

Abstract
α-Fetoprotein (AFP) is known to be highly produced in fetal liver despite its barely detectable level in normal adult liver. On the other hand, hepatocellular carcinoma often shows high expression of AFP. Thus, AFP seems to be an oncogenic marker. In our present study, we investigated how TGF-β signaling cooperates with AT motif-binding factor-1 (ATBF1) to inhibit AFP transcription. Indeed, the expression of AFP mRNA in HuH-7 cells was negatively regulated by TGF-β signaling. To further understand how TGF-β suppresses the transcription of the AFP gene, we analyzed the activity of the AFP promoter in the presence of TGF-β. We found that the TGF-β signaling and ATBF1 suppressed AFP transcription through two ATBF1 binding elements (AT-motifs). Using a heterologous reporter system, both AT-motifs were required for transcriptional repression upon TGF-β stimulation. Furthermore, Smads were found to interact with ATBF1 at both its N-terminal and C-terminal regions. Since the N-terminal (ATBF1N) and C-terminal regions of ATBF1 (ATBF1C) lack the ability of DNA binding, both truncated mutants rescued the cooperative inhibitory action by the TGF-β signaling and ATBF1 in a dose-dependent manner. Taken together, these findings indicate that TGF-β signaling can act in concert with ATBF1 to suppress the activity of the AFP promoter through direct interaction of ATBF1 with Smads.
1. Introduction
The oncofetal glycoprotein α-fetoprotein (AFP) is a major serum protein expressed at high levels in the yolk sac and liver during embryonic development [1, 2]. However, AFP in adult serum is undetectable except in patients who suffer from hepatocellular carcinoma (HCC). Thus, AFP is a useful tumor marker for measuring the malignancy grade of HCC [3]. The human AFP gene is activated by hepatocyte nuclear factor-1 (HNF-1), which can bind to an AT-motif in the proximal and/or distal promoter region of AFP [4–6].
AT motif-binding factor-1 (ATBF1) encodes a protein comprising multiple zinc fingers and homeodomains [7, 8]. ATBF1 was originally discovered as a negative transcriptional regulator of the human AFP gene, which competes with HNF-1 for binding to the AT-motifs [4]. Thus, ATBF1 seems to act as a transcriptional repressor of the AFP gene [4, 9, 10]. Besides the AFP gene, ATBF1 can also negatively regulate the transcription of the Myb gene [11]. When ATBF1 was overexpressed in C2C12 cells, the expression of Id3 and cyclin D increased, whereas that of MyoD and myogenin did not [12]. Thus, ATBF1 seems to play a key role not only as a negative, but also as a positive transcriptional regulator. Recently, ATBF1 was suspected to be a candidate tumor suppressor gene because it is frequently mutated or deleted in prostate, breast, and gastric tumors, and its expression is also suppressed in some tumors [13–17].
Transforming growth factor-β (TGF-β) regulates a great number of cellular responses including proliferation, differentiation, apoptosis, extracellular matrix production, motility, and immunosuppression. TGF-β elicits its cellular effects by making a heteromeric complex between TGF-β type I (TβRI, also termed activin receptor-like kinase 5 [ALK5]) and TGF-β type II receptors (TβRII). After TGF-β binds to TβRII, TβRI is recruited by TβRII to be phosphorylated within its GS domain by the constitutively active TβRII kinase. The activated TβRI kinase initiates signaling through phosphorylation of specific receptor-regulated Smads (R-Smads, i.e., Smad2 and Smad3). Subsequently, two activated R-Smads form ternary complexes with a common-partner Smad (Co-Smad), Smad4. These complexes can regulate TGF-β-responsive genes either directly via their binding to the promoters of these genes or indirectly via their association with a large number of transcription factors, coactivators, and/or corepressors. Of interest, Co-Smad and R-Smads except for Smad2 possess the ability to bind to specific DNA sequences, whereas Smad2 has 30-aa insertion, immediately prior to obvious DNA binding region in its MH1 domain, which prevents Smad2 from binding to DNA [18–20].
TGF-β is known to negatively regulate AFP expression [21, 22]. Furthermore, TGF-β-mediated repression of AFP involves recruitment of Smad to p53 together with SnoN and mSin3A in the AFP promoter [22, 23]. Although ATBF1 also contributes to negative regulation of AFP, how TGF-β signaling cooperates with ATBF1 to inhibit AFP transcription remains veiled. In this study, we could confirm that the TGF-β/Smad signaling pathway plays an inhibitory role in the AFP transcription via the interaction of Smads with ATBF1.
2. Materials and Methods
2.1. Cell Culture
COS7 cells, monkey kidney fibroblast-like cells, were cultured in Dulbecco's modified Eagle's medium (Nacalai) containing 10% fetal calf serum (FCS; Invitrogen). HepG2 and HuH-7 cells, hepatocellular carcinoma cells, were maintained in Eagle's minimum essential medium (Wako) containing 10% FCS, 1% nonessential amino acids, and 1% sodium pyruvate. All media were supplemented with 100 IU/mL penicillin and 100 μg/mL streptomycin.
2.2. Plasmid Constructions
The expression constructs for HA-ATBF1, Myc-ATBF1, HA-ATBF1N, HA-ATBF1M, and HA-ATBF1C have been previously described [24, 25]. The AFP luciferase reporters and Flag-Smad2, Flag-Smad3, and Flag-Smad4 have been documented [6, 26, 27]. Various lengths of the 5′promoter region of human AFP were cloned by PCR and inserted into the luciferase gene with either TK (TK.Luc) or CMV minimal promoter (CMV Luc). Mutation in the AFP promoter was performed using a KOD-Plus-Mutagenesis kit (TOYOBO). Adenoviral ALK5ca was generously provided by Fujii et al. [28].
2.3. Luciferase Reporter Assay
HepG2 cells were seeded at 1.5 × 105 cells/well in a 12-well plate one day before transfection. The cells were transfected with reporter constructs using polyethyleneimine (PEI). After 24 h of transfection, cells were stimulated with 5 ng/mL TGF-β for 18 h. In all luciferase assays, β-galactosidase activity in cells transfected with 100 ng of pCH110 (GE Healthcare) was measured to normalize the transfection efficiency. The results were the averages of three independent transfections and were repeated at least twice. The representative data were shown. All values represent mean ± S.D. Each t-test between two columns was performed: *P < 0.05, **P < 0.01, and ***P < 0.001.
2.4. RT-PCR Analysis
Total RNAs from HuH-7 cells were extracted using an RNeasy Plus Mini kit (Qiagen). Reverse transcription was carried out using a High-Capacity RNA to cDNA kit (Applied Biosystems). PCR was performed using GoTaq (Promega) according to the manufacturer's instructions.
