptosis. Western blot analysis and siRNA silencing studies in mutant as well as p53 null cells highlighted a mechanism involving p73 which is also known to be under the regulation of MDM2, and unlike p53, it is rarely mutated in PC. Down regulating MDM2 using siRNA enhanced p73 reactivation and increased cell death. Further, the combination effectively reduced tumour growth in both wt-p53 and mut-p53 tumour xenograft models (50% Capan-2 animals were tumour free). Consistent with our in vitro results, remnant tumour tissue analysis showed up-regulation of p73 and the cell cycle regulator p21. In conclusion, this study highlights a new role of MDM2 inhibitors in combination with cisplatin, and thus warrants further clinical investigation in human pancreatic tumours containing both wt-p53 and mut-p53.
Keywords: MDM2 and p53, Small molecule inhibitors, Cisplatin, Apoptosis, Pancreatic cancer
Introduction
PC is a deadly disease and is considered to be among the most intractable malignancies as it is refractory to conventional chemotherapeutics and radiation 1. Among the various genetic alterations in PC, mutation in the tumour suppressor p53 (mut-p53) gene has been reported to be about 50% 2. In the remaining 50%, the p53 is normal (wild type), but its function is inhibited by MDM2 (murine double minute 2) protein whose key role is to promote ubiquitination and proteasomal-dependent degradation of p53 3. Therapies that are based on restoring p53 function by blocking MDM2 using small molecule inhibitors have by far been effective in inducing growth arrest and apoptosis in cells in culture as well as in different animal model system via activation of wt-p53 pathway 4–6. Our laboratory has been working on a new class of MDM2 inhibitors (MI-319 and MI-219) and assessing its effects on PC showing induction of cell growth inhibition, apoptosis and tumour growth arrest. However, the therapeutic applicability of blocking MDM2 by such inhibitors including Nutlin-3 has been focused on its activity in wt-p53 tumours and very modest information is available on the role of other MDM2 regulated proteins such as the role of p73 especially in PC. Interestingly, recent evidences suggest that all p53 family proteins may cooperate in preventing tumour formation and this is proven by the observation that p63- and p73-deficient cells are resistant to p53-induced apoptosis 7–9.
P73 appears to be an important protein, which is also under the influence of MDM2 and shares a high degree of sequence homology to p53 10. Although p73 has been implicated in developmental biology, there is ample evidence suggesting that, at least in part, the function of p73 may closely resemble that of p53 11;12. Indeed, like p53, p73 induces G1 cell growth arrest 13, activates the transcription of some endogenous p53 target genes such as p21Waf1/Cip1, mdm2, bax, cyclin G 14–16, and induces apoptosis irrespective of p53 status 1;4. Most significantly, p73 has been shown to be a key determinant of cellular sensitivity to anticancer therapeutics, and is widely induced by chemotherapeutic agents in a variety of tumour cell lines particularly in tumours lacking p53 17. Several studies have shown that p73 is activated by DNA-damaging agents, including anthracyclines, topoisomerase I inhibitors and cisplatin 18–21. Importantly, not all cells demonstrate activation of p73 in response to each of these DNA-damaging agents, and thus the p73-dependent response to drugs is likely dependent on the cellular context including the presence or absence of functional p53. Among chemotherapeutic agents, cisplatin, docetaxel (Taxotere), and doxorubicin (Adriamycin) have been frequently used for treatment of cancers including prostate, breast, lung, and pancreatic cancers, alone or in combination with other agents 22–26. Several clinical trials have reported that these agents, used in combination with other drugs, show improved outcomes in objective response rates and survival in pancreatic cancer (El-Reyes and Philip 2003) 27. Since both p53 and p73 have been implicated in cell response to cisplatin, the aim of the present study was to explore role of p73 in MDM2 inhibitor-cisplatin mediated anti-tumour effects in PC cells lacking functional p53 compared to PC cells having wt-p53.
Materials and Methods
Cell culture, experimental reagents and chemicals
Human PC cell lines Capan-2, BxPC-3, Colo-357 and Panc-28 were purchased from ATCC. The cell lines have been tested and authenticated in core facility Applied Genomics Technology Center at Wayne State University on March 13, 2009. The method used for testing was short tandem repeat (STR) profiling using the PowerPlex® 16 System from Promega (Madison, WI). Primary antibodies for p53, p73 and p21 were purchased from Cell Signaling. All secondary antibodies were obtained from Sigma (Saint Louis). MI-219 and MI-319 were synthesized by using our previously published methods 28;29.
Cell growth inhibition studies by MTT assay
The cells Capan-2, Colo-357, BxPC-3 and Panc-28 (3 × 103) were seeded in a 96-well culture plate and treated with MI-319 (0 or 15 μM) or cisplatin (1 μM) or combination of both for 72 hrs and MTT assay was done as described earlier 30. The results were plotted as means ± SD of three separate experiments having six determinations per experiment for each experimental condition. Clonogenic assay for cell survival on Colo-357, HPAC and Capan-2 was performed according to previously described methods 31.
Trypan blue exclusion test
Panc-28, Colo-357 and Capan-2 cells were treated with wither MI-319 (15 μM), cisplatin (1μM) or their combination for 24 hrs. On completion of incubation, viability was assessed after adding 50 μL trypan blue solution (0.4% in PBS) in culture medium.
siRNA and transfections
The p73 siRNA, p21WAF1 siRNA, MDM2 siRNA and control siRNA were obtained from Cell Signaling. Colo-357 cells were transfected with respective siRNAs for 5 hrs using LipofectAMINE 2000 as described in the manufacturers protocol (Cell Signaling).
