

Abstract
A major challenge to nanomaterial-based medicine is the ability to release drugs on-command. Here, we describe an innovative drug delivery system based on carbon nanotubes (CNTs), in which compounds can be released inside cells from within the nanotube “on-command” by inductive heating with an external alternating current or pulsed magnetic field. Without inductive heating the drug remains safely inside the CNTs, showing no toxicity in cell viability tests. Similar to the “Trojan-Horse” in function, we demonstrate the delivery of a combination of chemotherapeutic agents with low aqueous solubility, paclitaxel (Taxol), and C6-ceramide, to multidrug resistant pancreatic cancer cells. Nanotube encapsulation permitted the drugs to be used at a 100-fold lower concentration compared to exogenous treatment yet achieve a comparable ∼70% cancer kill rate.
Keywords: Drug delivery, cancer therapy, on-command release, stimuli responsive
Nanomaterials, including polymeric micelles, liposomes, dendrimers, nanoparticles, and nanotubes have long been investigated for their use in the targeted delivery of therapeutic agents, plasmids, siRNAs, genes and growth factors.1,2 With a size of less than several hundred nanometers, these nanoma-terials have been shown to accumulate in solid tumors via leaky tumor blood vessels, making them suitable for delivery of anticancer drugs.3,4 The nanomaterials can be further modified to enhance targeting to specific sites.5 However, for most nanoparticles, the drugs are attached to the outside of the structure or blended into a biodegradable polymer matrix, which can result in drug degradation over the delivery process or produce off-target toxicity.6 Thus, drug leakage, degradation, the limited selection of disparate cargos due to their biophysical properties, inefficient drug attachment, and most importantly “on-command” release remain major challenges.
Carbon nanotubes (CNTs) as drug delivery carriers have many useful physiochemical properties. They are biocompatible hollow structures with a large surface area and a high aspect ratio, with metallic or semimetallic behavior.7,8 The outer surface of the CNT can be chemically modified, while the inside cavity of the nanotubes can be accessed with drugs.9 Although the development of carbon nanotubes for drug delivery is highly promising, there remain several challenges to engineering a nanotube drug carrier with: (1) high loading efficiency; (2) high drug retention rate over the delivery process; and (3) “on-command” release. Metal halides and small molecules have previously been encapsulated in carbon nanotubes; however, encapsulation typically involves high-temperature molten-phase loading, for example, 900 °C.10 Moreover, the selection of cargo is often limited to molecules with low surface tension to facilitate their uptake into the nanotubes by capillary or van der Waals forces.11 Additionally, the interior space of conventional CNTs is small and difficult to access.9 Thus, while fullerenes and other molecules can be encapsulated into the nanotube, the encapsulation process is often irreversible due to the large energy barrier at the nanotube open end,9 which can make it inherently difficult to release the agents controllably from the CNTs. Here we describe a new strategy that facilitates the controlled loading of therapeutic agents into carbon nanotubes and their controlled release by deploying an alternating current (a.c.) magnetic field as an external command. The external stimulation with an a.c. magnetic field pulse, which can reach deep into any part of the body, may provide a promising new approach for triggering on-command drug release from the nanomaterials.
In contrast to conventional CNTs, the CNTs used here have a relatively large diameter (∼40 nm inner diameter). The CNTs are fabricated in an array with a perfectly aligned vertical orientation by growing them in a highly ordered and uniform anodic aluminum oxide (AAO) nanopore array template by chemical vapor deposition (CVD).12 After fabrication the CNTs undergo either a mechanical or chemical treatment to open both ends while embedded in the template, which provides access to the interior space for en-masse loading of molecules. Drug loading was accomplished by depositing droplets of the drug solution on top of the vertically aligned nanotube array membrane while applying vacuum suction at the bottom.