2.5. Immunoprecipitation and Western Blot Analysis
To detect interactions among the proteins, plasmids were transfected into COS7 cells (5 × 105 cells/6 cm dish) using PEI. Forty hours after the transfection, cells were lysed in 500 μL of TNE buffer (10 mM Tris (pH 7.4), 150 mM NaCl, 1 mM ethylenediamine-N′,N′,N′,N′-tetraacetic acid (EDTA), 1% NP-40, 1 mM phenylmethylsulfonyl-l-fluoride (PMSF), 5 μg/mL leupeptin, 100 U/mL aprotinin, 2 mM sodium vanadate, 40 mM NaF, and 20 mM β-glycerophosphate). The cell lysates were precleared with protein G-Sepharose beads (GE Healthcare) for 30 min at 4°C and then incubated with anti-Myc or anti-Flag antibody (Sigma) for 2 h at 4°C. The protein complexes were immunoprecipitated by incubation with protein G-Sepharose beads for 30 min at 4°C followed by three washes with TNE buffer. The immunoprecipitated proteins and aliquots of the total cell lysates were boiled for 5 min in sample buffer, separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a Hybond-C Extra membrane (GE Healthcare). The membranes were probed with primary antibodies. The primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies and chemiluminescent substrate (Thermo Scientific). Protein expression in the total cell lysates was evaluated by Western blot analysis.
2.6. DNA Affinity Precipitation (DNAP)
COS7 cells were seeded at 1.5 × 106 cells/well in a 10 cm dish one day before transfection. Ten micrograms of indicated expression vectors were transfected into COS7 cells using PEI. Forty hours after the transfection, the cells were lysed in 1 mL of TNE buffer. Then, each cell lysate was divided and mixed for DNAP. The combined cell lysates were precleared with 12 μg/mL poly (dI · dC) and streptavidin agarose (Sigma) for 30 min and then incubated with 24 μM biotinylated (AT-motif)3 for 2 h at 4°C. Subsequently, streptavidin agarose was added to the reaction mixture and incubated for 30 min at 4°C. After the precipitates had been washed with TNE buffer three times, the precipitates and aliquots of the total lysates were separated by SDS-PAGE. The proteins were then transferred to the membrane. The membrane was incubated with the indicated primary antibodies. The primary antibodies were detected as described above. The sequences of the biotinylated (AT-motif)3 were as follows: 5′-biotinylated CTCGAGGCTGTTAATTATTGGGGCTGTTAATTATTGGGGCTGTTAATTATTGAATTC-3′/3′-GAATTCAATAATTAACAGCCCCAATAATTAACAGCCCCAATAATTAACAGCCTCGAG-5′.
3. Results
3.1. AFP mRNA Is Inhibited upon TGF-β Stimulation
It has been reported that TGF-β contributes to the suppression of the activity of the AFP promoter [21, 22]. Indeed, we examined if the expression of AFP mRNA was affected in HuH-7 cells upon ALK5 activation. AFP mRNA was considerably reduced in cells infected with constitutively active ALK5 (ALK5ca) expressing adenovirus, whereas TMEPAI mRNA which has been known to be a direct target gene of TGF-β/Smad signal [29–31] was remarkably induced (Figure 1(a)). Furthermore, AFP mRNA slightly decreased with time when HuH-7 cells were stimulated with TGF-β. On the other hand, ATBF1 mRNA did not change upon TGF-β stimulation (Figure 1(c)).
Figure 1.
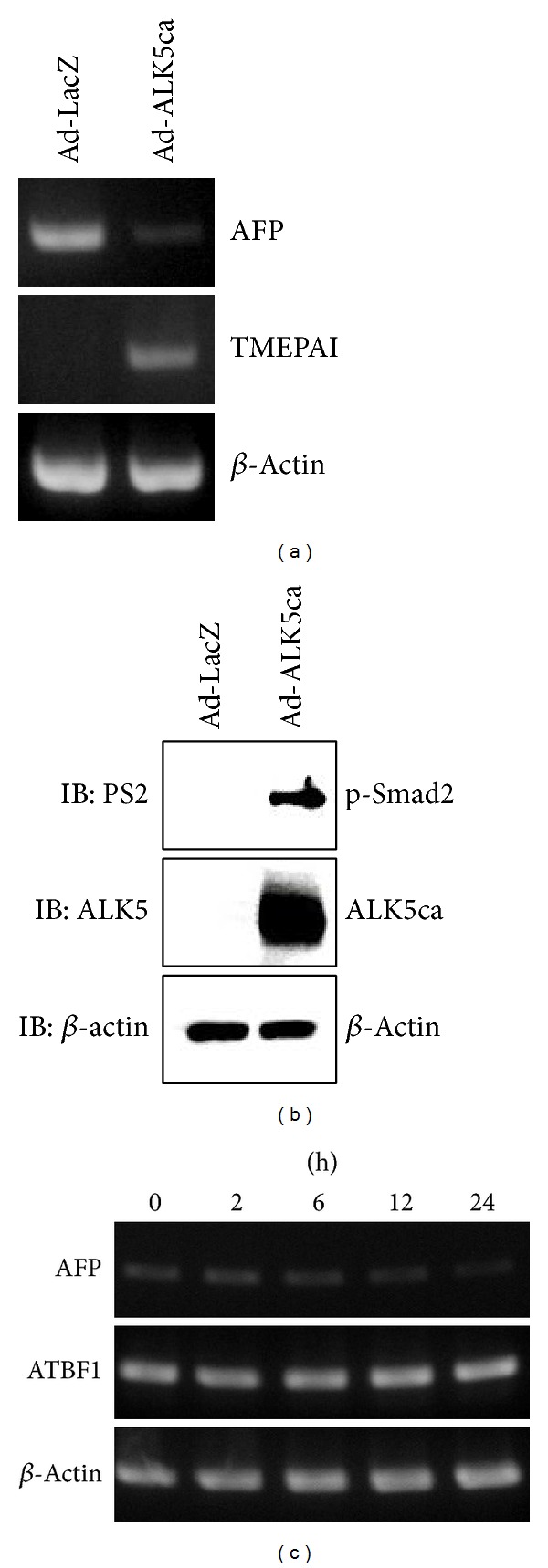
TGF-β represses AFP mRNA. (a) AFP mRNA expression was inhibited by ALK5ca in HuH-7 cells. The cells were infected with LacZ (left lane) or ALK5ca-expressing adenoviruses (right lane) at a dose of 250 multiplicity of infection. After 42 hours of infection, total RNA and cell lysates were prepared for RT-PCR analysis (a) and Western blot analysis (IB) (b), respectively. (a) Expression of AFP, TMEPAI, and β-actin mRNA was detected by RT-PCR. TMEPAI which is a direct target gene of TGF-β signal [29–31] was used as a positive control. (b) For IB, antiphosphorylated Smad2 (PS2), anti-ALK5, and anti-β-actin antibodies were used. (c) Expression of AFP mRNA upon TGF-β stimulation. HuH-7 cells were treated with 5 ng/mL TGF-β for the indicated times. Then, RT-PCR was performed as described in (a). Each PCR condition is described in Table 1.
