

Abstract
Nicotine binds to nicotinic acetylcholine receptors (nAChR), which can exist as many different subtypes. The α4β2 nAChR is the most prevalent subtype in the brain and possesses the most evidence linking it to nicotine seeking behavior. Herein we report the use of mixture based combinatorial libraries for the rapid discovery of a series of α4β2 nAChR selective compounds. Further chemistry optimization provided compound 301, which was characterized as a selective α4β2 nAChR antagonist. This compound displayed no agonist activity but blocked nicotine-induced depolarization of HEK cells with an IC50 of approximately 430 nM. 301 demonstrated nearly 500-fold selectivity for binding and 40-fold functional selectivity for α4β2 over α3β4 nAChR. In total over 5 million compounds were assessed through the use of just 170 samples in order to identify a series of structural analogues suitable for future optimization toward the goal of developing clinically relevant smoking cessation medications.
INTRODUCTION
Neuronal nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels that mediate cation flux and are activated by nicotine, the major constituent of tobacco leading to addiction.1,2 In mammalian neural tissue, the nAChR is a pentamer usually made up of a combination of α and β subunits,3 although there are also fully functional homomeric receptor proteins, particularly the prominent α7 nAChR. The α4β2* nAChR (where * denotes the possible presence of an additional subunit) is by far the most prevalent in the CNS,4,5 being expressed in large numbers throughout the brain. Addiction to cigarettes is primarily mediated by nicotine-induced activation of nAChR and, as with other abused drugs, nicotine addiction is thought to be primarily due to the activation of the dopaminergic mesocorticolimbic pathway.6,7 The α4β2* nAChR is the most prevalent nicotinic receptor subtype in this region.4,8
Despite the prevalence of the α4β2* nAChR in the drug reward pathway, a considerable amount of research has been conducted on many different nAChR receptor types and individual subunits, with many different receptor types and subunits having been demonstrated to be involved in acquiring or maintaining addiction to nicotine or other abused drugs. Pharmacological and genetic evidence has suggested that the α3β4* nAChR, found primarily in the medial habenula (MHb) and intepedunclear nucleus, is involved in addiction to cigarettes and other abused drugs. The relatively nonselective α3β4 antagonist 18-methoxycoronaridine (18-MC) blocks the self-administration and conditioned place preference of many abused drugs.9 The more selective and higher affinity α3β4 nAChR ligand AT-1001 potently blocks nicotine self-administration and reinstatement.10 These data are consistent with Genome Wide Association Studies (GWAS) that have implicated variants in the α3-α5-β4 gene cluster on chromosome 15 to be associated with an increased risk of whether a smoker becomes nicotine dependent and to smoking a greater number of cigarettes per day.11,12 The GWAS, therefore, also suggest a possible involvement of the α5 subunit in tobacco dependence. This is consistent with behavioral experiments. In α5 null mice, there is an increase in nicotine conditioned place preference and nicotine self-administration at higher nicotine levels.13,14 Furthermore, viral vector-mediated reintroduction of α5 subunits into the MHb recovers wild-type nicotine self-administration behavior.14 The studies indicate that nAChRs containing the α5 subunit may be involved in limiting the reward mediated by higher nicotine doses. There is also considerable evidence that the α6 subunit, which is also found in high concentrations in the mesolimbic dopamine pathway, is involved in nicotine reward. α6 knockout (KO) mice do not self-administer nicotine, and inhibition of α6-containing nAChR using conotoxin also blocks nicotine self-administration.15
Nevertheless, the greatest body of evidence accumulated for the involvement of a particular nAChR in smoking pertains to the α4β2* nAChR. Neither β2- nor α4-knockout mice self-administer nicotine.16,17 In addition, transgenic mice with a sensitized α4 subunit self-administer nicotine at lower doses,18 indicating that changes in the α4 alone is sufficient to modify nicotine self-administration. Pharmacological evidence for the involvement of the α4β2* nAChR also exists. The selective α4β2 antagonist dihydro-β-erythroidine hydrobromide (DHβE) blocks nicotine self-administration in rats,19 and most important of all, the α4β2 partial agonist varenicline is clinically used as a smoking cessation medication.
Unfortunately, although varenicline exhibits binding selectivity, this selectivity is not observed in functional assays. In functional assays, it is a full agonist at both α3β4 and α7 nAChR with comparable potency to that observed at α4β220,21 Varenicline users haves also experienced significant adverse psychiatric effects, including depression and suicidal thoughts, limiting its clinical usefulness.22 Although other high affinity α4β2 agonists and antagonists have been reported, generally, the selectivity compared to α3β4 nAChR and functional activity at α3β4 nAChR were generally not characterized.23–25 Here we report novel selective α4β2 nAChR antagonists, derived from compounds originally identified from combinatorial libraries containing mixtures of small molecules.
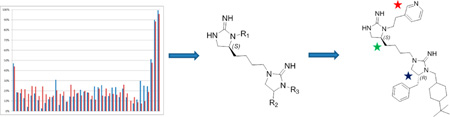
Starting from a small molecule mixture-based combinatorial scaffold ranking library with more than 5 million individual compounds arranged into 34 samples, we selected a mixture-based library comprised of 45864 compounds systematically arranged into 110 mixtures for further screening. Upon screening of this positional scanning library, a number of mixtures showed affinity and selectivity for α4β2 nAChR. From this single library, we identified 18 individual compounds with modest affinity but some with excellent selectivity for α4β2 nAChR over α3β4 nAChR. These lead compounds were modified to produce antagonists with high affinity and impressive selectivity in both receptor binding studies and in functional activity in vitro. The success of this approach shown herein demonstrates the ability of mixture-based combinatorial libraries to identify individual novel structures in the context of any low- to medium-throughput assay (Figure 1).
Figure 1.

Approach used to identify potent and selective (over α3β4) α4β2 ligands. Starting with just 34 samples comprised of >5 million compounds in the Scaffold Ranking Library, a specific Positional Scanning Library (1169) was identified for screening. This library contained 110 samples, and the results from screening the library were used in the selection of 18 compounds that were synthesized and tested (Deconvolution). On the basis of these results, eight new optimized compounds were synthesized and tested and found to be potent and selective (over α3β4) α4β2 ligands. In total, only 170 samples were tested; this shows the broad utility of this approach for many assay formats, especially those limited for whatever reason by throughput.
RESULTS AND DISCUSSION
Scaffold Ranking
To reduce the number of samples to test, we began by using our scaffold ranking library.26,27 As reported elsewhere, we have successfully utilized the scaffold ranking approach in efforts directed toward the identification of metalloprotease ADAM 17 ligands,28 antimicrobials that inhibit tyrosine recombinases and Holliday junction-resolving enzymes,29,30 and novel antinoceptives through the use of a direct in vivo assay.31 We have described in some detail the method for constructing a scaffold ranking library,26 as well as the advantages and limitations of the approach, elsewhere.27 The scaffold ranking library utilized in this work contained 34 mixture samples that in total contained more than 5 million individual compounds (Supporting Information Table S1). As previously described,26 each mixture sample contains only compounds with the same specific core scaffold, so only close structural analogues are contained in each mixture sample. The mixture samples tested in this work contained anywhere from as few as 10800 individual compounds (Library 1409 Supporting Information Table S1) to as many as 738192 (Libraries 1343–1347 Supporting Information Table S1). The median complexity was 45864 compounds per sample. Each of these individual compounds is represented at an approximately equal molar concentration within a given sample. Because the samples are organized to contain only structural analogues, if a particular individual compound is active then the mixture sample containing that compound may possesses many more structural analogues that are also active. Therefore the effective concentration of active compounds in a given mixture is, in general, significantly higher than it would be if only one compound in the mixture was active.32 In this manner, we are able to utilize these 34 mixture samples to prioritize which chemical scaffolds are likely to contain active individual compounds.
These 34 mixtures were screened at the four different concentrations 10, 5, 1, and 0.5 µg/mL for their ability to block [3H]epibatidine binding to α4β2 and α3β4 nAChR, as described in Materials and Methods. Shown in Figure 2 are the results from the 10 µg/mL screening (data from all doses is shown in Supporting Information Table S2).
Figure 2.

Scaffold Ranking Library data for the 10 µg/mL dose (approximately 20 µM assuming an average molecular weight of 500 Da for each compound in a given mixture). Values are shown as percent inhibition of [3H]epibatidine binding to α4β2 (red) and α3β4 (blue) nAChR. The numbers along the x-axis represent the specific Scaffold Ranking Library sample, sc = solvent control (0.1%DMF), and the EPI (no.) are different concentrations of epibatidine (EPI(1) = 1 nM, (10) = 10 nM, (100) = 100 nM, (1000) = 1000 nM).