The template-synthesized carbon nanotubes are intrinsically conductive but possess higher electrical resistivity than those synthesized by arc-discharging. This property is beneficial for our purpose as the electrical resistivity will generate eddy currents and consequential resistive heat via magnetic field induction. The extent of heating generated in each nanotube can be regulated by the applied magnetic field strength, frequency, and the conductivity and physical dimensions of the tube, the former two parameters being controlled via the source of induction, whereas the latter two are controlled through the fabrication process (see the Supporting Information). By incorporating a temperature-sensitive hydrogel mixed with the drug of interest, the surface tension and viscosity maintains the gel-drug payload inside the tube. Furthermore, the surface tension of water prevents external water from entering the nanotube to displace the drug payload. After exposure to an a.c. magnetic field, we theorize the inductive heating causes the temperature sensitive hydrogel to undergo a gel–sol phase transformation and discharge into the surrounding fluid, where the hydrogel dissolves releasing the payload.
To assess the loading and encapsulation efficiency we loaded an aqueous solution of quantum dots (QD, CdSe/ZnS nanocrystals) into the CNT array (Figure 1a) and verified encapsulation under transmission electron microscopy (TEM). After loading, individual CNTs were released by dissolving away the alumina template while the chemical inertness of the CNTs protected their contents. TEM images of QD-loaded CNTs clearly show QDs inside the CNT (Figure 1b), confirming successful encapsulation. To demonstrate the feasibility of the “Trojan-Horse” nanotube system, we initially tested the release of gel-QD loaded nanotubes subjected to an a.c. magnetic field (25 kHz) in water. Green luminescence was observed in the solution withdrawn from the CNT array surrounding solution only after the a.c. magnetic field was applied (Figure 1c), indicative of QD release by inductive heating.
Figure 1.
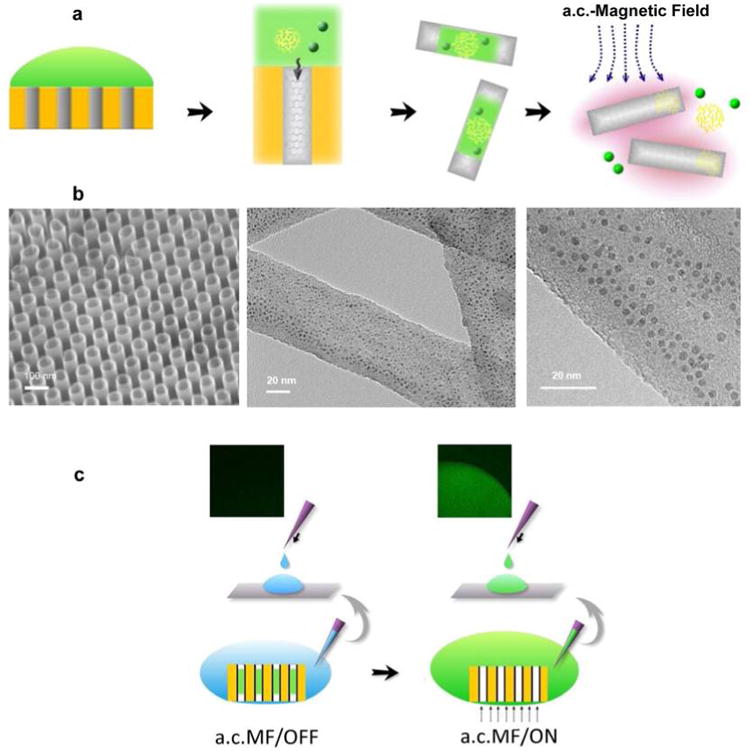
Loading and on-command release of Trojan-Horse nanotubes by the induction heating of CNTs. (a) Schematic illustration of filling QDs/gelatin/drug into nanotubes. The payload is loaded into the nanotubes array by the vacuum suction at the back side of the template followed by the release of Trojan-Horse nanotube from template by chemical dissolution of AAO template. Finally, the payload is released from the Trojan-Horse nanotubes by exposing Trojan-Horse nanotubes to the a.c. magnetic field. (b, left) SEM image of highly ordered carbon nanotube array. (center) TEM image showing the high loading yield of QDs into nanotubes. (right) A magnified TEM micrograph showing the QDs with diameter ∼4 nm are inside the nanotubes. (c) QD loaded nanotube array immersed in water and subjected to an a.c. magnetic field for 30 min. Thereafter, the solution was placed onto a glass slide and visualized by epifluorescence microscopy. (left) In the absence of the a.c. magnetic field induction, luminescence was not observed. (right) a.c. magnetic field induction results in a significant green luminescent signal corresponding to the QD luminescence emission wavelength.