3.2. TGF-β/Smad Signaling Inhibits the Activity of the AFP Promoter Together with ATBF1
It has already been demonstrated that enhancer elements (E A, E B), silencer elements (S D, S P), and a promoter (P) are present in the 5′ flanking region of AFP gene [5, 32–34]. ATBF1 is known to bind to E B and P to repress the activity of the AFP promoter [4, 6, 26]. To evaluate the involvement of these elements in TGF-β-mediated repression of the activity of the AFP promoter, each luciferase reporter shown in Figure 2(a) was transfected into HepG2 cells. The cells were then stimulated with TGF-β. As shown in Figure 2(b), both 4.9Luc and Δ2.7Luc, which possessed a high basal level of the reporter activity, obviously showed TGF-β-mediated repression of the reporter activity. 3Luc, 1.6Luc, and 0.9Luc contain Smad binding element/p53 response element (SBE/p53RE) which has been reported to confer TGF-β-mediated repression of the AFP promoter [22, 23]. Indeed, these reporter activities were also reduced upon TGF-β stimulation in spite of a very low basal activity of their luciferase reporters. Like these three reporters, 0.2Luc, which possesses one AT-motif, could exhibit TGF-β-mediated repression of the luciferase activity as well (Figure 2(b)). These results supported the notion that the region between −4995 and −2953 concededly includes cis-element(s) for TGF-β to suppress the activity of the AFP promoter. To test the possibility that ATBF1 can suppress the activity of the AFP promoter upon TGF-β stimulation, Δ2.7Luc was cotransfected with or without ATBF1. Subsequently, the cells were stimulated with TGF-β. As seen in Figure 2(c), ATBF1 could further reduce the luciferase activity that was repressed by TGF-β. Since Smad3 and Smad4 play key roles in the canonical TGF-β signaling pathway [18, 20], we investigated whether Smad3 and/or Smad4 are implicated in TGF-β-mediated repression of the AFP promoter together with ATBF1. As shown in Figure 2(d), Smad3 alone or the combination of Smad3 with Smad4 suppressed the reporter activity. Moreover, ATBF1 further diminished the reporter activity that was restrained by both Smad3 and Smad4 upon TGF-β stimulation.
Figure 2.
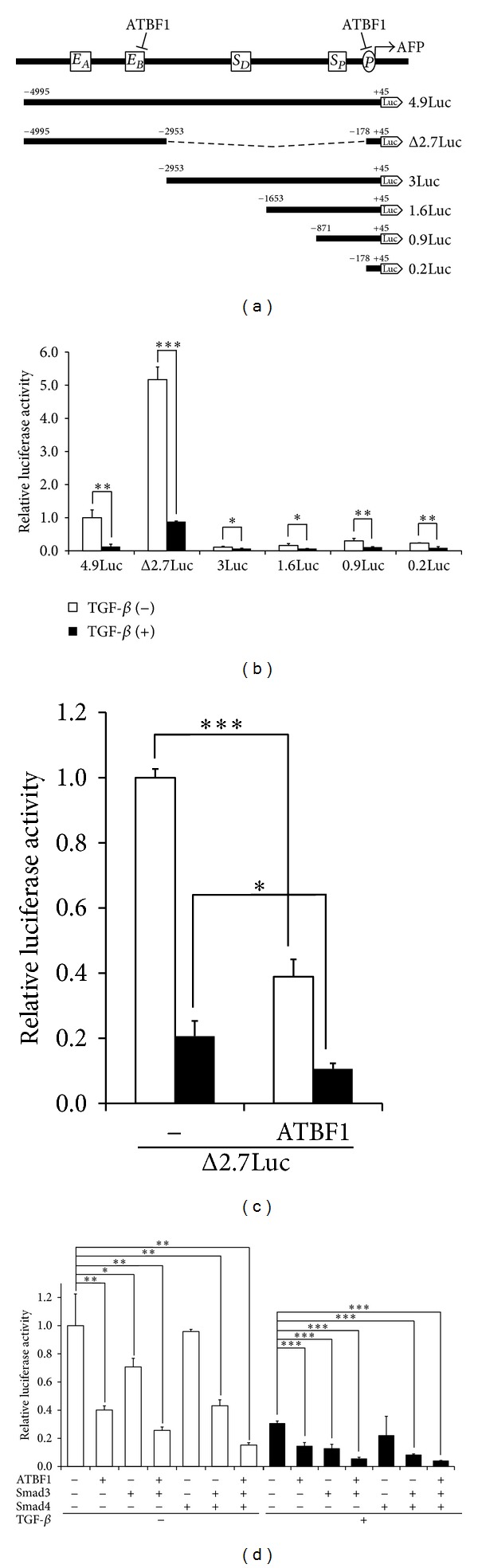
TGF-β/Smad signaling inhibits the activity of the AFP promoter together with ATBF1. (a) Schematic presentation of deletion mutants of the AFP promoter. E A between −4120 and −3756 and E B between −3492 and −3300 are enhancer elements. S D between −1807 and −1725 and S P between −317 and −300 are silencer elements. P indicates a promoter. (b) Effect of TGF-β on the activity of the AFP promoter. The examined luciferase reporters (500 ng) were transfected into HepG2 cells. The cells were then stimulated with 5 ng/mL TGF-β. (c) Cooperation of ATBF1 with the TGF-β signaling for suppression of the activity of the AFP promoter. ATBF1 expression plasmid (200 ng) was transfected into HepG2 cells with Δ2.7Luc (500 ng). The cells were then stimulated with 5 ng/mL TGF-β. (d) Effect of Smads and ATBF1 on the transcriptional activity of Δ2.7Luc. HepG2 cells were transfected with Smads (100 ng), ATBF1 (200 ng), or their combination together with Δ2.7Luc (500 ng). The cells were then stimulated with 5 ng/mL TGF-β.
3.3. Requirement of ATBF1 Binding Sites for Transcriptional Repression of the AFP Gene by TGF-β
Because the region between −4995 and −2953 is important for the TGF-β/Smad signaling to cooperate with ATBF1 for the suppression of the activity of the AFP promoter, we speculated that E B, which includes the ATBF1 binding site, is involved in an inhibitory action via the TGF-β/Smad signaling pathway. To test this possibility, the region between −4990 and −2968 was divided into three pieces. Then, each fragment was ligated to the luciferase reporter driven by the herpes virus thymidine kinase promoter (TK.Luc) (Figure 3(a)). As shown in Figure 3(b), TGF-β suppressed the reporter activity of fTK.Luc. Thus, the region between −4990 and −2968 possesses an inhibitory cis-element(s) upon TGF-β stimulation. Within this region, the fragment between −3658 and −2968 (termed 3′TK.Luc) revealed a remarkably inhibitory cis-element by TGF-β, although TGF-β-mediated repression of the reporter activity could also be seen in mTK.Luc. Intriguingly, E B containing the AT-motif is present between −3492 and −3300 (Figure 3(c)). To further narrow down a TGF-β-responsive inhibitory cis-element(s), we made four luciferase reporters that were linked to the AFP minimal promoter (Figure 3(c)). TGF-β dramatically suppressed the luciferase activity of 3′1Luc, 3′5Luc, and 3′52Luc although 3′3Luc activity was also inhibited by TGF-β to some extent in spite of its low basal luciferase activity, indicating that the region from −3562 to −3422 possesses a TGF-β-mediated inhibitory cis-element if not more (Figures 3(d) and 3(e)). Thus, an AT-motif and/or its adjacent sequences in E B were highly suggestive of a TGF-β-mediated inhibitory cis-element. In addition, ATBF1 could further inhibit the 3′52Luc activity suppressed by TGF-β (Figure 3(f)). Therefore, it is possible that the TGF-β/Smad pathway interferes via the region between −3562 and −3422.
Figure 3.