1169 Library Screening
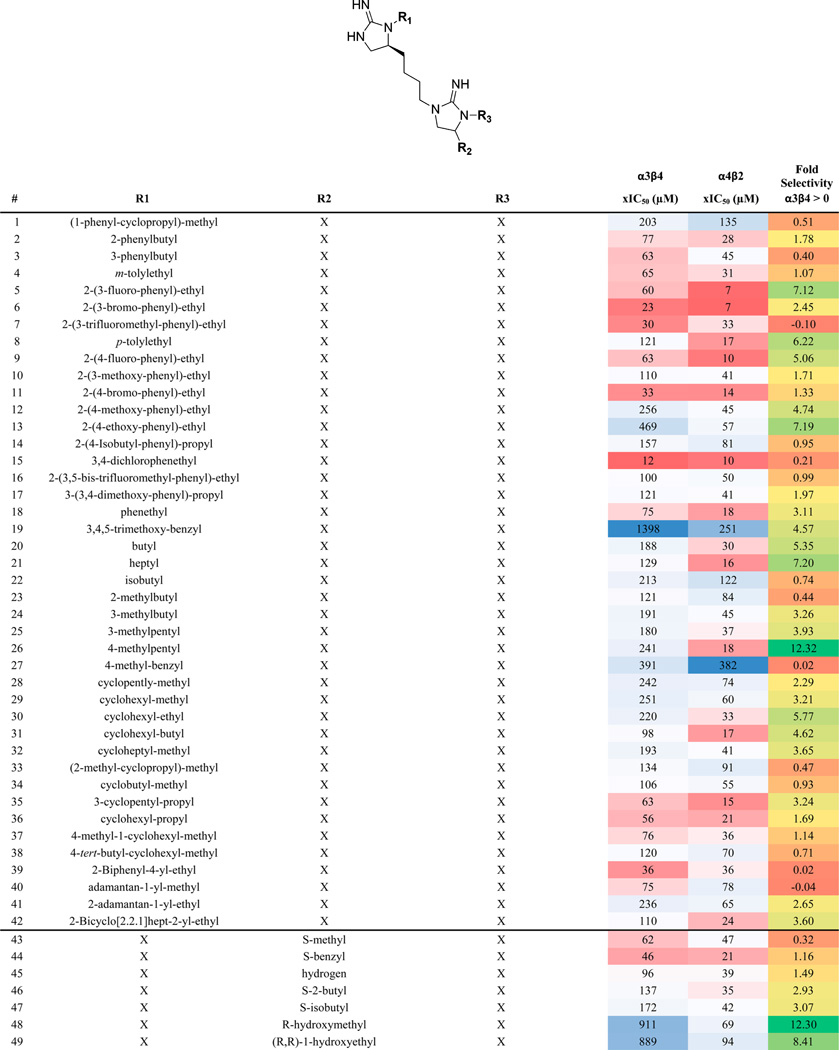
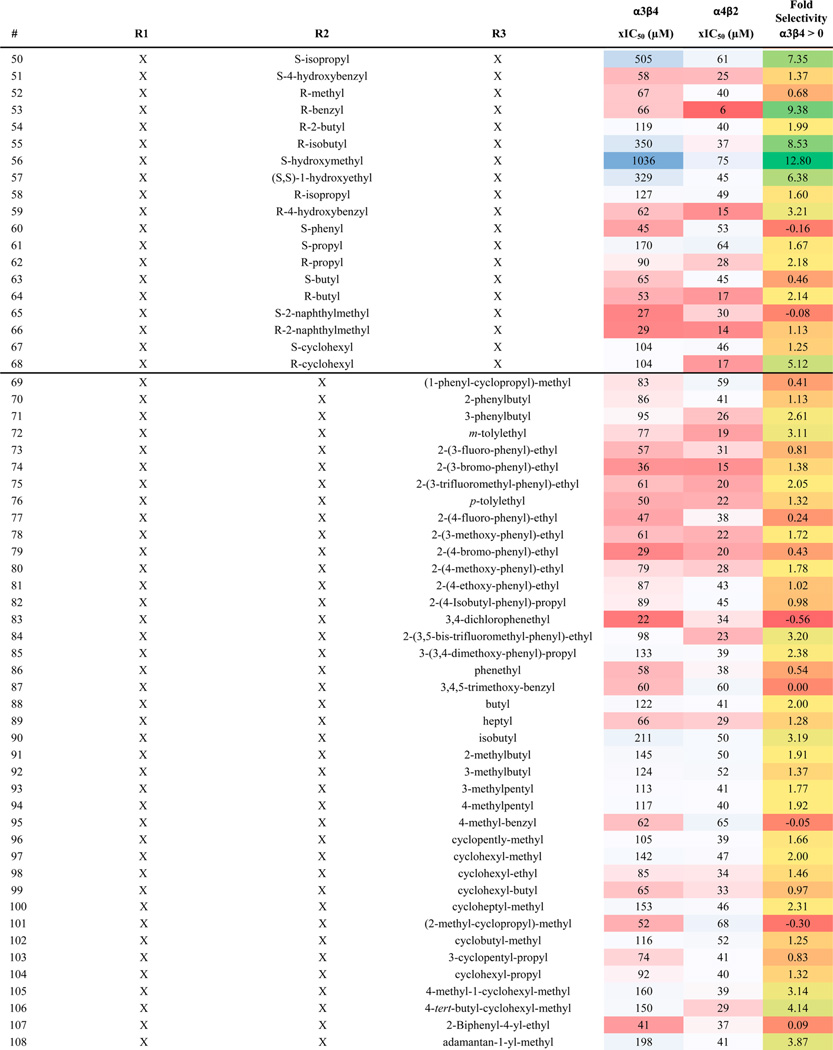
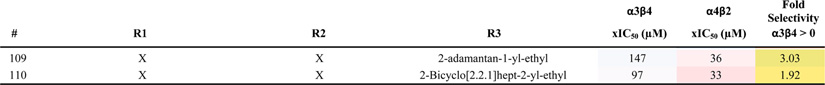
We identified positional scanning library 1169 for screening based on the results of the scaffold ranking library. Library 1169 contains 45864 individual bis-cyclic guanidines (see Scheme 1) systematically formatted into a positional scanning library containing 110 mixture samples (Table 1). For example, sample 1 contains in approximately equal molar amounts the 1092 individual compounds that have (1-phenyl-cyclopropyl)-methyl fixed at the R1 position (Table 1). In this manner, the first 42 samples of library 1169 contain all 45864 compounds in the library sorted by R1 functionality. Samples 43–68 contain the same 45864 compounds sorted by R2 functionality, and samples 69–110 are sorted by R3 functionality.
Scheme 1.

Synthesis of Bis-cyclic Guanidines
Table 1.
The 110 Samples That Make up Library 1169, Their Associated xIC50 Values for α4β2 and α3β4 nAChR, and Fold Selectivity Are Reporteda
|
Library 1169 is made up of 45864 bis-cyclic guanidines all containing the same central scaffold shown at the top of Table 1. The compounds are systematically synthesized into a positional scanning library containing the 110 samples shown in the table. By way of example, sample 1 is an approximate equal molar mixture of 1092 compounds. The 1092 compounds contain (1-phenyl-cyclopropyl)-methyl fixed in the R1 position, and all 1092 combinations of the 26 R2 (shown in Table 1 43–68) and 42 functionalities at R3 (Table 1 69–110). Similarly, sample 83 contains the 1092 compounds generated from fixing R3 with 3,4-dichlorophenethyl and utilizing all 1092 combinations of the 42 R1 functionalities and 26 R2 functionalities. The xIC50 and fold selectivity values were calculated as described in the Materials and Methods section. The xIC50 values are further color coded using a three-color scale: red (most active), white, blue (least active). The fold selectivity values are also color coded using a three-color scale: green (more selective for α4β2), yellow, red (less selective for α4β2). A value of 0 for fold selectivity means a sample produced the same activity in both receptors.