Functionalized nanotubes can be internalized by tumor cells and may provide a vehicle with which to delivery chemo-therapeutic drugs to the interior of cells, causing apoptosis.13 To investigate the Trojan-Horse nanotube as a viable controllable delivery strategy, we tested the delivery of the chemotherapeutic drugs paclitaxel and C6-ceramide, to an in vitro cell model of pancreatic cancer, an aggressive malignancy with a five-year mortality greater than 95%.14 Although Paclitaxel (Taxol), a microtubule stabilizing agent, is an extensively used neoplastic drug,15 its application has been limited due to water insolubility. In addition, this drug can produce severe hypersensitivity reactions, and many tumors acquire chemoresistance.16
However, recent studies suggest that short-chain C6-ceramide can dramatically increase the efficacy of many chemotherapeutic agents including Taxol,17 doxorubicin,18 and histone deacetylase inhibitors (HDACi)19 in vitro, but the utility of C6-ceramide is limited because of its hydrophobicity. It precipitates as a fine lipid suspension when administered in aqueous solution,20,21 and the presence of a second aliphatic chain hinders its cellular permeability.22 Furthermore, free ceramides are susceptible to metabolic pathways which generate less toxic pro-oncogenic metabolites.23 Nanoliposomes have been tested to deliver short-chain ceramides both in vitro and in vivo, but their use is limited by the relatively high concentration of nanoliposome ceramide (∼1–4 μg/mL in vitro22,24 and ∼10–50 mg/kg in vivo20,25,26) required to achieve antitumor effects, leading to possible off-target toxicity. However, as demonstrated by our previous studies,17,27,28 the levels of ceramide required to kill cancer cells can be substantially reduced by combination therapy with paclitaxel, which interacts with ceramide in a supra-additive manner to promote apoptosis. As carbon nanotubes allow the encapsulation of multiple drugs, they may facilitate the delivery of ceramide at a smaller dosage, resulting in less toxic effects than current delivery methods.
Pancreatic cancer has proven to be extremely difficult to treat due to multidrug resistance, making it the fourth most common cause of cancer-related deaths.29 We first tested the ability of C6-ceramide and Taxol to kill three different pancreatic cancer cell lines, L3.6, PANC-1, and MIA PaCa-2(MIA). Even at high doses, Taxol alone or C6-ceramide alone had a limited effect on killing the three pancreatic cancer cells lines, whereas combining these two agents at a relatively low dose potently produced cell death (Figure 2). These results confirm that C6-ceramide sensitizes Taxol-induced pancreatic cell death and therefore could be a useful adjuvant for treating pancreatic cancer.
Figure 2.
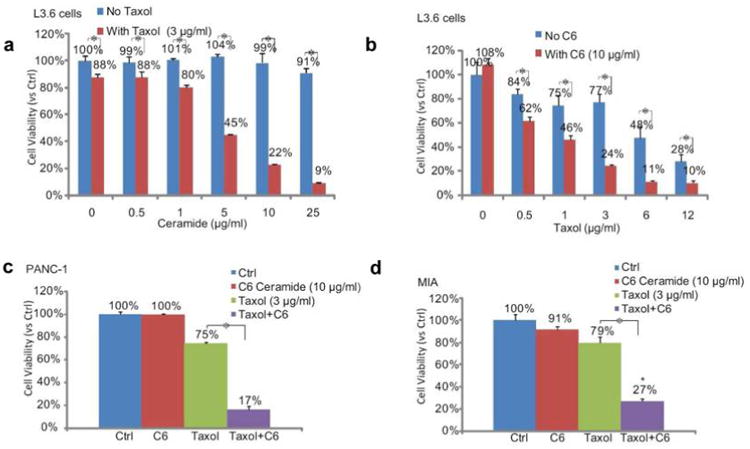
Cell-permeable short-chain C6-ceramide sensitizes Taxol induced cell death in pancreatic cancer cells in vitro. A cell viability MTT assay performed 48 h after treatment confirmed C6-ceramide dramatically sensitizes Taxol induced cell death in three different pancreatic cancer cell lines: (a) L3.6, (b) L3.6, (c) PANC-1, and (d) MIA PaCa-2. Cell death was reflected by reduced cell viability/MTT OD. Note that a relative high concentration of C6-ceramide (5–10 μg/mL) is needed to reach the chemo-sensitization effect. *p < 0.05.