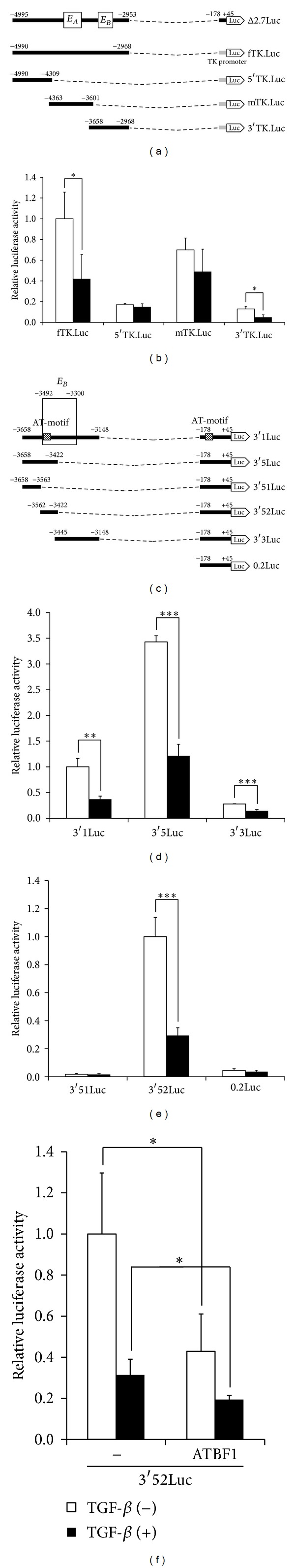
Requirement of a distal AT-motif for TGF-β-mediated repression of the activity of the AFP promoter. (a) Schematic presentation of TK.Luc conjugated with several fragments of the AFP promoter between −4990 and −2968. (b) Effect of TGF-β on the activity of TK.Luc reporters conjugated with several truncated AFP promoter regions. Each reporter (200 ng) was transfected into HepG2 cells. The cells were then stimulated with 5 ng/mL TGF-β. (c) Schematic presentation of 0.2Luc conjugated with several fragments of the AFP promoter between −3658 and −3148. (d) Effect of TGF-β on the activity of the luciferase reporters described in Figure 3(c). Each reporter (200 ng) was transfected into HepG2 cells. The cells were then stimulated with 5 ng/mL TGF-β. (e) Requirement of the fragment between −3562 and −3422 for TGF-β-mediated suppression of the AFP promoter. HepG2 cells were transfected with the indicated plasmids (200 ng). The cells were then stimulated with 5 ng/mL TGF-β. (f) Cooperation of ATBF1 with the TGF-β signaling for 3′52Luc activity. HepG2 cells were transfected with either 3.52Luc reporter (200 ng) in the presence or absence of ATBF1 (200 ng). The cells were then stimulated with 5 ng/mL TGF-β.
3.4. A Role of the Proximal AT-Motif in the AFP Promoter
Since there are two functional AT-motifs within the AFP promoter between −4995 and +45 (Figure 2(a)), it is possible that the proximal AT-motif also influences TGF-β-mediated inhibition of the activity of the AFP promoter. To confirm that the proximal AT-motif can also contribute to TGF-β-mediated repression of the activity of the AFP promoter, the 141nt-length fragment from −3562 to −3422 was ligated to the 64nt-length region between −155 and −92 of the AFP promoter. Then, this fragment was further placed at the 5′port of CMV Luc (3′52_65CMVLuc) because the CMV promoter shows the high level of luciferase activity. Figure 4(b) demonstrates that both the distal and the proximal AT-motifs are needed for TGF-β to suppress the activity of the AFP promoter.
Figure 4.
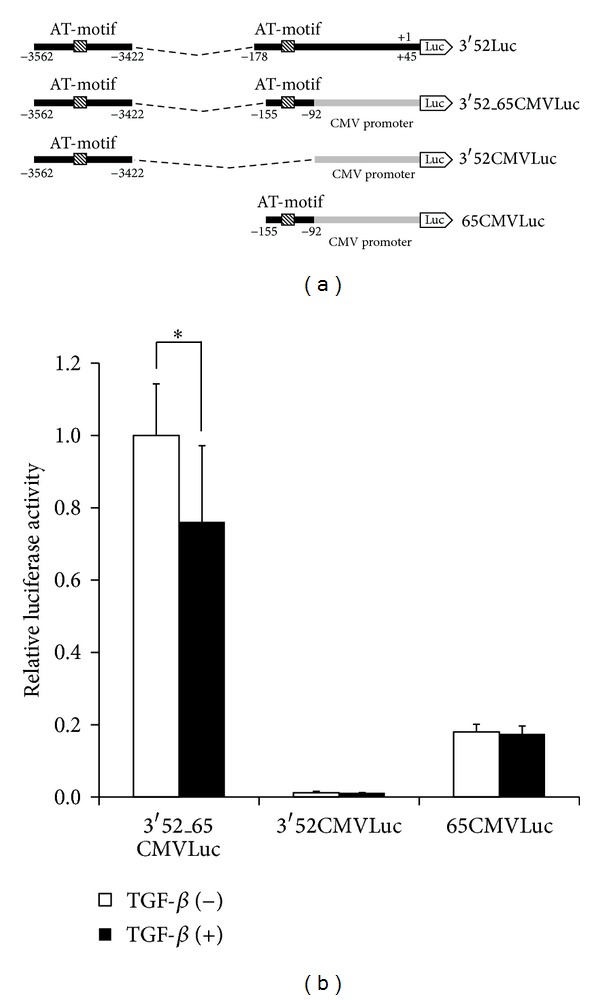
Importance of both distal and proximal AT-motifs for TGF-β-mediated repression of the AFP promoter. (a) Schematic presentation of CMV Luc conjugated with distal and proximal AT-motifs. (b) HepG2 cells were transfected with each reporter (200 ng) and then stimulated with 5 ng/mL TGF-β.
3.5. Association of Smad Proteins with ATBF1
Since ATBF1 cooperates with Smad3 for inhibition of the activity of the AFP promoter, we tried to determine whether ATBF1 could interact with R-Smads. Expectedly, ATBF1 could associate with Smad2 and Smad3, although ALK5 activation was not involved in its interaction (Figure 5(a) and data not shown). In these experiments, we could observe multiple bands corresponding to ATBF1. These multiple bands might be due to calpain-1-dependent proteolysis in COS7 cells [35]. To identify the domain(s) of ATBF1 that interact with Smads, we divided ATBF1 into three pieces (Figure 5(b)). ATBF1N as well as ATBF1C had the ability to interact with Smad2 and Smad3, although ATBF1M no longer possessed the ability to bind to either Smad2 or Smad3 (Figures 5(c)–5(e)). It has been reported that DNA-binding domains are present in ATBF1M, including zinc finger domains and homeodomains [12]. When either ATBF1N or ATBF1C was cotransfected with ATBF1, TGF-β-mediated inhibition of the activity of the AFP promoter was improved owing to their competition with ATBF1 for Smad binding (Figures 6(a) and 6(b)). On the other hand, ATBF1M could inhibit the reporter activity as a dominant negative form of ATBF1 because ATBF1M possesses the ability to bind to AT-motif via its DNA binding domain (data not shown). These results indicate that the N-terminal and C-terminal domains are needed for ATBF1 to interact with R-Smads.
Figure 5.