The 110 samples were screened for binding affinity exactly as conducted on the original scaffold ranking library. To use Harmonic Meaning to confirm the integrity of the screening,32 percent inhibition values were converted to xIC50 values, assuming a Hill coefficient of one (see Materials and Methods). The Harmonic Mean was then taken in each position for each receptor. The calculated µM harmonic mean values at the R1, R2, and R3 positions were (respectively) 26.6, 28.0, and 34.1 for the α4β2 receptor, and 78.6, 82.4, and 73.2 for the α3β4 receptor; the deviation of at most 27% at each receptor represents excellent evidence of the integrity of the synthesis and screening. The xIC50 values for each sample are shown in Table 1 as well as the fold selectivity. In general, the samples produced a stronger response against α4β2 than against α3β4. The data shows significant differentiation in activity levels among the samples tested at each position, a key feature for deconvoluting a positional scanning library.27 Moreover, some preliminary SAR can be seen from the library screening. For example when an aromatic group with a halogen substitution is fixed in the R1 position (Table 1: 5, 6, 9, 11, and 15), the sample produced strong inhibition for α4β2. However, while these same fixed functionalities provided activity at the R3 position (Table 1: 73, 74, 77, 79, and 83) against α4β2, a number of other samples with different functionalities fixed at this position also produced strong inhibition. At the R2 position, the profile indicates that for activity an R configuration could be preferred over S for α4β2 (Table 1: 53 over 44, 59 over 51, 62 over 61, 66 over 65, and 68 over 67). The screening data also identifies several active samples that are highly selective for α4β2 over α3β4 (>4-fold selectivity, such as Table 1: 5, 21, 26, 53, 68, and 106). There were some samples that showed activity against both targets providing low levels of selectivity (<1.5-fold selectivity, Table 1: 15, 65, 66, 74, 79, and 83).
Selection of Individual Compounds for Synthesis
On the basis of the 1169 library screening data, 18 individual compounds were selected for synthesis (Scheme 1). These 18 compounds were selected based on three different criteria. The first set was chosen by selecting functionalities that were active at both α4β2 and α3β4 nAChR, showing little selectivity for one receptor over the other. For the R1 position, 3,4-dichlorophenethyl (15) was selected. While two other samples that also have aromatic functionalities with halogen groups 2-(3-fluoro-phenyl)-ethyl (5) and 2-(3-bromo-phenyl)-ethyl (6) were actually more active at α4β2 than 15, they were also more selective for α4β2 in the library screening (Table 1). From the R2 position, R-2-naphthylmethyl (66) and S-2-naphthylmethyl (65) were selected due to the fact they showed activity at both receptors. Additionally, by using these chiral isomers, the preference for the R configuration for activity at α4β2 could be confirmed with individual compounds. From the R3 position, three different halogen substituted aromatic functionalities that showed activity and low selectivity in the library screening were selected: 2-(3-bromo-phenyl)-ethyl (74), 2-(4-bromo-phenyl)-ethyl (79), and 3,4-dichlorophenethyl (83). There are therefore only six compounds in this set (1 × 2 × 3), identified as compounds 201 through 206 in Table 2. While this set of individual compounds was chosen with the purpose of reducing receptor selectivity, it should also be noted that this is the set of compounds that would have been made if the sole purpose was to choose the compounds most likely to be active at α3β4. In contrast to the first set of compounds, the second set was chosen in order to maximize affinity at α4β2 as well as selectivity for α4β2 over α3β4. Additionally, consideration was made to explore chemical diversity where appropriate. For the R1 position in this set, three different functionalities were chosen: 2-(3-fluoro-phenyl)-ethyl (5), heptyl (21), and 4-methylpentyl (26). While sample 5 (Table 1) was not the only sample in the library with a substituted aromatic functionality at R1 that showed activity and selectivity at α4β2, it was the most active/selective sample and therefore was selected. The two aliphatic functionalities, heptyl and 4-methylpentyl, were chosen because they showed strong selectivity for α4β2 (7.20- and 12.32-fold, respectively) and provided a different chemical characteristic. Likewise for the R2 position in this set, two chemically diverse functionalities were selected: R-benzyl and R-cyclohexyl. Both mixture samples with these defined functionalities, 53 and 68, respectively, showed affinity for α4β2 and strong selectivity. For the R3 position, in general, mixture samples with aliphatic groups defined showed slightly less potency but more selectivity for α4β2 than samples defined with aromatic groups. Therefore, for this set, 4-tert-butyl-cyclohexyl-methyl was utilized, as it was among the most active mixtures with an aliphatic defined R3 group and the most selective R3 sample (R3 samples are samples 69–110 in Table 1). This second set of individuals is shown in Table 2 as compounds 207–212. A final set of six compounds was selected for synthesis from the library screening, compounds 213–218 shown in Table 2. This set contains the same functionalities at R1 and R2 as the previous set but replaces the R3 group with 2-(3-bromo-phenyl)-ethyl (Table 1: 74). This substitution changes the functionality used in the R3 group from the most selective R3 functionality in the library at this position to the most active at α4β2 regardless of selectivity. In so doing, this set contains among it, the compound that combines the most active functionaities at each position (Table 2: 213) against α4β2.
Table 2.
The Functionalities, Ki Values, and Selectivity (Both Experimentally Determined and Projected) for the 18 Individual Compounds Synthesized Based on the Data Generated from the Screening of 1169 are Showna
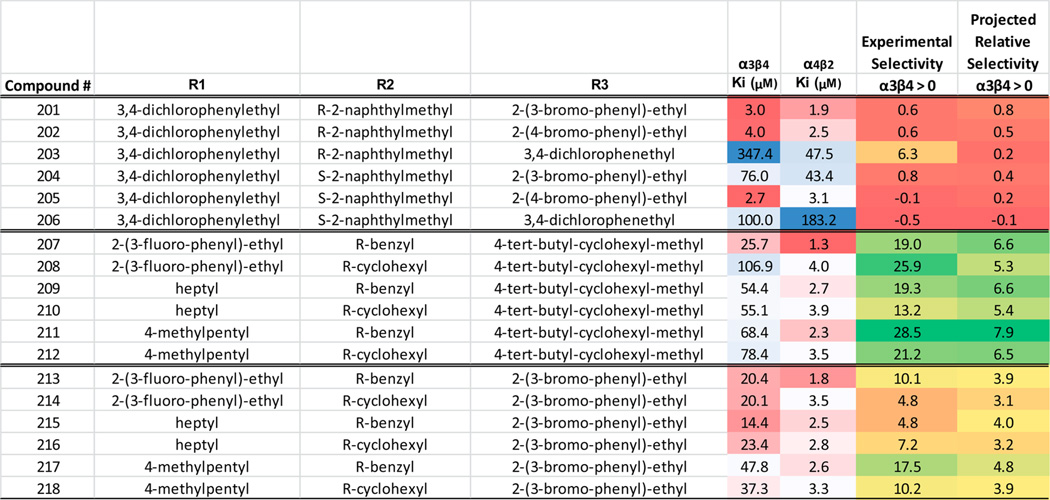 |
The functionalities correspond to the core scaffold shown in Table 1. The activity data is presented as a Ki (µM) for each individual compound against each of the two targets. The experimental selectivity is the value obtained from (Ki α3β4 - Ki α4β2)/(Ki α4β2). The projected relative selectivity value is generated from using only library 1169 data as described in the Materials and Methods section. The Ki values are further color coded using a three-color scale: red (most active), white, blue (least active). The fold selectivity values are also color coded using a three-color scale: green (more selective for α4β2), yellow, red (less selective for α4β2). A value of 0 for fold selectivity means a sample produced the same activity in both receptors. Note the strong correlation between the experimental and projected selectivity values (R2 = 0.831), also shown in Figure 3.
Screening of Individual Compounds
The 18 individual compounds were synthesized and screened against α4β2 and α3β4 nAChR (Table 2). The results further confirmed the SAR identified from the library screening. The first set of six compounds proved to be the least selective of the three sets, the second set the most selective, and the third set of six showed moderate selectivity. The predicted selectivity versus experimental results is further shown in Figure 3. The experimental selectivity exhibited a high correlation of R = 0.91 to predicted values (R2 = 0.83), demonstrating the validity of choosing compounds to synthesize based upon the affinities of the individual functionalities in positions 1, 2, and 3 derived from mixture screening as well as the validity of our methodology of predicting binding affinities. Of the 18 individual compounds tested in these sets, the highest affinity compounds against α3β4 came from the first set (201, 202, and 205); this was to be expected as this set contained the compounds that matched the most active functionalities against α3β4 in the library screening. From the first set of six compounds, 201–206, the apparent preference for R over S absolute configuration at the R2 position that was seen at the library level for α4β2 is also observed. When comparing the affinities of the second set of six compounds (207–212) against the last set of six (213–218), we note that the respective affinities against α4β2 remain similar but the selectivity in general decreases significantly. The only difference in these two sets is the functionality used in the R3 position. For compounds 207–212, the aliphatic functionality 4-tert-butyl-cyclohexyl-methyl was utilized whereas the halogen substituted aromatic 2-(3-bromo-phenyl)-ethyl group was used for compounds 213–218. The results indicate that the R3 group change has little effect on the affinity of the compounds against α4β2 but has a dramatic effect on their affinity for α3β4. We sought to further exploit these observations in order to produce a set of compounds with increased affinity for α4β2 as well as selectivity for α4β2 over α3β4.
Figure 3.