Quantification of apoptosis by annexin V FITC flow cytometry and ELISA
The cell apoptosis in Capan-2, Colo-357, BxPC-3 and Panc-28 post MI-319 (15 μM) alone; cisplatin (1 μM) alone or their combination treatment (for 72 hrs) was determined using Annexin V FITC apoptosis kit (Biovision Research Products) and ELISA detection kit (Roche, Palo Alto, CA) according to manufacturer’s protocol.
Western blot analysis
Panc-28, Colo-357, Capan-2 and BxPC-3 cells were treated with either MI-319 (15 μM) or cisplatin (1μM) or their combination for 20 hrs followed by extraction of protein for western blot analysis. Procedure for cells lyses, protein concentration determination and SDS-PAGE analysis has been described in our previous publication 30.
Animal Preclinical Efficacy Trail Design
All in vivo studies were conducted in accordance with Wayne Sate University approved animal care and ethics committee guidelines and procedures. Capan-2 and BxPC-3 xenograft were generated using our well established methods 30. To ensure randomness, 32 animals that were transplanted bilaterally with 30 mg tumour fragments (one week earlier) were pooled in a single cage. 4 groups, each containing 8 animals were assigned as follows; Control (Vehicle only), MI-319 treated 200mg/Kg orally twice a day for three weeks, Cisplatin 4 mg/kg (i.v.) twice a week for two weeks-treated and combination (MI-319 200 mg/Kg orally + Cisplatin 4 mg/kg). Tumour weight was recorded throughout the treatment period using previously described methods 30. At the end of the treatment period, animals were euthanized and their tumours harvested for protein isolation and western blot analysis.
Statistical analysis
Statistics was evaluated using GraphPad StatMate software (GraphPad Software, Inc.). Comparisons were made between control and treated groups and transfections. P < 0.05 or P < 0.01 was used to indicate statistical significance.
Results
MI-319 mediated effects on PC cells were enhanced by cisplatin in reducing cell viability and inhibition of cell growth/survival irrespective of p53 function
The combination studies of MI-319 with cisplatin have never been done on PC cells with mut-p53, we therefore tested whether MI-319 could synergize with cisplatin leading to enhanced suppression of cell viability and survival as assessed by trypan blue, MTT and clonogenic assays. As can be seen from results of Figure 1 A in Panc-28 and colo-357 cells MI-319 or cisplatin (at 15 μM and 1 μM respectively) alone did not induce any appreciable loss of cell viability (only 10–15% in Panc-28 and Colo-357). However in the combination we observed drastic growth inhibition (greater than 60%). As expected capan-2 that is wt-p53 was responsive to MI-319 alone at the concentrations tested and the combination resulted in even more pronounced loss of viability. We then tested growth inhibition using MTT assay and our results presented in Figure 1B clearly show that MI-319 alone or cisplatin alone do not show appreciable inhibition of cell viability (except for Capan-2 which contains wt-p53). However, in the combination group, we observed more pronounced suppression of cell viability, and isobologram analysis revealed a synergistic combination effect between MI-319-cisplatin (Capan-2 CI=0.44; Colo-357 CI=0.43; BxPC-3 CI=0.84 and Panc-28 CI=0.64) (Figure 1 B lower panel).
Figure 1. MI-319-cisplatin combination induces cell growth inhibition in PC cells irrespective of p53 functional status.
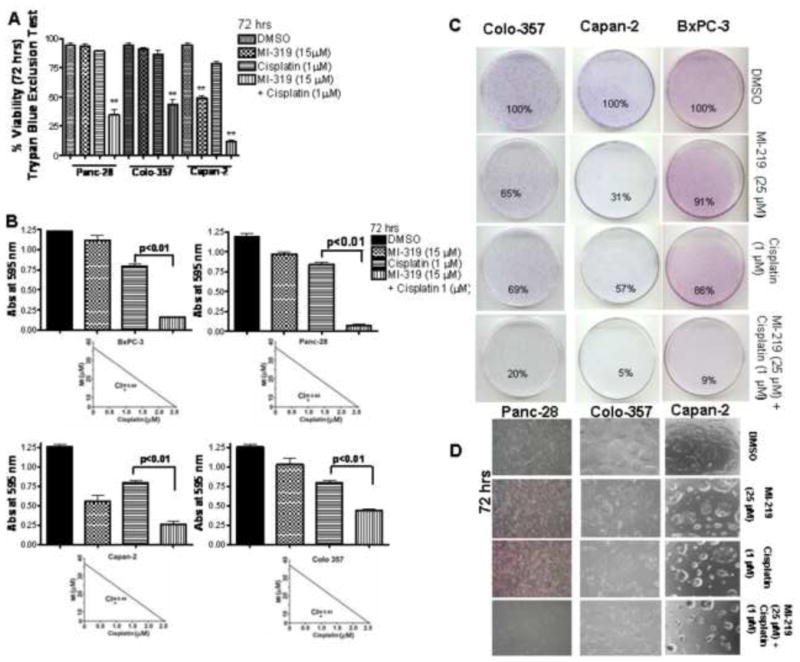
A. Trypan blue exclusion assay for loss of viability in Panc-28, Colo-357 and Capan-2 cells treated for 72 hrs at indicated concentrations. B. Evaluation of effect of MI-319-cisplatin combination on cell viability by MTT assay in BxPC-3, Panc-28, Capan-2 and Colo-357 cells after 72 hr treatment at indicated concentrations. Lower Panels Isobologram analysis of MI-319-cisplatin combination. (CI< 1 is considered synergistic). C. Microphotographs of cell survival of PC cell lines (Colo-357, BxPC-3 and Capan-2) at indicated treatments and evaluated by the clonogenic assay. In all the cell lines tested there was a significant reduction in the colony formation in the combination compared to cells treated with either drug alone. D. Microphotograph of Colo-357, Capan-2 and BxPC-3 cells post indicated treatments for 72 hrs. *, P < 0.05; **, P < 0.01.