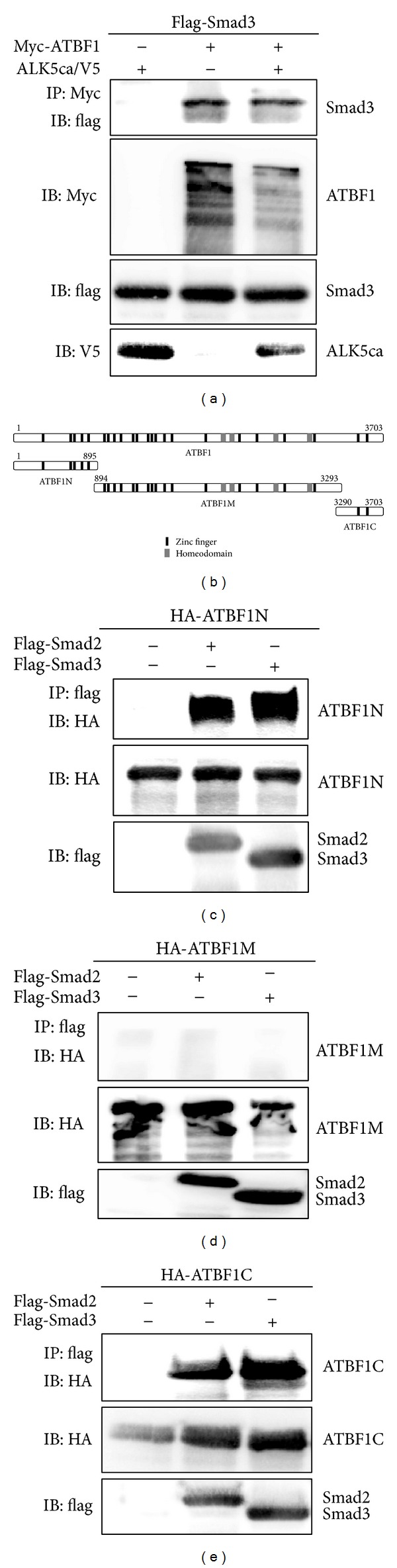
Association of Smads with ATBF1. (a) Interaction of Smad3 with ATBF1. COS7 cells were transfected with Flag-Smad3 (1 μg) with Myc-ATBF1 (2 μg) in combination with ALK5ca/V5 (0.5 μg) or without it. Cell lysates were subjected to immunoprecipitation (IP) with anti-Myc antibody followed by IB with anti-Flag antibody to show the interaction of Flag-Smad3 with Myc-ATBF1. (b) Schematic presentation of ATBF1 mutants. (c–e) Interaction of each ATBF1 mutant with Smad2 or Smad3. COS7 cells were transfected with Flag-Smad2 or Flag-Smad3 (1.5 μg) with HA-ATBF1N (2 μg) (c), HA-ATBF1 M (2 μg) (d), or HA-ATBF1C (2 μg) (e). Cell lysates were subjected to IP with anti-Flag antibody followed by IB with anti-HA antibody to show the interaction of Smad2 and Smad3 with ATBF1 mutants.
Figure 6.
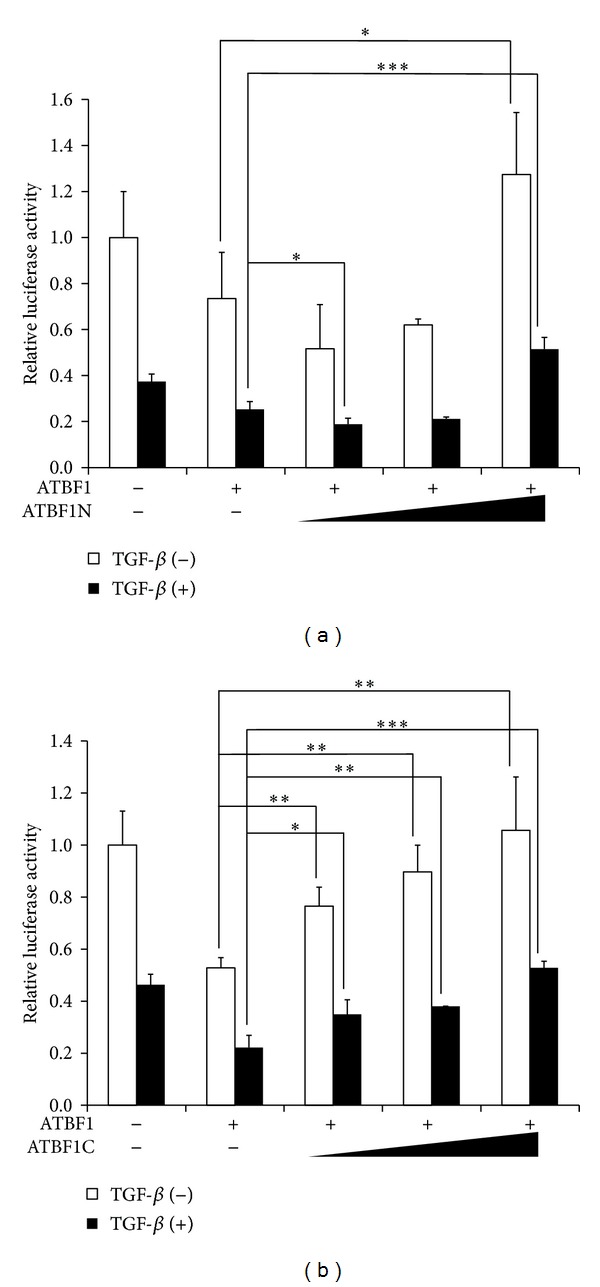
ATBF1N and ATBF1C perturb ATBF1-mediated suppression of the activity of the AFP promoter upon TGF-β stimulation. ATBF1N (a) and ATNF1C (b) rescue ATBF1-mediated repression of the activity of the AFP promoter. HepG2 cells were transfected with increased amounts of either ATBF1N (50, 100, or 200 ng) (a) or ATBF1C (50, 100, or 200 ng) (b) together with ATBF1 (200 ng) and Δ2.7Luc (200 ng). The cells were then stimulated with 5 ng/mL TGF-β.
3.6. Indirect Binding of Smads to the AFP Promoter via ATBF1
We could find only one Smad binding element (SBE) [36, 37] in 3′52Luc (Figure 7(a)). Thus, we examined whether this SBE is necessary for TGF-β-mediated repression of the AFP promoter. For that purpose, we made one reporter construct that had mutations within the SBE of 3′52Luc (3′52mSBELuc) (Figure 7(a)). However, 3′52mSBELuc activity was still inhibited by TGF-β, although its basal level became weaker than that of 3′52Luc (Figure 7(b)). Thus, the SBE in the AFP promoter is not essential for TGF-β/Smad signaling to repress the activity of the AFP promoter. Indeed, a DNAP assay showed that Smad3 can be detected in the presence of ATBF1 using three repeats of an AT-motif as the probe (Figure 7(c)). Thus, Smad3 probably binds to the promoter region of the AFP gene indirectly through its interaction with ATBF1.
Figure 7.