The values for “Projected Selectivity” and “Experimental Selectivity” shown in Table 2 for the 18 compounds are graphed in the scatter plot. Each dot corresponds to one of the 18 compounds. The Experimental Selectivity value is calculated using the following equation (Ki α3β4 – Ki α4β2)/(Ki α4β2). The Projected Selectivity value is a value “projected” from using the 1169 library data. The methodology used to determine the Projected Selectivity is detailed in Materials and Methods. In brief, first a projected activity value for each compound is determined for both receptors. This is done by taking the product of the activity value of the 1169 positional scanning sample that corresponds with the functionality used in the Table 2 individual compounds. Once this value is obtained for each of the compounds and for both receptors, the Projected Selectivity value can be determined using the same selectivity formula used for the Experimental Selectivity values.
Optimization of Individuals
Utilizing the acquired data, compounds 207 and 208 were selected for further optimization. Compound 207 was selected because it exhibits the highest affinity for α4β2, is among the most selective for α4β2, and has an aromatic functionality at the R1 position that we identified for modification. Compound 208 was selected, as it is the α4β2 selective analogue of 207 containing the same aromatic group in the R1 position. The focus of the optimization was the modification of the R1 functionality. While the 1169 library contains a diverse set of aromatic functionalities in the R1 position, it does not contain any pyridyl groups. A number of reported nAChR ligands contain pyridyl groups, and some of the most potent, including nicotine, contain a 3-pyridyl group or substituted 3-pyridyl group such as epibatidine.33 Using the same two-carbon spacer off the core cyclic guanidine scaffold, the 3-fluoro-phenyl group of 207 was replaced with a 3-pyridyl group in 301 (see Figure 4, Table 3). This substitution drastically increased its affinity at α4β2, from 1587 to 118 nM while remaining relatively inactive at α3β4, 61750 nM for 207 and 56480 nM for 301, producing a high affinity and highly selective compound. The same R1 modification to compound 208 also dramatically increased its α4β2 affinity and selectivity, 306 (see Figure 4, Table 3). Modifying the R2 chirality of 301 and 306 from R to S, compounds 302 and 307, respectively, decreases the affinity at α4β2 in line with what is observed in both the 1169 library samples (Table 1) as well as the individual compounds (Table 2) with regards to this change in absolute configuration at the R2 position. Eliminating the R2 position entirely from 301 and 306 provides the same analogue, 303, which has a lower affinity to α4β2 and selectivity than either parent compound. A change in absolute configuration in the core scaffold of 301 from S to R, compound 304, also decreased its affinity at α4β2, however, the same change to 306 provided a slightly more potent α4β2 ligand, 308. Combining the change in absolute configuration in the core scaffold from S to R with the elimination of the R2 functionality provides the same analogue, 305, which has the lowest affinity at α4β2 of all the analogues tested in this optimization phase. Therefore, from this set of optimized hits, compound 301 was selected for further evaluation.
Figure 4.

Hit optimization of compounds 207 and 208. The colored stars and arrows correspond to site specific analogue modifications.
Table 3.
Binding Affinity, Selectivity, and Antagonist Activity of Selected Individual Compoundsa
| [3H]epibatidine binding |
||||
|---|---|---|---|---|
| compd | α3β4 Ki (nM) | α4β2 Ki (nM) |
selectivity (fold) (α3β4 Ki/α4β2 Ki) |
α4β2 antagonist potency IC50 (nM) |
| 207 | 61750 ± 36030 | 1587 ± 301 | 39 | 26320 ± 980 |
| 208 | 88895 ± 18005 | 3731 ± 237 | 24 | 5319 ± 346 |
| 301 | 56480 ± 8920 | 118 ± 2 | 478 | 514 ± 84 |
| 302 | 23765 ± 5345 | 323 ± 152 | 74 | 1465 ± 43 |
| 303 | 27550 ± 7150 | 433 ± 79 | 64 | 2820 ± 82 |
| 304 | 63320 ± 3560 | 307 ± 78 | 206 | 3024 ± 125 |
| 305 | 51893 ± 27353 | 2217 ± 1194 | 23 | 2953 ± 594 |
| 306 | 59678 ± 15253 | 340 ± 148 | 176 | 2830 ± 75 |
| 307 | 78085 ± 8355 | 611 ± 220 | 128 | 3405 ± 125 |
| 308 | 33353 ± 4373 | 257 ± 113 | 130 | 1686 ± 373 |
| DHβE | >10000 | 485 ± 33 | >20 | 624 ± 24 |
Experiments were conducted a minimum of two times by methods described in Materials and Methods. Values shown are mean ± SEM for experiments conducted in triplicate at least twice.
Biological Profiling of 301
Compound 301 was further evaluated for binding affinity against α3β4α5 nAChR and found to be weakly active with a Kd/Ki of 9062 nM. Compound 301 was tested for functional activity by measuring the ligand-induced change in membrane potential in intact HEK cells transfected with α3β4 and α4β2 nAChR Membrane potential changes were determined using a FlexStation, as described previously10 and in Materials and Methods. Compound 301 displayed no agonist activity in either cell line. As seen in Figure 5 and Table 3, 301 blocks nicotine stimulation at α4β2 nAChR with an IC50 of approximately 500 nM, which is slightly more potent than the selective α4β2 nAChR antagonist DHβE. It is also very selective for α4β2 nCAhR, as it did not inhibit α3β4 nAChR activation by 50% up to 10 µM, the highest concentration tested (data not shown). 301 has no apparent agonist activity, so there is no reason to believe that this compound induces desensitization rather than direct inhibition of nicotine activity. To determine if antagonist activity is competitive, nicotine dose response curves were constructed in the presence of various concentrations of 301. As seen in Figure 6, 301 causes a small rightward shift in the nicotine dose response curve but to a greater extent a decrease in maximal activity. These results appear to suggest a noncompetitive inhibition of nicotine’s activity.
Figure 5.

Antagonist activity of compound 301. 301 has no agonist activity but potently blocks nicotine-induced increase in membrane potential in α4β2-containing cells. 301 has potential inverse agonist activity at very high concentrations.
Figure 6.

Compound 301 inhibits nicotine-induced α4β2 nAChR stimulation in a dose-dependent manner. Increasing concentrations of 301 induce a progressive decrease in maximal activity with a slight change in the EC50. EC50 and maximum stimulation were: 779 nM and 100%, 999 nM and 83%, 1809 nM and 38%, EC50 not measurable, maximum 0, for 0, 0.3, 1.0, and 3.0 µM 301, respectively.
DISCUSSION AND CONCLUSION
Screening to identify novel structures with high affinity to nAChR started with a “scaffold ranking library,” which is a collection of 34 samples. Each of these 34 samples contains a mixture of individual compounds arranged so that compounds with the same structural core “scaffold” are contained in the same sample, overall more than 5 million individual compounds are contained in the 34 mixture samples. From the initial 34 mixtures, we identified a sample that demonstrated moderate affinity for α4β2 nAChR. Therefore the complete positional scanning library associated with this sample was chosen for further screening. This positional scanning library, 1169, contains 45864 individual bis-cyclic guanidines systematically formatted into 110 samples allowing for the identification of individual compounds of high affinity, by methods that were discussed above and described previously.26,34–36 The series of individual compounds identified by the library provided useful SAR that was utilized to produce a series of further optimized compounds with high affinity and selectivity for nAChR subtypes. The highest affinity and most selective compounds appear to be pure antagonists when measuring membrane potential changes in HEK cells transfected with α4β2 nAChR. These compounds have low affinity (IC50 > 10 µM) for α3β4 nAChR.
As we have demonstrated previously, mixture-based libraries provide a method for the efficient and rapid identification of novel structures that can be used directly from the libraries or for further chemical modification.26,37 Of the compounds described here, 301 is a novel compound, never previously demonstrated to bind to nAChR. In FlexStation assays, 301 appears to have no agonist activity at α4β2 nAChR up to 10 µM. When cells are pretreated with 301, the increase in membrane potential induced by nicotine is blocked at concentrations of 301 similar to binding affinity. 301 has an IC50 for inhibition of nicotine activity of approximately 430 nM. This is at least 20-fold selective over its ability to block nicotine activity in cells transfected with α3β4 nAChR. This 20-fold functional selectivity is encouraging because some very selective compounds, such as varenicline, have excellent binding selectivity but very poor functional selectivity.21
Interestingly, inhibition of nicotine stimulation appears to be noncompetitive, as increasing concentrations of 301 induce a progressive decrease in the maximal activity rather than a parallel shift to the right of a nicotine dose response curve. It seems unlikely that this would be due to desensitization of the receptor because 301 does not activate the receptor in these experiments, although it is possible that desensitization of the α4β2 nAChR might occur. The α4β2 partial agonist sazetidine A was originally described as a nonactivating desensitizer, but it was later determined that it was in fact a partial or full agonist in other cellular assays.38,39 Additional studies of 301 will be required to determine if it has agonist activity under some circumstances.