In order to further determine the effect of MI-319 and cisplatin on cell growth, we performed clonogenic assay. The combination of MI-319 and cisplatin resulted in a significant inhibition of colony formation in Colo-357, Capan-2 and BxPC-3 cells when compared with either agent alone (Figure 1 C). Further, microphotographs were taken post MI-319, cisplatin or combination treatment which also showed similar trend (Figure 1D). The consistent results from trypan blue, MTT and clonogenic assay data as shown in Fig. 1A, B, C and D confirm that MI-319 in combination with cisplatin is effective against PC cells irrespective of p53 function. We then studied the combination effects of MI-319 and cisplatin on apoptotic cell death in Capan-2, Colo-357, BxPC-3 and Panc-28 cells.
Induction of Apoptosis by MI-319, cisplatin and the Combination
The underlying mechanism on the inhibition of cell viability was investigated by determining apoptosis. Results of Histone DNA ELISA concur with our earlier data, and we found that MI-319 (15 μM at 72 hrs) alone induces apoptosis only in Capan-2 cells (Figure 2A; Left panel) and not in BxPC-3, Panc28 or Colo-357 cells. However, when combined with cisplatin, we found synergistic induction of apoptosis in all the cell lines tested (CI<1 in all cases Fig 2A; right panels). We also verified apoptosis by Annexin V FITC flow cytometry assay and found similar results as shown in Fig 2B. Subsequently, we sought to find the molecular mechanism for the induction of apoptosis Panc-28, Colo-357, Capan-2 and Colo-357 cells as presented below. It is interesting to note that MI-319 also synergized with oxaliplatin that resulted in enhanced growth inhibition and apoptosis in the combination treatment in mut-p53 PC. However, this manuscript focuses on MI-319-cisplatin combination.
Figure 2. MI-319-cisplatin combination induces apoptosis in PC cells.
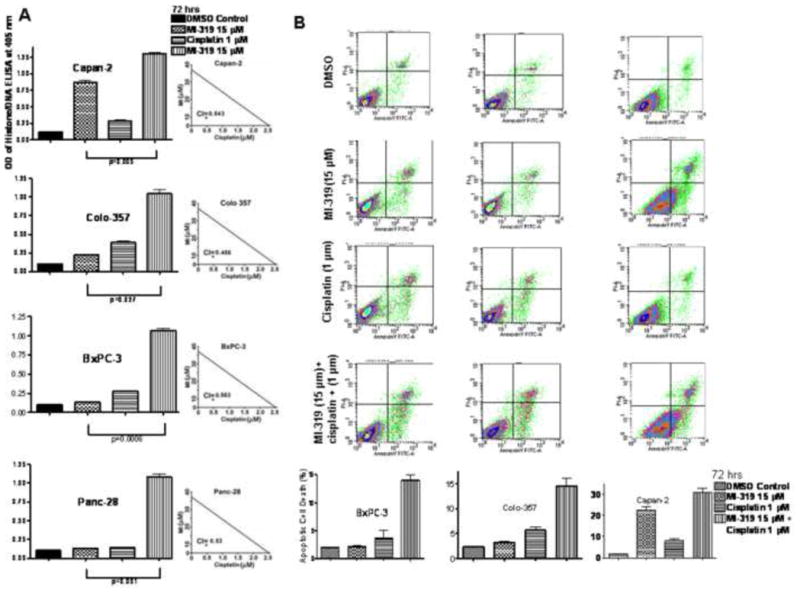
A. (Left Panel) Histone DNA/ELISA analysis of Capan-2, Colo-357, BxPC-3 and Panc-28 cells post indicated treatments for 72 hrs. (Right Panel) Isobologram analysis of MI-319-cisplatin combination (CI<1 synergistic). B. (Upper panel) Apoptosis analysis by Annexin V FITC flow cytometry analysis at 72 hrs under indicated treatments in BxPC-3, Colo-357 and Capan-2 cells. (Lower panel) Representative % apoptosis by MI-319-cisplatin combination.