To verify that CNTs can be taken up by pancreatic cancer cells, we labeled the CNTs with Texas Red by conjugating Texas Red-X succinimidyl ester to CNTs that were non-covalently functionalized with amine-terminated polyethylene glycol phospholipids (PL-PEG-NH2).30 The fluorescently labeled CNTs were added to pancreatic cancer L3.6 cells overnight, and then Texas Red fluorescence was examined using confocal microscopy. From the planar (Figure 3a) and cross-sectional (Figures 3b and c) views, Texas Red fluorescence was clearly observed inside the pancreatic cancer cells, demonstrating that the CNTs were taken up by these cells (Figure S1 in the Supporting Information).
Figure 3.
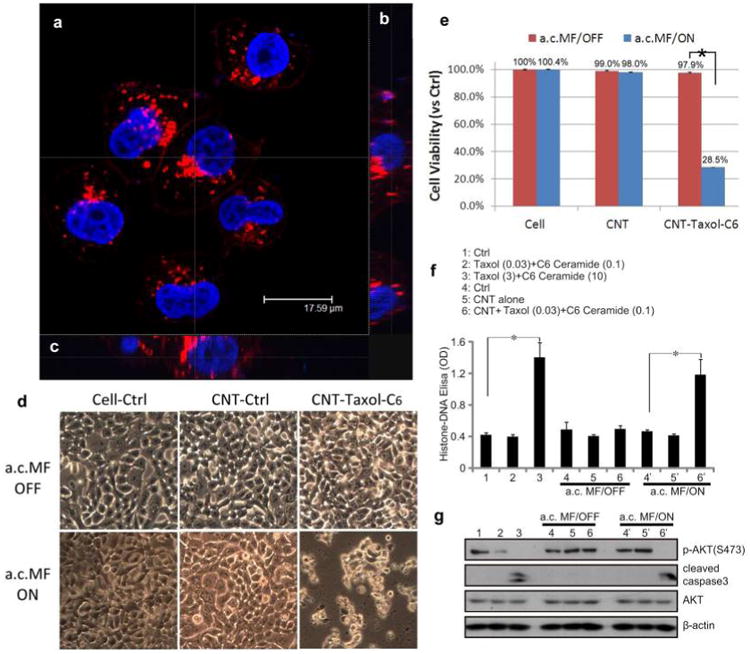
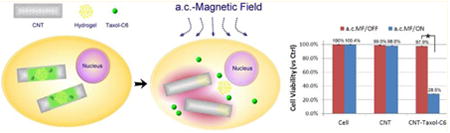
Internalization of Trojan-Horse nanotubes and on-command therapeutic delivery of CNT-occluded material to pancreatic cancer cells in vitro. (a) A confocal fluorescence image in plan-view of L3.6 pancreatic cancer cells taken after overnight incubation with Texas Red-labeled Trojan-Horse nanotubes. The Trojan-Horse nanotubes accumulated within the perinuclear space (red signal) indicating the nanotubes were internalized by cells. Cell nuclei were stained in blue with Hoechst 33342. (b) Using Z-axis stacks of images the intracellular three-dimensional distribution of Texas Red-labeled CNTs was demonstrated. The Z-stack was sampled in vertical (b) and horizontal orientation (c) with these oriented images confirmed the Trojan-Horse CNTs to be internalized for these pancreatic cancer cells. (d) CNT-exposed cells were subjected to a.c. magnetic field for on-command drug (Taxol/C6-ceramide) release. The cell morphology of pancreatic cancer cells (L3.6) was observed under different conditions. Significant cell death was observed only when Trojan-Horse CNTs were subjected to the flux of the a.c. magnetic field. There was no effect of “empty” CNTs under any condition tested. (e) An in vitro cytotoxicity assay (MTT) was used to determine cell viability 48 h after CNT treatment. The a.c. magnetic field resulted in on-command release of Taxol/C6-ceramide from the Trojan-Horse CNTs, and this therapeutic drug delivery resulted in over 70% cell death. (f) A histone-DNA ELISA assay was performed to examine L3.6 cell apoptosis under different conditions following a 36 h treatment. Increased apoptosis was observed in the cells treated with a.c. magnetic field activation of [CNT + Taxol (0.03 μg/mL) + C6-ceramide (0.1 μg/mL)] (group #6′) delivery, an outcome achieved only when cells received a 100-fold higher dose of exogenous Taxol/C6-ceramide (group #3). (g) L3.6 cell apoptosis was reflected by increased cleavage of caspase-3 and reduced phosphorylation of Akt (Ser 473). These biochemical surrogate markers confirm the pattern of apoptosis observed in panel f.