In conclusion, through the use of mixture-based libraries a total over 5 million compounds were assessed through the use of only 170 samples in order to provide a series of nAChR-selective compounds. One compound, 301, has moderately high affinity and binding and functional selectivity for α4β2 over α3β4 nAChR; additional studies are underway on congeners and fragments of 301. The success of this study demonstrates conclusively that mixture-based combinatorial libraries utilizing the scaffold ranking and positional scanning approaches can successfully be used in assays of low to medium throughput to develop novel lead compounds for further research.
MATERIALS AND METHODS
Chemistry General
The 1H and 13C NMR spectra were obtained utilizing the Bruker 400’54 Ascend (400 and 100 MHz, respectively). NMR chemical shifts were reported in δ (ppm) using the δ 7.26 signal of CHCl3 (1H NMR), the δ 4.79 signal of D2O (1H NMR), and the δ 77.16 signal of CDCl3 (13C NMR) as internal standards. 13C NMR spectra in D2O were not adjusted (representative NMR is available in the Supporting Information). Mass spectra data was acquired using Shimatzu LCMS-2010. The reported final purity of the compounds were verified by Shimadzu HPLC and mass spectra under the following conditions: column, Phenomenex Luna 150 mm × 21.20 mm, 5 µm, C18; mobile phase, (A) H2O (+0.1% formic acid)/(B) MeCN (+0.1% formic acid). Three gradient methods were used based on hydro-phobicity (2% B to 20% B, 11 min) (25% B to 45% B, 31 min) (45% B to 65% B, 21 min); flow rate, 12 mL/min; detection, UV 214 nm. The purity of all final compounds were >95%. All chirality was generated from the corresponding amino acids. Under the reaction conditions described, no epimerization was observed and, for those compounds with multiple chiral centers, a single diastereomer was obtained.
Synthesis of Library 1169 and Individual Compounds and Construction of Scaffold Ranking Plate. General Synthesis of Bis-cyclic Guanidines F
Library 1169 and all individual compounds reported (201–218 and 301–308) were synthesized following the same general scheme (Scheme 1).40,41 The solid phase synthesis was performed using the “tea-bag” methodology.42 Initially, 100 mg of p-methylbenzdrylamine (MBHA) resin (1.1 mmol/g, 100–200 mesh) was sealed in a mesh “tea-bag,” neutralized with 5% diisopropylethylamine (DIEA) in dichloromethane (DCM) and subsequently swelled with additional DCM washes. Fmoc-Lys(Boc)-OH (Fmoc-L-Lys(Boc) for all compounds reported within except 304, 305, and 308 where Fmoc-d-Lys(Boc) was used) (6 equiv) was coupled in dimethylfor-mamide (0.1 M DMF) for 120 min in the presence of diisopropylcarbodiimide (DIC, 6 equiv) and 1-hydroxybenzotriazole hydrate (HOBt, 6 equiv) (A, Scheme 1). The Boc protecting group was removed with 55% TFA/DCM for 30 min and subsequently neutralized with 5% DIEA/DCM (3×). Boc-amino acids were coupled utilizing standard coupling procedures (6 equiv) with DIC (6 equiv) and HOBt (6 equiv) in DMF (0.1M) for 120 min (B, Scheme 1). The Boc protecting group was removed with 55% trifluoroacetic acid (TFA) in DCM for 30 min and subsequently neutralized with 5% DIEA/DCM (3×). Carboxylic acids are coupled using (6 equiv) in the presence of DIC (10 equiv) and HOBt (10 equiv) in DMF (0.1M) for 120 min (C, Scheme 1). The Fmoc protecting group was removed with 20% piperidine in DMF for 20 min, and the R1 carboxylic acids were coupled in 10 equiv under the same solvent and activating reagent conditions. All coupling reactions were monitored for completion by the ninhydrin test. The reduction was performed in a 4000 mL Wilmad LabGlass vessel under nitrogen. A borane in 1.0 M tetrahydrofuran complex solution was used in 40-fold excess for each amide bond. The vessel was heated to 65 °C and maintained at temperature for 72 h. The solution was then discarded, and the bags were washed with THF and methanol. Once completely dry, the bags were treated overnight with piperidine at 65 °C and washed several times with methanol, DMF, and DCM (D, Scheme 1). Before proceeding, completion of reduction was monitored by a control cleavage and analyzed by LCMS. Urea cyclization (E, Scheme 1) was performed with a 5-fold excess of cyanogen bromide (CNBr) in a 0.1 M anhydrous DCM solution for each of the cyclization sites overnight. Following the cyclization, the bags were rinsed with DMF and DCM. The resin was cleaved with HF in the presence of anisole in an ice bath at 0 °C for 90 min (F, Scheme 1).
(R)-3-(3-Bromophenethyl)-1-(4-((S)-3-(3,4-dichlorophenethyl)-2-iminoimidazolidin-4-yl)butyl)-4-(naphthalen-1-ylmethyl)-imidazolidin-2-imine (201)
Using general scheme for the synthesis of F, compound 201 was synthesized using the following reagents: 3,4-dichlophenylacetic acid (R1), Boc-d-2-naphthylalanine (R2), 3-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.51 (s, 2H), 7.86 (d, J = 7.6 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.55 (t, J = 7.2 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 8.0 Hz, 2H), 7.32–7.29 (m, 3H), 7.20 (d, J = 6.8 Hz, 2H), 7.15–7.10 (m, 2H), 4.15–4.08 (m, 1H), 3.85–3.80 (m, 2H), 3.65–3.58 (m, 2H), 3.49 (dd, J = 13.6, 4.4 Hz, 1H), 3.40–3.28 (m, 4H), 3.24–3.14 (m, 3H), 2.94–2.88 (m, 3H), 2.82–2.72 (m, 2H), 1.65–1.54 (m, 1H), 1.50–1.35 (m, 3H), 1.25–1.10 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 158.97, 156.94, 140.78, 138.74, 134.10, 132.31, 132.01, 131.67, 131.27, 131.09, 130.71, 130.63, 130.61, 130.10, 129.36, 129.04, 128.44, 127.87, 127.81, 126.92, 126.23, 125.74, 122.74, 58.99, 57.37, 50.83, 46.14, 44.97, 44.65, 43.00, 35.58, 33.39, 32.72, 31.23, 26.52, 20.64. MS (ESI) m/z [M + H]+: 719.0.
(R)-3-(4-Bromophenethyl)-1-(4-((S)-3-(3,4-dichlorophenethyl)-2-iminoimidazolidin-4-yl)butyl)-4-(naphthalen-1-ylmethyl)-imidazolidin-2-imine (202)
Using general scheme for the synthesis of F, compound 202 was synthesized using the following reagents: 3,4-dichlophenylacetic acid (R1), Boc-d-2-naphthylalanine (R2), 4-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.52 (s, 1H), 8.34 (s, 1H), 7.86 (d, J = 7.2 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.55–7.49 (m, 2H), 7.41 (s, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.35 (d, J = 7.6 Hz, 2H), 7.30 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 7.2 Hz, 2H), 7.08 (d, J = 7.6 Hz, 2H), 4.15–4.12 (m, 1H), 3.92–3.84 (m, 2H), 3.65–3.57 (m, 2H), 3.51 (dd, J = 13.6, 4.0 Hz, 1H), 3.42–3.25 (m, 5H), 3.22–3.15 (m, 2H), 2.94–2.90 (m, 1H), 2.88–2.75 (m, 4H), 1.65–1.56 (m, 1H), 1.50–1.37 (m, 3H), 1.27– 1.13 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 158.89, 157.01, 138.81, 137.38, 134.12, 132.28, 131.95, 131.67, 131.33, 131.11, 130.91, 130.71, 130.60, 129.35, 129.10, 128.42, 127.78, 126.90, 126.24, 125.75, 122.73, 120.83, 59.05, 57.14, 50.87, 46.09, 44.92, 44.45, 43.01, 35.55, 33.25, 32.72, 31.15, 26.51, 20.57. MS (ESI) m/z [M + H]+: 718.9.