MI-319-cisplatin combination activates p73 in cells with dysfunctional p53
In principle, MDM2 inhibitors are not effective in tumours with dysfunctional p53. This is proven by our results as we do not find any cell growth inhibition and apoptosis with MI-319 alone in all the cell lines tested (BxPC-3, Colo-357 and Panc-28). Therefore, mechanism behind the observed cell growth inhibitory and apoptotic activity of the MI-319-cisplatin combination was further investigated using Western blot analysis. As can be seen from the results, MI-319 induces the MDM2 regulated p73 in cells with mut-p53, and that the induction of p73 was further enhanced in the combination treatment with cisplatin (Figure 3A). The p21WAF1 is a key cell cycle regulator which is under the transcriptional control of p53 family members of proteins, and we found that p21WAF1 was up-regulated in the combination treatment group. Further, we also used siRNA to down regulate MDM2 and see the effect on p73. As expected siRNA silencing of MDM2 resulted in upregulation of p73 (Figure 3B). This proved that the observed upregulation of p73 was indeed a consequence of MDM2 downregulation by MI-319. These results clearly show that MDM2 inhibitor can reactivate p73, which is responsible for cell growth inhibition and induction of apoptosis induced by MI-319 and cisplatin. We subsequently tested whether blocking p73 using siRNA could abrogate the cell growth inhibitory and apoptotic inducing potential of MI-319-cisplatin combination treatment.
Figure 3. MI-319-cisplatin combination induces p73 in cells with functional and non functional p53.
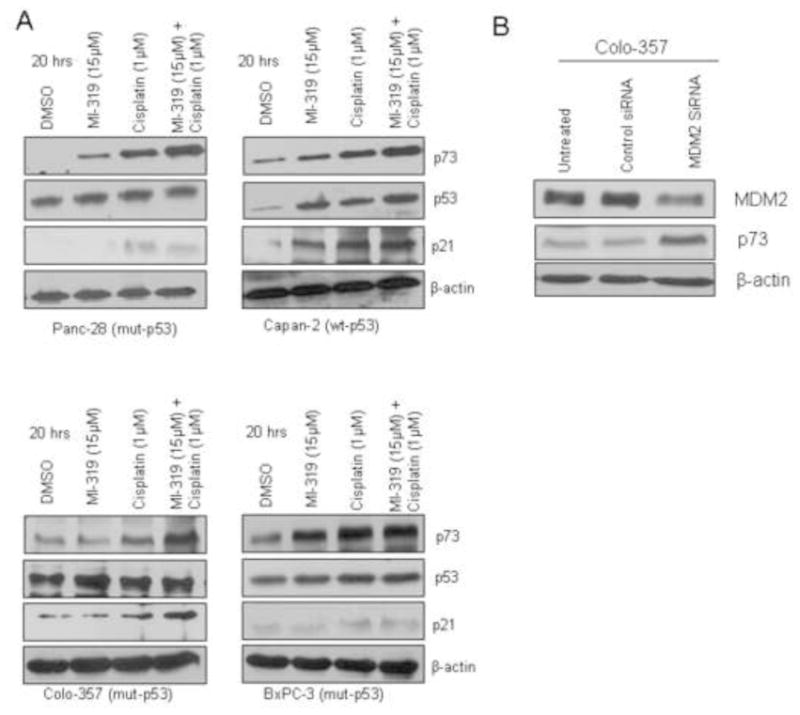
A. Protein levels of p73, p53 and p21 in Panc-28, Capan-2, Colo-357 and BxPC-3 cells detected by western blot analysis post 20 hrs at indicated treatments. B Western blot analysis of lysates of Colo-357 cells were treated with either DMSO, control siRNA or MDM2 siRNA for 5 hrs. Note MDM2 downregulation results in p73 activation.
siRNA against p73 and p21 blocks apoptotic activity of MI-319-cisplatin in mut-p53 Colo-357 cells
To verify the role of p73 and downstream cell cycle regulator p21 in the induction of cell growth inhibition and apoptosis by MI-319-cisplatin combination, we first incubated mut-p53 Colo-357 cells with p73 or p21 siRNA for 5 hours followed by incubation with MI-319-cisplatin combination and assayed cell growth inhibition and apoptosis using MTT, Histone DNA ELISA and Annexin V FITC assay. As can be seen from results of Figure 4 A growth inhibition by MI-319-cisplatin combination could be abrogated by p73 and p21 siRNA.
Figure 4. siRNA against p73 and p21 abrogates MI-319-cisplatin mediated cell growth inhibition and apoptosis.