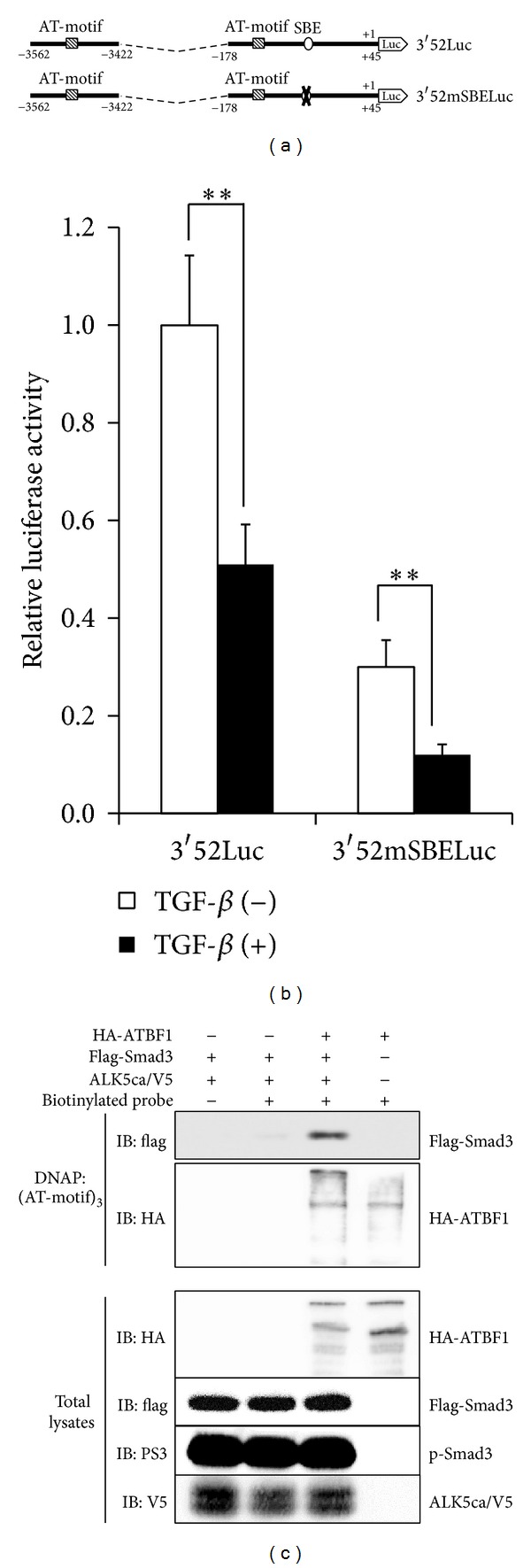
SBE in the proximal region of the AFP promoter is not essential for TGF-β-mediated suppression of the AFP promoter. (a) Schematic presentation of 3′52mSBELuc. A putative SBE, 5′-CAGATA-3′, was mutated to 5′-TTTTTT-3′. (b) Effect of TGF-β on 3′52Luc and 3′52mSBELuc activities. HepG2 cells were transfected with each reporter plasmid (200 ng). The cells were then stimulated with 5 ng/mL TGF-β. (c) Indirect binding of Smad3 to an AT-motif via ATBF1. The indicated cell lysates were mixed with biotinylated AT-motifs. Then, streptavidin-agarose was added to the mixture. Subsequently, a protein(s) bound to streptavidin-agarose was purified. After SDS-PAGE, IB was carried out using anti-Flag or anti-HA antibody. Using the total lysates, each protein was detected with anti-Flag, anti-HA, anti-V5, or anti-phosphorylated Smad3 (PS3) antibody.
4. Discussion
The hepatoma cells HuH-7 and HepG2 cells are known to secrete a large and a low amount of AFP, respectively [38]. A growing body of evidence indicates that the level of AFP in serum from patients is linked to HCC tumorigenicity [3]. p53 has been reported to cooperate with the TGF-β/Smad pathway to repress AFP expression via induction of SnoN [22]. However, it remains veiled whether ATBF1, which is known to block AFP transcription [4, 10], can also regulate the activity of the AFP promoter in consort with the TGF-β/Smad pathway because both ATBF1 and TGF-β/Smad might possess an antioncogenic function [13–17, 20, 39, 40]. ATBF1 could inhibit the activity of the AFP promoter via its regions from −4995 to −2953 and/or from −178 to +45 [4, 26]. Since Δ2.7Luc had a remarkably basal promoter activity compared with 0.2Luc (Figure 2(b)), we initially focused on the distal region of the AFP promoter for involvement of the TGF-β/Smad signaling with ATBF1. As expected, the TGF-β-mediated repressive element in the AFP promoter lay near E B, which contains a distal AT-motif. The luciferase reporter conjugated with the inhibitory element including a distal AT-motif (3.52Luc) showed coordinated inhibitory action between the TGF-β signaling and ATBF1 (Figure 3(f)). Therefore, we were struck with the notion that ATBF1 physically interacts with Smads, which play key roles in the canonical TGF-β signaling pathway [18–20]. Not surprisingly, ATBF1 could interact with Smad2 and Smad3, although C-terminal phosphorylation of R-Smads was dispensable (Figure 5(a) and data not shown). In addition, both the N-terminal (ATBF1N) and the C-terminal mutants of ATBF1 (ATBF1C), but not the middle domain of ATBF1 (ATBF1M) which possesses DNA-binding activity [12], could interact with R-Smads (Figures 5(c)–5(e)). We hypothesized that both ATBF1N and ATBF1C might compete with wildtype ATBF1 for Smad binding to rescue ATBF1-mediated repression of the AFP promoter upon TGF-β stimulation because both of them have the ability to bind to R-Smads. According to our expectation, both ATBF1N and ATBF1C improved the reporter activity decreased by the cooperative ATBF1 and TGF-β signaling (Figures 6(a) and 6(b)). As shown in Figure 2(b), 0.2Luc activity could also be reduced upon TGF-β stimulation despite very low basal promoter activity. Because of the presence of a proximal AT-motif within this region, we speculated that a proximal AT-motif also contributes to TGF-β-mediated-repression of the activity of the AFP promoter. The deletion of a proximal AT-motif from 3′52_65CMVLuc led us to recognize the importance of a proximal AT-motif as well as a distal AT-motif for TGF-β-mediated repression of the activity of the AFP promoter (Figure 4(b)). A Smad binding element (SBE) composed of consensus 5′-AGAC-3′ is flanked to a proximal AT-motif. However, the mutation of the SBE in 3′52Luc showed reduced basal promoter activity but still maintained TGF-β-mediated repression of the AFP promoter (Figure 7(b)). Thus, Smad might not be necessary to bind to a promoter element(s) in the AFP gene directly when ATBF1 cooperates with TGF-β signaling, whereas p53-mediated repression of the AFP promoter activity needs direct binding of Smads to DNA [22, 23]. The requirement of direct DNA binding for R-Smads is probably dependent on which transcriptional regulator(s) they cooperate or synergize with. Indeed, Smad3 could bind to AT-motifs in the presence of ATBF1, but not in its absence (Figure 7(c)). Although we do not know if the ATBF1/Smad complex at a distal AT-motif can physically interact with that at a proximal AT-motif directly or indirectly through another protein(s) (“X”; Figure 8), both AT-motifs are definitely important for cooperation between ATBF1 and the TGF-β signaling to suppress the activity of the AFP promoter.
Figure 8.