(R)-3-(3,4-Dichlorophenethyl)-1-(4-((S)-3-(3,4-dichloropheneth-yl)-2-iminoimidazolidin-4-yl)butyl)-4-(naphthalen-1-ylmethyl)-imidazolidin-2-imine (203)
Using general scheme for the synthesis of F, compound 203 was synthesized using the following reagents: 3,4-dichlophenylacetic acid (R1), Boc-d-2-naphthylalanine (R2), 3,4-dichlorophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.61 (s, 2H), 7.83 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.54–7.46 (m, 2H), 7.40–7.38 (m, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.27–7.25 (m, 2H), 7.21 (d, J = 5.6 Hz, 2H), 7.18–7.13(m, 1H), 7.03 (d, J = 6.4 Hz, 1H), 4.15–4.02 (m, 1H), 3.94–3.85 (m, 2H), 3. 56–3.47 (m, 3H), 3.35–3.25 (m, 5H), 3.20–3.13 (m, 2H), 2.97–2.92 (m, 1H), 2.88–2.70 (m, 4H), 1.58–1.50 (m, 1H), 1.42–1.25 (m, 3H), 1.17–1.05 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 159.17, 157.03, 138.83, 138.57, 134.06, 132.42, 132.21, 131.60, 131.31, 131.05, 130.91, 130.82, 130.64, 130.50, 129.35, 129.02, 128.77, 128.42, 127.77, 126.90, 126.24, 125.72, 122.65, 58.79, 57.20, 50.99, 46.26, 45.11, 44.48, 43.01, 35.66, 32.75, 31.29, 26.52, 24.80, 20.72. MS (ESI) m/z [M + H]+: 709.0.
(S)-3-(3-Bromophenethyl)-1-(4-((S)-3-(3,4-dichlorophenethyl)-2-iminoimidazolidin-4-yl)butyl)-4-(naphthalen-1-ylmethyl)-imidazolidin-2-imine (204)
Using general scheme for the synthesis of F, compound 204 was synthesized using the following reagents: 3,4-dichlophenylacetic acid (R1), Boc-L-2-naphthylalanine (R2), 3-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.64 (s, 2H), 7.84 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.55–7.47 (m, 2H), 7.39 (s, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.38 (d, J = 3.2 Hz, 1H), 7.30–7.26 (m, 2H), 7.19 (d, J = 7.2 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 7.12–7.08 (m, 2H), 4.13–4.10 (m, 1H), 3.92–3.84 (m, 1H), 3.82–3.75 (m, 1H), 3.65–3.53 (m, 2H), 3.46 (dd, J = 13.6, 4.0 Hz, 1H), 3.38–3.25 (m, 4H), 3.19–3.12 (m, 3H), 2.91 (dd, J = 13.6, 9.2 Hz, 1H), 2.84–2.78 (m, 4H), 1.57–1.48 (m, 1H), 1.46–1.35 (m, 3H), 1.20–1.10 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 159.25, 157.01, 140.73, 138.88, 134.06, 132.28, 131.96, 131.65, 131.35, 131.08, 130.68, 130.61, 130.55, 130.07, 129.33, 129.00, 128.40, 127.86, 127.81, 126.90, 126.21, 125.73, 122.71, 58.78, 57.35, 50.86, 46.21, 45.07, 44.71, 42.98, 35.58, 33.32, 32.73, 31.30, 26.54, 20.66. MS (ESI) m/z [M + H]+: 719.0.
(S)-3-(4-Bromophenethyl)-1-(4-((S)-3-(3,4-dichlorophenethyl)-2-iminoimidazolidin-4-yl)butyl)-4-(naphthalen-1-ylmethyl)-imidazolidin-2-imine (205)
Using general scheme for the synthesis of F, compound 205 was synthesized using the following reagents: 3,4-dichlophenylacetic acid (R1), Boc-L-2-naphthylalanine (R2), 4-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.62 (s, 2H), 7.88–7.85 (m, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.56–7.50 (m, 2H), 7.41–7.39 (m, 2H), 7.36 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 6.8 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.06 (d, J = 8.4 Hz, 2H), 4.20–4.16 (m, 1H), 3.91–3.89 (m, 2H), 3.58–3.55 (m, 2H), 3.51 (dd, J = 14.0, 4.8 Hz, 1H), 3.38–3.31 (m, 4H), 3.21–3.18 (m, 3H), 2.95 (dd, J = 13.6, 8.8 Hz, 1H), 2.85– 2.80 (m, 4H), 1.61–1.50 (m, 1H), 1.50–1.38 (m, 3H), 1.25–1.13 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 159.28, 157.01, 138.76, 137.18, 134.13, 132.38, 132.00, 131.66, 131.26, 131.10, 130.86, 130.72, 130.69, 129.38, 128.98, 128.49, 127.81, 126.94, 126.29, 125.76, 122.69, 120.92, 58.76, 57.20, 50.96, 46.19, 45.17, 44.66, 43.07, 35.64, 33.22, 32.81, 31.05, 26.53, 20.56. MS (ESI) m/z [M + H]+: 718.9.
(S)-3-(3,4-Dichlorophenethyl)-1-(4-((S)-3-(3,4-dichloropheneth-yl)-2-iminoimidazolidin-4-yl)butyl)-4-(naphthalen-1-ylmethyl)-imidazolidin-2-imine (206)
Using general scheme for the synthesis of F, compound 206 was synthesized using the following reagents: 3,4-dichlophenylacetic acid (R1), Boc-L-2-naphthylalanine (R2), 3,4-dichlorophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.62 (s, 2H), 7.83 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.55–7.47 (m, 2H), 7.38–7.35 (m, 2H), 7.28–7.25 (m, 2H), 7.22 (d, J = 7.2 Hz, 1H), 7.20 (s, 1H), 7.15 (d, J = 7.6 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 4.13–4.02 (m, 1H), 4.00–3.90 (m, 1H), 3.87–3.77 (m, 1H), 3. 56–3.47 (m, 3H), 3.29–3.26 (m, 5H), 3.20– 3.17 (m, 2H), 2.97 (q, J = 8.8 Hz, 1H), 2.85–2.70 (m, 4H), 1.58–1.50 (m, 1H), 1.47–1.28 (m, 3H), 1.17–1.05 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 159.19, 157.03, 138.84, 138.54, 134.09, 132.48, 132.26, 131.63, 131.36, 131.07, 130.91, 130.88, 130.82, 130.67, 130.55, 129.38, 129.00, 128.76, 128.45, 127.80, 126.93, 126.27, 125.75, 122.66, 58.79, 57.21, 51.03, 46.20, 45.11, 44.50, 43.00, 35.73, 32.74, 31.31, 26.52, 20.68. MS (ESI) m/z [M + H]+: 709.0.
(R)-4-Benzyl-3-((4-tert-butylcyclohexyl)methyl)-1-(4-((S)-3-(3-flu-orophenethyl)-2-iminoimidazolidin-4-yl)butyl)imidazolidin-2-imine (207)
Using general scheme for the synthesis of F, compound 207 was synthesized using the following reagents: 3-fluorophenylacetic acid (R1), Boc-d-phenylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.53 (s, 1H), 8.25 (s, 2H), 7.30 (t, J = 7.2 Hz, 2H), 7.24 (d, J = 7.6 Hz, 1H), 7.20 (t, J = 6.8 Hz, 1H), 7.12 (d, J = 7.2 Hz, 2H), 7.07 (d, J = 7.6 Hz, 1H), 7.03 (d, J = 10.0 Hz, 1H), 6.84 (t, J = 8.4 Hz, 1H), 4.04–3.99 (m, 1H), 3.92–3.84 (m, 1H), 3.81–3.61 (m, 1H), 3. 53–3.42 (m, 3H), 3.37–3.26 (m, 2H), 3.24–3.17 (m, 3H), 3.11–2.95 (m, 2H), 2.90–2.80 (m, 2H), 2.73–2.65 (m, 1H), 1.83–1.75 (m, 1H), 1.73– 1.68 (m, 2H), 1.65–1.45 (m, 4H), 1.43–1.30 (m, 3H), 1.20–1.11 (m, 2H), 1.08 (d, J = 12.8 Hz, 1H), 1.03–0.98 (m, 1H), 0.95–0.90 (m, 2H), 0.81 (s, 9H). MS (ESI) m/z [M + H]+: 589.2.
(R)-3-((4-tert-Butylcyclohexyl)methyl)-4-(cyclohexylmethyl)-1-(4-((S)-3-(3-fluorophenethyl)-2-iminoimidazolidin-4-yl)butyl)-imidazolidin-2-imine (208)
Using general scheme for the synthesis of F, compound 208 was synthesized using the following reagents: 3-fluorophenylacetic acid (R1), Boc-d-cyclohexylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, D2O): δ 7.44 (q, J = 7.6 Hz, 1H), 7.19 (t, J = 7.6 Hz, 1H), 7.12 (t, J = 8.0 Hz, 2H), 4.06–4.00 (m, 1H), 3.80 (q, J = 9.6 Hz, 2H), 3.72–3.66 (m, 2H), 3.62–3.55 (m, 1H), 3.48–3.38 (m, 1H), 3.36–3.31 (m, 2H), 3.29–3.21 (m, 1H), 3.16 (d, J = 7.6 Hz, 1H), 2.99 (t, J = 6.4 Hz, 2H), 2.75–2.73 (m, 1H), 1.86–1.71 (m, 8H), 1.66– 1.54 (m, 8H), 1.43–1.37(m, 2H), 1.31–1.25 (m, 5H), 1.10–1.08 (m, 2H), 1.04–1.03 (m, 2H), 0.99–0.93 (m, 2H), 0.89 (s, 9H). MS (ESI) m/z [M + H]+: 595.2.