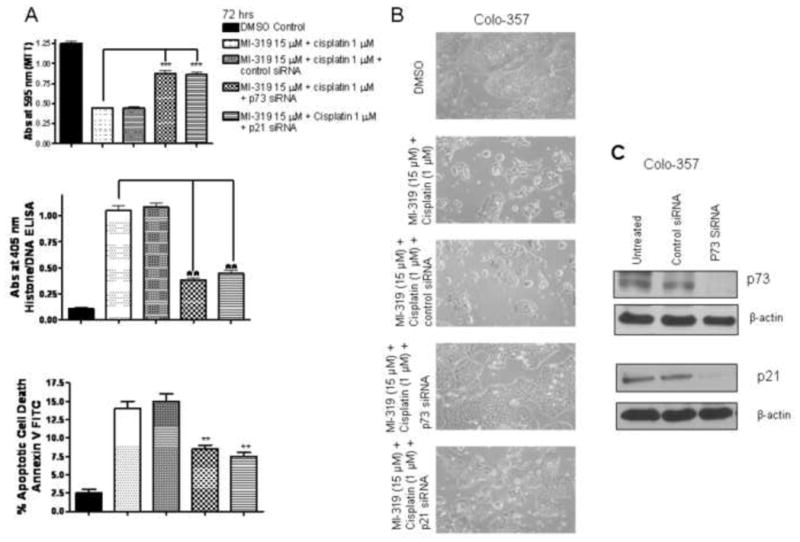
A. Cell growth inhibition and apoptosis analysis by MTT, histone DNA/ELISA and Annexin V FITC flow cytometry analysis of Colo-357 cells treated with DMSO; MI-319 15 μMol/L + cisplatin 1 μMol/L combination; combination + control siRNA; combination + p73 siRNA and combination + p21 siRNA. B. Microphotograph of Colo-357 cells treated for 72 hrs under similar conditions and C. Western blot analysis showing down regulation of p73 and p21 post respective siRNA treatment in Colo-357 cells. *** P <0.01; ** P < 0.05
Furthermore, histone DNA ELISA and Annexin V FITC flow cytometry confirmed that indeed treatment of cell with p73 or p21 siRNA could block the apoptotic potential of MI-319-cisplatin combination (Figure 4A middle and lower panels). Further, microphotograph of Colo-357 cells were in line with MTT and apoptosis results of Colo-357 where we observed enhanced cell death in combination group which was suppressed by siRNA treatment. To verify p73 and p21 downregulation by siRNA, western blot analysis was performed on lysates from respective treatments and as can be seen from Figure 4C indeed both p73 and p21 were effectively down regulated post siRNA treatment. In order to gain further insight and provide direct proof in support of p53 independent mechanism of action of the combination of MI-319 and cisplatin, we tested the effect of MI-319-cisplatin combination in a non-pancreatic cancer cell line lacking p53 (HCT116−/− obtained from Dr. Vogelstein at Johns Hopkins). As can be seen from MTT, Histone/DNA ELISA and annexin V FITC apoptosis results on HCT-116 (p53 null) cells post p73 siRNA presented in Figure 5 A, the combination was effective in inducing growth inhibition and apoptosis but was ineffective in the p73 siRNA treated groups. Western blot analysis confirmed up-regulation of p73 and p21 by MI-319, cisplatin and combination treatment. Together, these results provided direct evidence, for the first time, showing that p73 is involved in cell growth inhibition and induction of apoptosis induced by MI-319-cisplatin combination treatment but not p53. We subsequently tested whether this combination could work in animal xenograft models induced by both Capan-2 and BxPC-3 cells.
Figure 5. MI-319-cisplatin induces cell growth inhibition and apoptosis in p53 null cells through activation of p73 pathway.
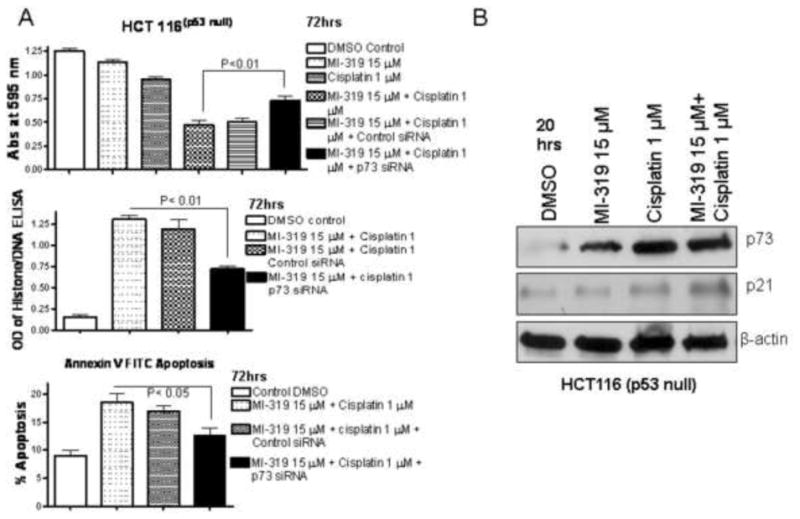
A. HCT116 (p53 null) cells were exposed to indicated treatments. Cell growth inhibition was assessed by MTT assay at 72 hrs (top panel). Apoptosis was assessed by Histone/DNA ELISA at 72 hrs and Annexin V FITC assay at 72 hrs (middle and lower panel). B. Western blot analysis of of HCT116 (p53-null) cells exposed to indicated treatments for 20 hrs.
Oral administration of MI-319 enhanced the therapeutic effect of cisplatin mediated via p73 dependent mechanism in PC xenografts
In order to study the clinical relevance of our MI-319-cisplatin combination, we used two PC xenograft tumour models (Capan-2 wt-p53 and BxPC-3 mut-p53). As expected, oral administration of MI-319 alone suppressed tumour growth only in Capan-2 and not BxPC-3 xenografts. Intravenous administration of cisplatin (4 mg/kg twice a week for two weeks) resulted in a nominal suppression of tumour growth in both models. Although the antitumour efficacy was more pronounced in wt-p53 xenografts yet MI-319-cisplatin could significantly suppress tumour growth in mut-p53 model (p=0.001 when compared to cisplatin alone treated group). Of greater importance is the observation that 50% Capan-2 mice were tumour free (Figure 6A). To further elucidate the molecular mechanism involved in MI-319-cisplatin combination, Western blot on tumour tissue lysates were performed. Figure 6B shows that tissue p73 was elevated along with its downstream cell cycle regulator p21WAF1 in both Capan-2 and BxPC-3 tumour samples. These results provide in vivo support which is consistent with in vitro results in support of the involvement of p73 rather than p53 in mediating the anti-tumour activity of MI-319-cisplatin combination in PC.