Using the same pancreatic cancer cells (L3.6), we next tested the on-command release of the encapsulated Taxol and C6-ceramide from CNTs. The drug-loaded CNTs were added to L3.6 cells for 12 h to allow uptake, and then the cells were then washed to remove the CNTs left in the media. A 30 min a.c. magnetic field (25 kHz) was applied to these cells to release the encapsulated drugs (Taxol and C6-ceramide) from inside the cells. After 48 h, drug release was assessed by the degree of cell death, which resulted in 71.5% of cell death (p < 0.05 vs control) as reflected by cell viability loss (Figure 3e, MTT dye assay) and cell morphology change (Figure 3d). Without magnetic field induction, the cells had a viability of 97.9% (p > 0.05 vs control), indicating the exceptional stability of encapsulated drugs within the CNT throughout the 48-h long incubation. Thus in contrast to previous magnetic nanoparticle drug delivery carriers,31,32 the Trojan-Horse nanotubes provide complete protection of payload. Importantly, no noticeable drop in cell viability was observed in cells incubated with empty nondrug loaded-CNTs (Figure 3d and e, middle), indicating that both the empty CNTs and the a.c. magnetic field used here (25 kHz) was safe to cells, with a 98–99% viability (Figure 3e, p > 0.05 vs control). This contrasts the high magnetic fields (up to MHz frequency) required for magnetic nanoparticles to release their payload due to the superparamagnetic nature and small value of magnetization of saturation.
The cancer cell death caused by the release of the encapsulated drugs was the result of cell apoptosis (Figure 3f) and Akt inhibition (Figure 3g), which is identical to the cell fate when Taxol/C6-ceramide was exogenously added directly. Again, only group #6′ [CNT + Taxol (0.03 μg/mL) + C6-ceramide (0.1 μg/mL)] with the a.c. magnetic field had increased Histone-DNA ELISA OD, an indicator of cell apoptosis (Figure 3f). Importantly, the drug concentration loaded into the Trojan-Horse CNTs was 100 times lower (Taxol: 0.03 μg/mL/C6-ceramide: 0.1 μg/mL) than the concentration required for a comparable effect with the exogenously applied treatment (Taxol: 3 μg/mL/C6-ceramide: 10 μg/mL). The same low concentration of Taxol + C6-ceramide (Taxol: 0.03 μg/mL/C6-ceramide: 0.1 μg/mL) used in CNTs failed to cause cell death when added exogenously (no CNT carrier, group #2), indicating that the drugs cannot reach toxic levels inside the cell. The 100 fold difference between these two groups shows that Trojan-Horse CNTs effectively transport these drugs into the cells and can “on-demand” release the drugs to cause cell death. As the CNTs contain a 100-fold lower drug concentration, this delivery vehicle has the potential to greatly reduce drug side effects.