(R)-4-Benzyl-3-((4-tert-butylcyclohexyl)methyl)-1-(4-((S)-3-heptyl-2-iminoimidazolidin-4-yl)butyl)imidazolidin-2-imine (209)
Using general scheme for the synthesis of F, compound 209 was synthesized using the following reagents: heptanoic acid (R1), Boc-d-phenylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+ % (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.54 (s, 1H), 8.00 (s, 2H), 7.32 (t, J = 7.2 Hz, 2H), 7.27–7.25 (m, 1H), 7.14 (d, J = 7.2 Hz, 2H), 4.06–4.02 (m, 1H), 3.85–3.80 (m, 1H), 3.73–3.65 (m, 2H), 3.55–3.47 (m, 2H), 3.40–3.32 (m, 2H), 3.29–3.22 (m, 2H), 3.12–3.05 (m, 2H), 3.03– 2.97 (m, 1H), 2.76–2.68 (m, 1H), 1.83–1.75 (m, 1H),1.71–1.65 (m, 3H),1.57–1.41 (m, 7H), 1.27–1.25 (m, 11H), 1.13–1.04 (m, 2H), 1.00–0.95 (m, 2H), 0.86 (d, J = 6.4 Hz, 3H), 0.81 (s, 9H). MS (ESI) m/z [M + H]+: 565.2.
(R)-3-((4-tert-Butylcyclohexyl)methyl)-4-(cyclohexylmethyl)-1-(4-((S)-3-heptyl-2-iminoimidazolidin-4-yl)butyl)imidazolidin-2-imine (210)
Using general scheme for the synthesis of F, compound 210 was synthesized using the following reagents: heptanoic acid (R1), Boc-d-cyclohexylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.58 (s, 1H), 3.83–3.79 (m, 2H), 3.66 (t, J = 8.8 Hz, 2H), 3.50–3.44 (m, 4H), 3.37–3.30 (m, 1H), 3.19–3.13 (m, 1H), 3.09–3.03 (m, 1H), 2.96 (dd, J = 15.2, 6.0 Hz, 1H), 1.83–1.77 (m, 2H), 1.75–1.72 (m, 2H), 1.71– 1.67 (m, 4H), 1.66–1.64 (m, 2H), 1.62–1.59 (m, 3H), 1.57–1.48 (m, 4H), 1.37–1.32 (m, 2H), 1.30–1.22 (m, 11H), 1.20–1.16 (m, 2H), 1.14–1.11 (m, 1H), 1.06 (d, J = 8.8 Hz, 1H), 1.03–1.00 (m, 2H), 0.97–0.93 (m, 3H), 0.88–0.86 (m, 3H), 0.82 (s, 9H). MS (ESI) m/z [M + H]+: 571.3.
(R)-4-Benzyl-3-((4-tert-butylcyclohexyl)methyl)-1-(4-((S)-2-imino-3-(4-methylpentyl)imidazolidin-4-yl)butyl)imidazolidin-2-imine (211)
Using general scheme for the synthesis of F, compound 211 was synthesized using the following reagents: 4-methylvaleric acid (R1), Boc-d-phenylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.55 (s, 1H), 7.33 (t, J = 7.2 Hz, 2H), 7.28–7.25 (m, 1H), 7.13 (d, J = 7.2 Hz, 2H), 4.07–4.02 (m, 1H), 3.83–3.80 (m, 1H), 3.71–3.64 (m, 1H), 3.51–3.45 (m, 2H), 3.30–3.21 (m, 4H), 3.10–3.06 (m, 2H), 3.04–2.98 (m, 2H), 2.76–2.68 (m, 1H), 1.83–1.78 (m, 1H), 1.71 (d, J = 10.4 Hz, 1H), 1.66–1.58 (m, 3H), 1.56–1.50 (m, 5H), 1.44–1.38 (m, 2H), 1.27–1.23 (m, 2H), 1.16 (q, J = 7.2 Hz, 3H), 1.07–1.04 (m, 1H), 1.00–0.97 (m, 3H), 0.86 (d, J = 6.8 Hz, 6H), 0.83 (s, 9H). MS (ESI) m/z [M + H]+: 551.2.
(R)-3-((4-tert-Butylcyclohexyl)methyl)-4-(cyclohexylmethyl)-1-(4-((S)-2-imino-3-(4-methylpentyl)imidazolidin-4-yl)butyl)-imidazolidin-2-imine (212)
Using general scheme for the synthesis of F, compound 212 was synthesized using the following reagents: 4-methylvaleric acid (R1), Boc-d-cyclohexylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.54 (s, 1H), 8.28 (s, 2H), 3.84–3.78 (m, 2H), 3.66 (t, J = 9.2 Hz, 2H), 3.48–3.42 (m, 4H), 3.34–3.30 (m, 1H), 3.19–3.13 (m, 1H), 3.05 (dd, J = 14.4, 7.6 Hz, 1H), 2.95 (dd, J = 14.8, 5.6 Hz, 1H), 1.80–1.77 (m, 1H), 1.71 (d, J = 10.4 Hz, 6H), 1.64–1.63 (m, 2H), 1.60 (d, J = 8.0 Hz, 4H), 1.53 (q, J = 6.0 Hz, 4H), 1.49–1.44 (m, 2H), 1.33–1.29 (m, 3H), 1.27–1.24 (m, 1H), 1.23–1.21 (m, 3H), 1.20–1.18 (m, 1H), 1.17–1.15 (m, 2H), 1.13–1.10 (m, 1H), 1.07– 0.1.05 (m, 1H), 1.02–0.99 (m, 2H), 0.96–0.92 (m, 3H), 0.86 (d, J = 6.8 Hz, 6H), 0.82 (s, 9H). MS (ESI) m/z [M + H]+: 557.2.
(R)-4-Benzyl-3-(3-bromophenethyl)-1-(4-((S)-3-(3-fluoropheneth-yl)-2-iminoimidazolidin-4-yl)butyl)imidazolidin-2-imine (213)
Using general scheme for the synthesis of F, compound 213 was synthesized using the following reagents: 3-fluorophenylacetic acid (R1), Boc-d-phenylalanine (R2), 3-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.54 (s, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.31 (s, 1H), 7.28 (d, J = 7.2 Hz, 2H), 7.24 (d, J = 7.2 Hz, 1H), 7.22–7.19 (m, 2H), 7.15 (t, J = 7.6 Hz, 2H), 7.03 (d, J = 6.8 Hz, 3H), 6.85 (t, J = 8.0 Hz, 1H), 4.03–3.98 (m, 1H), 3.87–3.80 (m, 1H), 3.71–3.64 (m, 1H), 3.62–3.50 (m, 2H), 3.36 (t, J = 9.6 Hz, 2H), 3.32–3.20 (m, 4H), 3.12 (dd, J = 9.6, 6.4 Hz, 1H), 2.91 (d, J = 4.0 Hz, 1H), 2.87–2.80 (m, 4H), 2.60 (d, J = 13.2, 7.6 Hz, 1H), 1.62–1.50 (m, 1H), 1.45–1.30 (m, 3H), 1.21–1.14 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 162.98 (J = 244.2 Hz), 159.00, 156.85, 140.99 (J = 7.6 Hz), 140.77, 135.27, 132.02, 130.61, 130.35 (J = 7.1 Hz), 130.07, 129.19 (J = 4.0 Hz), 128.02, 127.50, 125.04, 122.67, 121.63, 115.95 (J = 20.9 Hz), 113.68 (J = 21.7 Hz), 58.94, 58.22, 50.64, 46.10, 45.02, 44.43, 43.12, 38.30, 33.20, 31.05, 26.42, 20.55. MS (ESI) m/z [M + H]+: 619.0.