Figure 6. MI-319-cisplatin combination suppresses tumour growth irrespective of p53 status.
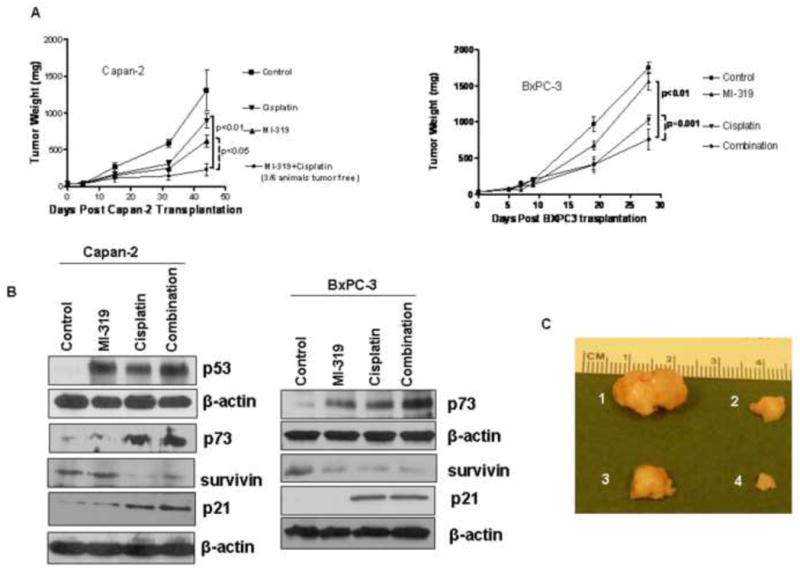
A. (Right and Left panels) Mice tumour weight post indicated treatment. B. Western blots analysis for p73 and p21 on lysates isolated from tumours harvested from mice of different treatment groups showing loss upregulation of p73 and p21. C. Photographs of isolated tumours post treatment (1) Vehicle only; (2) MI-319; (3) Cisplatin and (4) combination as indicated in Material and Methods section.
Discussion
To our knowledge this is the first report showing activity of MDM2 inhibitor in combination with cisplatin against PC cells irrespective of p53 status. The ability of p73 to be up-regulated by MDM2-cisplatin combination and mediating the p53-independent anti-tumour effects could be a reflection of its more pervasive role compared to mutant p53 in pancreatic tumourigenesis than has currently been believed. Our interesting and novel animal studies and supportive in vitro data provide confidence towards testing a potent anti-tumour combination (MI-319-cisplatin) for PC patients in the clinical setting.
In recent years small molecule inhibitors of MDM2 have been exhaustively pursued as anti-tumour agents for wt-p53 tumours 32–37. However, such inhibitors have their use restricted to only 50% of the tumour population harboring wt-p53 38;39. In the other half of tumours, mutations or complete absence of p53 renders MDM2 inhibitors of no use, and thus newer agents for this large tumour population is urgently needed. The protein p73, which is a functional homologue of p53 drew the attention of tumour biologists because it is rarely mutated in human cancers and can induce cell cycle arrest and apoptosis by activating genes that are also regulated by p53 40. Further, it is well established that p73 is also under the influence of MDM2, and thus making it an ideal target candidate by MDM2 inhibitors in tumours where p53 is deregulated 41. Thus we believe agents that induce p73 in combination with MDM2 inhibitors would be an attractive therapy. To that end cisplatin is an ideal candidate because it is a widely used chemotherapeutic agent and a known inducer of both p53 and p73 42. Its use in conjunction with an agent such MI-319 certainly holds logical promise because the mechanism of action of both drugs involves the activation of p53 (or its family members) effector pathways.
Indeed our results clearly showed, for the first time, that MI-319 in combination with cisplatin could induce drastic cell growth inhibition and apoptosis in PC cells irrespective of p53 function. This combination worked in a synergistic manner, which was confirmed by isobologram analysis (CI<1 in all cases). Mechanistically, MI-319-cisplatin combination induced apoptosis through up-regulation of p73 in PC cells harboring mutant p53. The MI-319-cisplatin combination also activated pathways that are downstream of p73 including the important cell cycle regulator p21WAF1. Most significantly the induction of cell growth inhibition and apoptosis in p53 null system (HCT116 cells with homozygous deletion of p53; p53−/−) through similar mechanism further reiterated the p53 independent mode of action of MI-319-cisplatin combination. We also used siRNA silencing studies to verify the exclusive role of p73 in cell killing and found that knockdown of p73 could block the cell growth inhibitory and apoptotic inducing potential of MI-319-cisplatin combination. Interestingly MDM2 siRNA knockdown also resulted in p73 upregulation which confirms that MDM2 blocks p73 function which can be re-activated by inhibitors such as MI-319. In order to substantiate the clinical relevance of our studies, we tested MI-319-cisplatin combination in two xenograft tumour models of PC consisting of both wild type and mutant p53. Consistent with our in vitro results, the combination suppressed tumour growth in both Capan-2 (50% tumour free) and BxPC-3 xenografts more potently compared to MI-319 or cisplatin alone. Further confirmation regarding the involvement of p73 came from the tumour tissue protein analysis which showed up-regulation of p73 and p21WAF1.