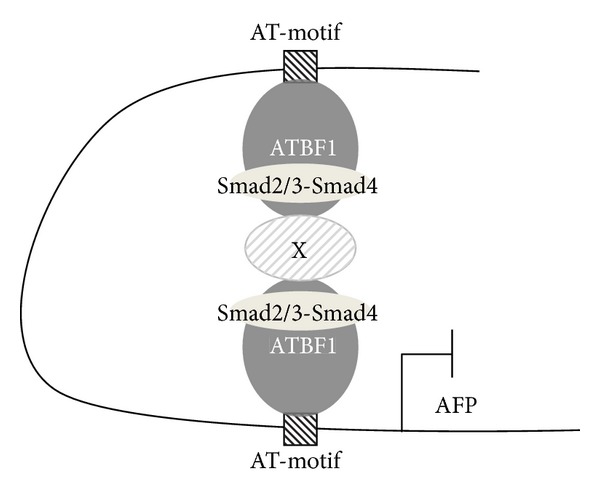
Proposed inhibitory mechanism by which the TGF-β/Smad signaling and ATBF1 cooperatively regulate the activity of the AFP promoter. The distal and proximal AT-motifs in the AFP promoter are bound by ATBF1. Upon TGF-β signaling, the R-Smads/Smad4 complex interacts with ATBF1 to make a repressive complex for the activity of the AFP promoter. However, whether another protein(s) (termed “X”) is essential for the R-Smad/Smad4/ATBF1 complex remains veiled.
ATBF-1 is known to compete with HNF-1 for binding to AT-motifs in the AFP promoter [4]. Thus, the following possibility can be speculated; upon TGF-β stimulation, Smads can not only bind to AT-motifs via ATBF-1 indirectly but also interact with HNF-1 to dissociate it from AT-motifs. Due to dual function of Smads, the AFP prompter activity might be inhibited. This possibility is interesting to be investigated.
p53-mediated repression of the AFP promoter activity needs direct binding of Smads to SBE/p53RE. Wilkinson et al. further demonstrated that mSin3A and SnoN which is a direct target gene of TGF-β signal are recruited by p53/Smads complex when the transcription of the AFP gene is inhibited [22, 23]. In our current study, the region including SBE/p53RE in the AFP promoter was not investigated because the basal reporter activities of 3Luc, 1.6Luc, and 0.9Luc were very low compared with that of Δ2.7Luc (Figure 2(b)). It is possible that ATBF1 might interplay with p53 via Smad complex to effectively repress the transcription of the AFP gene. Further experiments are needed in the future to confirm these hypotheses as well.
5. Conclusion
In summary, we report a novel interaction between ATBF1 and Smads. The interaction between ATBF1 and Smads appears to cooperatively inhibit the transcription of AFP gene upon TGF-β signaling. In particular, both proximal and distal AT-motifs are required for TGF-β/Smad signaling to counteract with the transcription of the AFP gene.
Table 1.
PCR primers to amplify human cDNAs.
| Human cDNA | Sequence | L | AT | Cycle | |
|---|---|---|---|---|---|
| AFP | (+) | ATCCAGGAGAGCCAAGCATT | 434 | 58°C | 20 |
| (−) | TTCATCGTTTGCAGCGCTAC | ||||
|
| |||||
| ATBF1 | (+) | TGGCATCAAGTACAGCGCTC | 392 | 53°C | 35 |
| (−) | GAACAGTTGTGCTGGGCAGA | ||||
|
| |||||
| TMEPAI | (+) | GATCATCATCATCGTGGTGG | 455 | 60°C | 35 |
| (−) | CACTGTCGAAGATGGTTCTG | ||||
|
| |||||
| β-actin | (+) | CACCCACACTGTGCCCATCTACGA | 410 | 63°C | 22 |
| (−) | TGGCGTACAGGTCTTTGCGGATGT | ||||
L: length of PCR fragment; AT: annealing temperature.
Acknowledgments
This research was supported by a Grant-in-Aid for Exploratory Research (A) from the Japan Society for the Promotion of Science (JSPS) (to Nobuo Sakata); the MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2013–2017) (to Susumu Itoh); the Takeda Science Foundation (to Susumu Itoh); the Smoking Research Foundation (to Susumu Itoh); the Daiichi-Sankyo Foundation of Life Science (to Susumu Itoh); the Naito Foundation (to Susumu Itoh); the Vehicle Racing Commemorative Foundation (to Susumu Itoh); and a Grant-in-Aid for Young Scientists of Showa Pharmaceutical University (to Souichi Ikeno). The authors were also supported by the Joint Usage/Research Program of the Medical Research Institute, the Tokyo Medical and Dental University, and by the Core-to-Core Program “Cooperative International Framework in TGF-β Family Signaling” of the JSPS. They thank Ms. F. Miyamasu for excellent English proofreading.
Conflict of Interests
The authors have no competing financial interests to declare.
References
- 1.Dziadek MA, Andrews GK. Tissue specificity of α-fetoprotein messenger RNA expression during mouse embryogenesis. The EMBO Journal. 1983;2(4):549–554. doi: 10.1002/j.1460-2075.1983.tb01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nahon JL, Tratner I, Poliard A, et al. Albumin and α-fetoprotein gene expression in various nonhepatic rat tissues. The Journal of Biological Chemistry. 1988;263(23):11436–11442. [PubMed] [Google Scholar]
- 3.Terentiev AA, Moldogazieva NT. α-fetoprotein: a renaissance. Tumor Biology. 2013;34(4):2075–2091. doi: 10.1007/s13277-013-0904-y. [DOI] [PubMed] [Google Scholar]
- 4.Yasuda H, Mizuno A, Tamaoki T, Morinaga T. ATBF1, a multiple-homeodomain zinc finger protein, selectively down-regulates AT-rich elements of the human α-fetoprotein gene. Molecular and Cellular Biology. 1994;14(2):1395–1401. doi: 10.1128/mcb.14.2.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawadaishi K, Morinaga T, Tamaoki T. Interaction of a hepatoma-specific nuclear factor with transcription-regulatory sequences of the human α-fetoprotein and albumin genes. Molecular and Cellular Biology. 1988;8(12):5179–5187. doi: 10.1128/mcb.8.12.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakabayashi H, Koyama Y, Suzuki H, et al. Functional mapping of tissue-specific elements of the human α-fetoprotein gene enhancer. Biochemical and Biophysical Research Communications. 2004;318(3):773–785. doi: 10.1016/j.bbrc.2004.04.096. [DOI] [PubMed] [Google Scholar]
- 7.Morinaga T, Yasuda H, Hashimoto T, Higashio K, Tamaoki T. A human α-fetoprotein enhancer-binding protein, ATBF1, contains four homeodomains and seventeen zinc fingers. Molecular and Cellular Biology. 1991;11(12):6041–6049. doi: 10.1128/mcb.11.12.6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miura Y, Tam T, Ido A, et al. Cloning and characterization of an ATBF1 isoform that expresses in a neuronal differentiation-dependent manner. The Journal of Biological Chemistry. 1995;270(45):26840–26848. doi: 10.1074/jbc.270.45.26840. [DOI] [PubMed] [Google Scholar]
- 9.Kataoka H, Miura Y, Joh T, et al. α-fetoprotein producing gastric cancer lacks transcription factor ATBF1. Oncogene. 2001;20(7):869–873. doi: 10.1038/sj.onc.1204160. [DOI] [PubMed] [Google Scholar]
- 10.Ninomiya T, Mihara K, Fushimi K, Hayashi Y, Hashimoto-Tamaoki T, Tamaoki T. Regulation of the α-fetoprotein gene by the isoforms of ATBF1 transcription factor in human hepatoma. Hepatology. 2002;35(1):82–87. doi: 10.1053/jhep.2002.30420. [DOI] [PubMed] [Google Scholar]
- 11.Kaspar P, Dvorakova M, Kralova J, Pajer P, Kozmik Z, Dvořák M. Myb-interacting protein, ATBF1, represses transcriptional activity of Myb oncoprotein. The Journal of Biological Chemistry. 1999;274(20):14422–14428. doi: 10.1074/jbc.274.20.14422. [DOI] [PubMed] [Google Scholar]
- 12.Berry FB, Miura Y, Mihara K, et al. Positive and negative regulation of myogenic differentiation of C2C12 cells by isoforms of the multiple homeodomain zinc finger transcription factor ATBF1. The Journal of Biological Chemistry. 2001;276(27):25057–25065. doi: 10.1074/jbc.M010378200. [DOI] [PubMed] [Google Scholar]