Overall, the use of Trojan-Horse CNTs as a therapeutic drug delivery carrier has a number of advantages. The unique structure of these CNTs makes them easily accessed by a variety of drugs, and once the drugs are loaded, they are stably encapsulated within the CNTs until their release by induction heating. A major advantage is the lower electromagnetic field required for release, which had no detectable adverse effect on the cells. Finally, the CNTs allow efficient delivery of membrane impermeant drugs, allowing the concentration of the drugs to be greatly reduced, potentially bypassing adverse effects. Although we focused on the delivery of chemotherapeutic drugs, the Trojan-Horse CNTs delivery system could be extended to other cargo such as plasmids, siRNAs, growth factors, and even metallic and atomic substances.
Supplementary Material
Acknowledgments
Support for this research was provided by a NINDS R21 NS061176 (J.M.) and the Intramural Research Program of the National Institute of Environmental Health Sciences, NIH and the Pathobiology Graduate Program at Brown University (C.C.). This research was also supported in part by Brown's Institute for Brain Science, AFOSR, and the WCU Program at Seoul National University during J.X.'s sabbatical leave. C.H.W., C.C., J.H.K., and C.H.H. performed the experiments and analyzed the results; W.D.B. and H.J.W. developed the general concept of ceramide-taxol synergy and provided the pancreatic cancer cell lines and Taxol; C.H.W. and J.X. conceived the Trojan-Horse nanotube idea, and with J.M. they devised the experimental demonstration method and prepared the manuscript. We thank Dr. Edith Mathiowitz for advice and comments on the manuscript.
Footnotes
Supporting Information: Materials and methods for Taxol/C6-ceramide loading into CNT arrays, cell apoptosis assay, viability assay and Western blot analysis. Supporting figures showing experimental and control data on the cellular uptake of CNT, time course study on cell viability after the treatment of CNT with drugs (Taxol/C6-ceramide) encapsulated, and plot of cell viability versus CNT-drugs concentration. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Chen CC, Liu YC, Wu CH, Yeh CC, Su MT, Wu YC. Adv Mater. 2005;17(4):404–407. [Google Scholar]
- 3.Barreto JA, O'Malley W, Kubeil M, Graham B, Stephan H, Spiccia L. Adv Mater. 2011;23(12):H18–40. doi: 10.1002/adma.201100140. [DOI] [PubMed] [Google Scholar]
- 4.Bae KH, Chung HJ, Park TG. Mol Cells. 2011;31(4):295–302. doi: 10.1007/s10059-011-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. Nat Mater. 2011;10(5):389–397. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petros RA, DeSimone JM. Nat Rev Drug Discovery. 2010;9(8):615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 7.Mintmire JW, Dunlap BI, White CT. Phys Rev Lett. 1992;68(5):631–634. doi: 10.1103/PhysRevLett.68.631. [DOI] [PubMed] [Google Scholar]
- 8.Kostarelos K, Bianco A, Prato M. Nat Nanotechnol. 2009;4(10):627–633. doi: 10.1038/nnano.2009.241. [DOI] [PubMed] [Google Scholar]
- 9.Hilder TA, Hill JM. Small. 2009;5(3):300–308. doi: 10.1002/smll.200800321. [DOI] [PubMed] [Google Scholar]
- 10.Hong SY, Tobias G, Al-Jamal KT, Ballesteros B, Ali-Boucetta H, Lozano-Perez S, Nellist PD, Sim RB, Finucane C, Mather SJ, Green ML, Kostarelos K, Davis BG. Nat Mater. 2010;9(6):485–490. doi: 10.1038/nmat2766. [DOI] [PubMed] [Google Scholar]