Recent findings clearly demonstrated that p53-dependent apoptosis in response to DNA damage is impaired in cells lacking p73, indicating that p73 is critical component of the apoptotic response to DNA damage (8). Moreover, emerging evidence strongly suggests that the pro-apoptotic activity of p73 is regulated through a pathway distinct from that used by p53. As p53 and p73 share extensive structural and functional similarities, they have overlapping as well as distinct biological functions. Similar to p53, p73 is induced and accumulated in response to a subset of DNA-damaging agents such as cisplatin; however, the regulatory mechanisms of the pro-apoptotic activity of p73 are distinct from those used by p53. In addition, p73 might enhance the chemosensitivity of tumour cells to conventional DNA damaging agents. Therefore, p73 alone or in combination with the other p53 family members might provide a clue in overcoming chemo-resistance in tumour cells, the typical feature of PC.
Pancreatic cancer is particularly resistant to apoptosis induced by anti-neoplastic agents such as gemcitabine and cisplatin, which is partly attributable to the lack of functional p53. To this end, it has been shown that E2F1 in combination with the most clinically efficient drug, gemcitabine, resulted in a strong induction of apoptosis independent of functional p53 43. This therapeutic effect was directly correlated with the induction of p73, suggesting that the E2F1/p73 pathway plays a critical role in PC therapy. Therefore, the data presented herein provide compelling evidence in support of the role of p73 rather than p53 in mediating the anti-proliferative and pro-apoptotic effects of MDM2 inhibitor when combined with cisplatin and, as such, provide useful information for the development of combinatorial therapy for the treatment of PC with both functional and non-functional p53. In conclusion, our studies clearly suggest that the combination of MI-319 with cisplatin would become a universal therapeutic strategy for the treatment of the majority of the human pancreatic tumours in the future.
Acknowledgments
Role of grant support and acknowledgements: National Cancer Institute, NIH grant R01CA109389 (R.M. Mohammad), NIH grant 5R01CA101870 (F.H. Sarkar), and NIH grant U19CA113317 (S. Wang). We sincerely acknowledge the Guido foundation for their support.
Footnotes
Conflict of Interest: The University of Michigan has filed a patent on MI-319, which has been licensed by Ascenta Therapeutics, Inc. The University of Michigan and S. Wang and Dajun Yang own equity in Ascenta.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Li J, Saif MW. Any progress in the management of advanced pancreatic cancer?. JOP 2009; Highlights from the 45th ASCO annual meeting; Orlando, FL, USA. May 29–June 2, 2009; pp. 361–365. [PubMed] [Google Scholar]
- 2.Sato Y, Nio Y, Song MM, Sumi S, Hirahara N, Minari Y, et al. p53 protein expression as prognostic factor in human pancreatic cancer. Anticancer Res. 1997;17(4A):2779–2788. [PubMed] [Google Scholar]
- 3.Chi SW, Lee SH, Kim DH, Ahn MJ, Kim JS, Woo JY, et al. Structural details on mdm2-p53 interaction. J Biol Chem. 2005;280(46):38795–38802. doi: 10.1074/jbc.M508578200. [DOI] [PubMed] [Google Scholar]
- 4.Bauer S, Muhlenberg T, Leahy M, Hoiczyk M, Gauler T, Schuler M, et al. Therapeutic Potential of Mdm2 Inhibition in Malignant Germ Cell Tumours. Eur Urol. 2009 doi: 10.1016/j.eururo.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Secchiero P, di Iasio MG, Gonelli A, Zauli G. The MDM2 inhibitor Nutlins as an innovative therapeutic tool for the treatment of haematological malignancies. Curr Pharm Des. 2008;14(21):2100–2110. doi: 10.2174/138161208785294663. [DOI] [PubMed] [Google Scholar]
- 6.Drakos E, Thomaides A, Medeiros LJ, Li J, Leventaki V, Konopleva M, et al. Inhibition of p53-murine double minute 2 interaction by nutlin-3A stabilizes p53 and induces cell cycle arrest and apoptosis in Hodgkin lymphoma. Clin Cancer Res. 2007;13(11):3380–3387. doi: 10.1158/1078-0432.CCR-06-2581. [DOI] [PubMed] [Google Scholar]
- 7.Flores ER. The roles of p63 in cancer. Cell Cycle. 2007;6(3):300–304. doi: 10.4161/cc.6.3.3793. [DOI] [PubMed] [Google Scholar]
- 8.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416(6880):560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 9.Senoo M, Manis JP, Alt FW, McKeon F. p63 and p73 are not required for the development and p53-dependent apoptosis of T cells. Cancer Cell. 2004;6(1):85–89. doi: 10.1016/j.ccr.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90(4):809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 11.Yang A, Kaghad M, Caput D, McKeon F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002;18(2):90–95. doi: 10.1016/s0168-9525(02)02595-7. [DOI] [PubMed] [Google Scholar]
- 12.Yang A, McKeon F. P63 and P73: P53 mimics, menaces and more. Nat Rev Mol Cell Biol. 2000;1(3):199–207. doi: 10.1038/35043127. [DOI] [PubMed] [Google Scholar]
- 13.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90(4):809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 14.Jost CA, Marin MC, Kaelin WG., Jr p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature. 1997;389(6647):191–194. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 15.Di Como CJ, Urist MJ, Babayan I, Drobnjak M, Hedvat CV, Teruya-Feldstein J, et al. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res. 2002;8(2):494–501. [PubMed] [Google Scholar]