- 13.Sun X, Frierson HF, Chen C, et al. Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nature Genetics. 2005;37(6):407–412. doi: 10.1038/ng1528. [DOI] [PubMed] [Google Scholar]
- 14.Sun X, Zhou Y, Otto KB, et al. Infrequent mutation of ATBF1 in human breast cancer. Journal of Cancer Research and Clinical Oncology. 2007;133(2):103–105. doi: 10.1007/s00432-006-0148-y. [DOI] [PubMed] [Google Scholar]
- 15.Cho YG, Song JH, Kim CJ, et al. Genetic alterations of the ATBF1 gene in gastric cancer. Clinical Cancer Research. 2007;13(15):4355–4359. doi: 10.1158/1078-0432.CCR-07-0619. [DOI] [PubMed] [Google Scholar]
- 16.Kai K, Zhang Z, Yamashita H, Yamamoto Y, Miura Y, Iwase H. Loss of heterozygosity at the ATBF1-A locus located in the 16q22 minimal region in breast cancer. BMC Cancer. 2008;8, article 262 doi: 10.1186/1471-2407-8-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cleton-Jansen AM, van Eijk R, Lombaerts M, et al. ATBF1 and NQO1 as candidate targets for allelic loss at chromosome arm 16q in breast cancer: absence of somatic ATBF1 mutations and no role for the C609T NQO1 polymorphism. BMC Cancer. 2008;8, article 105 doi: 10.1186/1471-2407-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes & Development. 2005;19(23):2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 19.Moustakas A, Heldin CH. The regulation of TGFβ signal transduction. Development. 2009;136(22):3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 20.Massagué J. TGFβ signalling in context. Nature Reviews Molecular Cell Biology. 2012;13(10):616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakao K, Nakata K, Mitsuoka S, et al. Transforming growth factor β 1 differentially regulates α-fetoprotein and albumin in HuH-7 human hepatoma cells. Biochemical and Biophysical Research Communications. 1991;174(3):1294–1299. doi: 10.1016/0006-291x(91)91562-q. [DOI] [PubMed] [Google Scholar]
- 22.Wilkinson DS, Ogden SK, Stratton SA, et al. A direct intersection between p53 and transforming growth factor β pathways targets chromatin modification and transcription repression of the α-fetoprotein gene. Molecular and Cellular Biology. 2005;25(3):1200–1212. doi: 10.1128/MCB.25.3.1200-1212.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkinson DS, Tsai WW, Schumacher MA, Barton MC. Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-β-mediated transcription repression. Molecular and Cellular Biology. 2008;28(6):1988–1998. doi: 10.1128/MCB.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung CG, Kim HJ, Kawaguchi M, et al. Homeotic factor ATBF1 induces the cell cycle arrest associated with neuronal differentiation. Development. 2005;132(23):5137–5145. doi: 10.1242/dev.02098. [DOI] [PubMed] [Google Scholar]
- 25.Dong XY, Sun X, Guo P, et al. ATBF1 inhibits estrogen receptor (ER) function by selectively competing with AIB1 for binding to the ER in ER-positive breast cancer cells. The Journal of Biological Chemistry. 2010;285(43):32801–32809. doi: 10.1074/jbc.M110.128330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kataoka H, Bonnefin P, Vieyra D, et al. ING1 represses transcription by direct DNA binding and through effects on p53. Cancer Research. 2003;63(18):5785–5792. [PubMed] [Google Scholar]
- 27.Nakao A, Imamura T, Souchelnytskyi S, et al. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. The EMBO Journal. 1997;16(17):5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujii M, Takeda K, Imamura T, et al. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Molecular Biology of the Cell. 1999;10(11):3801–3812. doi: 10.1091/mbc.10.11.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe Y, Itoh S, Goto T, et al. TMEPAI, a transmembrane TGF-β-inducible protein, sequesters smad proteins from active participation in TGF-β signaling. Molecular Cell. 2010;37(1):123–134. doi: 10.1016/j.molcel.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 30.Itoh S, Thorikay M, Kowanetz M, et al. Elucidation of Smad requirement in transforming growth factor-β type I receptor-induced responses. The Journal of Biological Chemistry. 2003;278(6):3751–3761. doi: 10.1074/jbc.M208258200. [DOI] [PubMed] [Google Scholar]
- 31.Nakano N, Itoh S, Watanabe Y, Maeyama K, Itoh F, Kato M. Requirement of TCF7L2 for TGF-β dependent transcriptional activation of the TMEPAI gene. The Journal of Biological Chemistry. 2010;285(49):38023–38033. doi: 10.1074/jbc.M110.132209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe K, Saito A, Tamaoki T. Cell-specific enhancer activity in a far upstream region of the human α-fetoprotein gene. The Journal of Biological Chemistry. 1987;262(10):4812–4818. [PubMed] [Google Scholar]
- 33.Nakabayashi H, Watanabe K, Saito A, Otsuru A, Sawadaishi K, Tamaoki T. Transcriptional regulation of β-fetoprotein expression by dexamethasone in human hepatoma cells. The Journal of Biological Chemistry. 1989;264(1):266–271. [PubMed] [Google Scholar]
- 34.Nakabayashi H, Hashimoto T, Miyao Y, Tjong K-K, Chan J, Tamaoki T. A position-dependent silencer plays a major role in repressing β-fetoprotein expression in human hepatoma. Molecular and Cellular Biology. 1991;11(12):5885–5893. doi: 10.1128/mcb.11.12.5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang S, Kim TS, Dong Y, et al. AT motif binding factor 1 (ATBF1) is highly phosphorylated in embryonic brain and protected from cleavage by calpain-1. Biochemical and Biophysical Research Communications. 2012;427(3):537–541. doi: 10.1016/j.bbrc.2012.09.092. [DOI] [PubMed] [Google Scholar]
- 36.Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W. Identification and functional characterization of a smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-β, activin, and bone morphogenetic protein-inducible enhancer. The Journal of Biological Chemistry. 1998;273(33):21145–21152. doi: 10.1074/jbc.273.33.21145. [DOI] [PubMed] [Google Scholar]
- 37.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier J. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. The EMBO Journal. 1998;17(11):3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ido A, Nakata K, Kato Y, et al. Gene therapy for hepatoma cells using a retrovirus vector carrying herpes simplex virus thymidine kinase gene under the control of human α-fetoprotein gene promoter. Cancer Research. 1995;55(14):3105–3109. [PubMed] [Google Scholar]
- 39.Roberts AB, Wakefield LM. The two faces of transforming growth factor β in carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(15):8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bierie B, Moses HL. Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nature Reviews Cancer. 2006;6(7):506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]