- 11.Dujardin E, Ebbesen TW, Hiura H, Tanigaki K. Science. 1994;265(5180):1850–1852. doi: 10.1126/science.265.5180.1850. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Papadopoulos C, Xu J, Moskovits M. Appl Phys Lett. 1999;75(3):367–369. [Google Scholar]
- 13.Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Cancer Res. 2008;68(16):6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hidalgo M. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 15.Kavallaris M. Nat Rev Cancer. 2010;10(3):194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 16.Singh S, Dash AK. Crit Rev Ther Drug Carrier Syst. 2009;26(4):333–372. doi: 10.1615/critrevtherdrugcarriersyst.v26.i4.10. [DOI] [PubMed] [Google Scholar]
- 17.Qiu L, Zhou C, Sun Y, Di W, Scheffler E, Healey S, Wanebo H, Kouttab N, Chu W, Wan Y. Oncol Rep. 2006;16(4):907–913. [PubMed] [Google Scholar]
- 18.Ji C, Yang B, Yang YL, He SH, Miao DS, He L, Bi ZG. Oncogene. 2010;29(50):6557–6568. doi: 10.1038/onc.2010.379. [DOI] [PubMed] [Google Scholar]
- 19.Zhu QY, Wang Z, Ji C, Cheng L, Yang YL, Ren J, Jin YH, Wang QJ, Gu XJ, Bi ZG, Hu G, Yang Y. Cell Death Dis. 2011;2:e117. doi: 10.1038/cddis.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stover TC, Sharma A, Robertson GP, Kester M. Clin Cancer Res. 2005;11(9):3465–3474. doi: 10.1158/1078-0432.CCR-04-1770. [DOI] [PubMed] [Google Scholar]
- 21.Radin NS. Eur J Biochem. 2001;268(2):193–204. doi: 10.1046/j.1432-1033.2001.01845.x. [DOI] [PubMed] [Google Scholar]
- 22.Shabbits JA, Mayer LD. Biochim Biophys Acta. 2003;1612(1):98–106. doi: 10.1016/s0005-2736(03)00108-1. [DOI] [PubMed] [Google Scholar]
- 23.Devalapally H, Duan Z, Seiden MV, Amiji MM. Int J Cancer. 2007;121(8):1830–1838. doi: 10.1002/ijc.22886. [DOI] [PubMed] [Google Scholar]
- 24.Stover T, Kester MJ. Pharmacol Exp Ther. 2003;307(2):468–475. doi: 10.1124/jpet.103.054056. [DOI] [PubMed] [Google Scholar]
- 25.Tagaram HR, Divittore NA, Barth BM, Kaiser JM, Avella D, Kimchi ET, Jiang Y, Isom HC, Kester M, Staveley-O'Carroll KF. Gut. 2011;60(5):695–701. doi: 10.1136/gut.2010.216671. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Ryland L, Yang J, Liao A, Aliaga C, Watts R, Tan SF, Kaiser J, Shanmugavelandy SS, Rogers A, Loughran K, Petersen B, Yuen J, Meng F, Baab KT, Jarbadan NR, Broeg K, Zhang R, Liao J, Sayers TJ, Kester M, Loughran TP., Jr Blood. 2010;116(20):4192–4201. doi: 10.1182/blood-2010-02-271080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehta S, Blackinton D, Omar I, Kouttab N, Myrick D, Klostergaard J, Wanebo H. Cancer Chemother Pharmacol. 2000;46(2):85–92. doi: 10.1007/s002800000140. [DOI] [PubMed] [Google Scholar]
- 28.Myrick D, Blackinton D, Klostergaard J, Kouttab N, Maizel A, Wanebo H, Mehta S. Leuk Res. 1999;23(6):569–578. doi: 10.1016/s0145-2126(99)00048-x. [DOI] [PubMed] [Google Scholar]
- 29.Hariharan D, Saied A, Kocher HM. HPB (Oxford) 2008;10(1):58–62. doi: 10.1080/13651820701883148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kam NW, Liu Z, Dai H. J Am Chem Soc. 2005;127(36):12492–12493. doi: 10.1021/ja053962k. [DOI] [PubMed] [Google Scholar]
- 31.Hu SH, Liu TY, Liu DM, Chen SY. Macromolecules. 2007;40(19):6786–6788. [Google Scholar]
- 32.Rovers SA, Hoogenboom R, Kemmere MF, Keurentjes JTF. Soft Matter. 2012;8(5):1623–1627. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.