- 16.Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol. 1999;19(2):1438–1449. doi: 10.1128/mcb.19.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG., Jr Chemosensitivity linked to p73 function. Cancer Cell. 2003;3(4):403–410. doi: 10.1016/s1535-6108(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 18.Irwin MS, Miller FD. p73: regulator in cancer and neural development. Cell Death Differ. 2004;11 (Suppl 1):S17–S22. doi: 10.1038/sj.cdd.4401452. [DOI] [PubMed] [Google Scholar]
- 19.Irwin MS. Family feud in chemosensitvity: p73 and mutant p53. Cell Cycle. 2004;3(3):319–323. [PubMed] [Google Scholar]
- 20.Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr, Levrero M, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399(6738):806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 21.Shimodaira H, Yoshioka-Yamashita A, Kolodner RD, Wang JY. Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin. Proc Natl Acad Sci U S A. 2003;100(5):2420–2425. doi: 10.1073/pnas.0438031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Rayes BF, Philip PA. A review of systemic therapy for advanced pancreatic cancer. Clin Adv Hematol Oncol. 2003;1(7):430–434. [PubMed] [Google Scholar]
- 23.El-Rayes BF, Zalupski MM, Shields AF, Ferris AM, Vaishampayan U, Heilbrun LK, et al. A phase II study of celecoxib, gemcitabine, and cisplatin in advanced pancreatic cancer. Invest New Drugs. 2005;23(6):583–590. doi: 10.1007/s10637-005-1028-z. [DOI] [PubMed] [Google Scholar]
- 24.El-Rayes BF, Zalupski MM, Shields AF, Vaishampayan U, Heilbrun LK, Jain V, et al. Phase II study of gemcitabine, cisplatin, and infusional fluorouracil in advanced pancreatic cancer. J Clin Oncol. 2003;21(15):2920–2925. doi: 10.1200/JCO.2003.03.022. [DOI] [PubMed] [Google Scholar]
- 25.Jacobs AD, Otero H, Picozzi VJ, Jr, Aboulafia DM. Gemcitabine combined with docetaxel for the treatment of unresectable pancreatic carcinoma. Cancer Invest. 2004;22(4):505–514. doi: 10.1081/cnv-200026392. [DOI] [PubMed] [Google Scholar]
- 26.Jacobs AD. Gemcitabine-based therapy in pancreas cancer: gemcitabine-docetaxel and other novel combinations. Cancer. 2002;95(4 Suppl):923–927. doi: 10.1002/cncr.10756. [DOI] [PubMed] [Google Scholar]
- 27.Philip PA, Zalupski MM, Vaitkevicius VK, Arlauskas P, Chaplen R, Heilbrun LK, et al. Phase II study of gemcitabine and cisplatin in the treatment of patients with advanced pancreatic carcinoma. Cancer. 2001;92(3):569–577. doi: 10.1002/1097-0142(20010801)92:3<569::aid-cncr1356>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 28.Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127(29):10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 29.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2006;49(12):3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 30.Azmi AS, Wang Z, Burikhanov R, Rangnekar VM, Wang G, Chen J, et al. Critical role of prostate apoptosis response-4 in determining the sensitivity of pancreatic cancer cells to small-molecule inhibitor-induced apoptosis. Mol Cancer Ther. 2008;7(9):2884–2893. doi: 10.1158/1535-7163.MCT-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali S, Banerjee S, Ahmad A, El-Rayes BF, Philip PA, Sarkar FH. Apoptosis-inducing effect of erlotinib is potentiated by 3,3′-diindolylmethane in vitro and in vivo using an orthotopic model of pancreatic cancer. Mol Cancer Ther. 2008;7(6):1708–1719. doi: 10.1158/1535-7163.MCT-08-0354. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105(10):3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popowicz GM, Czarna A, Rothweiler U, Szwagierczak A, Krajewski M, Weber L, et al. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle. 2007;6(19):2386–2392. doi: 10.4161/cc.6.19.4740. [DOI] [PubMed] [Google Scholar]
- 35.Binder BR. A novel application for murine double minute 2 antagonists: the p53 tumor suppressor network also controls angiogenesis. Circ Res. 2007;100(1):13–14. doi: 10.1161/01.RES.0000255897.84337.38. [DOI] [PubMed] [Google Scholar]
- 36.Vassilev LT. p53 Activation by small molecules: application in oncology. J Med Chem. 2005;48(14):4491–4499. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]
- 37.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 38.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 39.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253(5015):49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 40.Bell HS, Ryan KM. Targeting the p53 family for cancer therapy: ‘big brother’ joins the fight. Cell Cycle. 2007;6(16):1995–2000. doi: 10.4161/cc.6.16.4614. [DOI] [PubMed] [Google Scholar]
- 41.Lau LM, Nugent JK, Zhao X, Irwin MS. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene. 2008;27(7):997–1003. doi: 10.1038/sj.onc.1210707. [DOI] [PubMed] [Google Scholar]
- 42.Lapi E, Di AS, Donzelli S, Gal H, Domany E, Rechavi G, et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol Cell. 2008;32(6):803–814. doi: 10.1016/j.molcel.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 43.Rodicker F, Stiewe T, Zimmermann S, Putzer BM. Therapeutic efficacy of E2F1 in pancreatic cancer correlates with TP73 induction. Cancer Res. 2001;61(19):7052–7055. [PubMed] [Google Scholar]