(R)-3-(3-Bromophenethyl)-4-(cyclohexylmethyl)-1-(4-((S)-3-(3-flu-orophenethyl)-2-iminoimidazolidin-4-yl)butyl)imidazolidin-2-imine (214)
Using general scheme for the synthesis of F, compound 214 was synthesized using the following reagents: 3-fluorophenylacetic acid (R1), Boc-d-cyclohexylalanine (R2), 3-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.56 (s, 2H), 7.35 (s, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.22–7.19 (m, 2H), 7.14 (t, J = 7.6 Hz, 1H), 7.02 (t, J = 8.0 Hz, 2H), 6.84 (t, J = 8.0 Hz, 1H), 3.95–3.85 (m, 2H), 3.58–3.50 (m, 2H), 3.50 (d, J = 8.8 Hz, 1H), 3.46–3.42 (m, 2H), 3.37–3.30 (m, 3H), 3.25–3.17 (m, 1H), 3.04–3.00 (m, 1H), 2.87–2.78 (m, 4H), 1.67–1.62 (m, 4H), 1.50–1.45 (m, 6H), 1.21 (d, J = 6.4 Hz, 2H), 1.16–1.11 (m, 5H), 0.91 (t, J = 11.6 Hz, 1H), 0.76 (t, J = 6.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 162.96 (J = 244.2 Hz), 159.10, 156.94, 141.01 (J = 7.3 Hz), 140.80, 131.98, 130.53, 130.30 (J = 8.1 Hz), 130.01, 127.93, 124.98 (d, J = 2.0 Hz), 122.69, 115.90 (J = 20.7 Hz), 113.64 (J = 20.7 Hz), 58.83, 55.56, 51.89, 46.13, 45.14, 43.94, 43.07, 39.87, 34.28, 34.11, 33.20, 32.32, 31.30, 26.54, 26.27, 26.16, 20.66. MS (ESI) m/z [M + H]+: 625.0.
(R)-4-Benzyl-3-(3-bromophenethyl)-1-(4-((S)-3-heptyl-2-imino-imidazolidin-4-yl)butyl)imidazolidin-2-imine (215)
Using general scheme for the synthesis of F, compound 215 was synthesized using the following reagents: heptanoic acid (R1), Boc-d-phenylalanine (R2), 3-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.56 (s, 1H), 8.06 (s, 2H), 7.33 (d, J = 8.8 Hz, 2H), 7.27 (d, J = 7.6 Hz, 2H), 7.23 (t, J = 7.2 Hz, 2H), 7.15 (t, J = 7.6 Hz, 1H), 7.02 (d, J = 6.8 Hz, 2H), 4.17–4.05 (m, 1H), 3.85–3.83 (m, 1H), 3.65–3.63 (m, 2H), 3.52–3.48 (m, 1H), 3.37–3.30 (m, 5H), 3.11 (dd, J = 9.6, 6.4 Hz, 1H), 3.07–3.03 (m, 1H), 2.90 (d, J = 4.4 Hz, 1H), 2.87 (d, J = 4.0 Hz, 2H), 2.59 (dd, J = 13.6, 8.0 Hz, 1H), 1.67–1.65 (m, 1H), 1.49–1.41 (m, 5H), 1.26–1.23 (m, 10H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 158.93, 156.87, 140.78, 135.19, 132.00, 130.56, 130.02, 129.18, 129.13, 127.99, 127.48, 122.61, 58.22, 58.15, 50.57, 46.03, 45.01, 44.32, 41.97, 38.26, 33.17, 31.81, 31.22, 29.16, 27.19, 26.61, 22.68, 20.60, 14.17. MS (ESI) m/z [M + H]+: 595.0.
(R)-3-(3-Bromophenethyl)-4-(cyclohexylmethyl)-1-(4-((S)-3-hep-tyl-2-iminoimidazolidin-4-yl)butyl)imidazolidin-2-imine (216)
Using general scheme for the synthesis of F, compound 216 was synthesized using the following reagents: heptanoic acid (R1), Boc-d-cyclohexylalanine (R2), 3-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.56 (s, 1H), 8.31 (s, 1H), 7.35 (s, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 7.2 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 3.96– 3.94 (m, 1H), 3.85–3.83 (m, 1H), 3.71–3.64 (m, 1H), 3.53 (t, J = 8.8 Hz, 2H), 3.44–3.42 (m, 2H), 3.35–3.31 (m, 3H), 3.04 (t, J = 8.0 Hz, 2H), 2.87–2.82 (m, 2H), 1.70–1.65 (m, 4H), 1.55–1.44 (m, 8H), 1.26–1.23 (m, 11H), 1.18–1.10 (m, 5H), 0.95–0.89 (m, 1H), 0.84 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 159.19, 156.98, 140.80, 131.98, 130.53, 129.99, 127.94, 122.67, 58.09, 55.56, 51.90, 46.05, 45.21, 43.93, 41.91, 39.89, 34.28, 34.11, 33.19, 31.82, 31.32, 29.15, 27.21, 26.65, 26.26, 26.16, 25.96, 22.69, 20.66, 14.18. MS (ESI) m/z [M + H]+: 601.1.
(R)-4-Benzyl-3-(3-bromophenethyl)-1-(4-((S)-2-imino-3-(4-methylpentyl)imidazolidin-4-yl)butyl)imidazolidin-2-imine (217)
Using general scheme for the synthesis of F, compound 217 was synthesized using the following reagents: 4-methylvaleric acid (R1), Boc-d-phenylalanine (R2), 3-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.57 (s, 1H), 8.26 (s, 1H), 7.34 (s, 1H), 7.32–7.31 (m, 1H), 7.28 (d, J = 7.2 Hz, 2H), 7.24 (d, J = 7.2 Hz, 1H), 7.22–7.20 (m, 1H), 7.16 (t, J = 7.6 Hz, 1H), 7.02 (d, J = 7.2 Hz, 2H), 4.13–4.07 (m, 1H), 3.85–3.82 (m, 1H), 3.65–3.60 (m, 2H), 3.51–3.48 (m, 1H), 3.37–3.33 (m, 5H), 3.12 (dd, J = 9.6, 6.4 Hz, 1H), 3.06–3.03 (m, 1H), 2.90 (d, J = 4.4 Hz, 1H), 2.87 (d, J = 4.0 Hz, 2H), 2.59 (dd, J = 13.6, 8.0 Hz, 1H), 1.67–1.60 (m, 1H), 1.52–1.46 (m, 4H), 1.42–1.36 (m, 2H), 1.26–1.23 (m, 2H), 1.18–1.12 (m, 2H), 0.84 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 159.11, 156.90, 140.76, 135.21, 132.01, 130.58, 130.03, 129.19, 129.14, 127.99, 127.49, 122.62, 58.16, 50.61, 46.04, 45.05, 44.35, 42.11, 38.28, 35.58, 33.17, 31.25, 27.93, 26.60, 25.02, 22.67, 22.59, 20.58. MS (ESI) m/z [M + H]+: 581.0.
(R)-3-(3-Bromophenethyl)-4-(cyclohexylmethyl)-1-(4-((S)-2-imino-3-(4-methylpentyl)imidazolidin-4-yl)butyl)imidazolidin-2-imine (218)
Using general scheme for the synthesis of F, compound 218 was synthesized using the following reagents: 4-methylvaleric acid (R1), Boc-d-cyclohexylalanine (R2), 3-bromophenylacetic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.57 (s, 1H), 8.25 (s, 1H), 7.35 (d, J = 8.0 1H), 7.32 (s, 1H), 7.22 (d, J = 7.2 Hz, 1H), 7.15 (t, J = 8.0 Hz, 1H), 3.96–3.92 (m, 1H), 3.85–3.83 (m, 1H), 3.75–3.65 (m, 1H), 3.53 (t, J = 8.8 Hz, 2H), 3.45–3.41 (m, 2H), 3.38–3.31 (m, 3H), 3.03 (t, J = 8.0 Hz, 2H), 2.86–2.84 (m, 2H), 1.70–1.63 (m, 4H), 1.54– 1.43 (m, 10H), 1.38–1.25 (m, 2H), 1.20–1.09 (m, 8H), 0.84 (d, J = 6.4 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 159.13, 156.95, 140.86, 131.99, 130.54, 129.98, 127.97, 122.66, 58.09, 55.57, 51.89, 46.05, 45.23, 43.96, 42.11, 39.88, 35.59, 34.28, 33.21, 32.31, 31.30, 27.93, 26.71, 26.16, 25.96, 25.02, 22.67, 20.63. MS (ESI) m/z [M + H]+: 587.1.
(R)-4-Benzyl-3-((4-tert-butylcyclohexyl)methyl)-1-(4-((S)-2-imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)butyl)imidazolidin-2-imine (301)
Using general scheme for the synthesis of F, compound 301 was synthesized using the following reagents: 3-pyridinylacetic acid (R1), Boc-d-phenylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, D2O): δ 8.55–8.45 (m, 2H), 7.87 (d, J = 8.0 Hz, 1H), 7.51 (dd, J = 7.2, 5.2 Hz, 1H), 7.42 (t, J = 7.2 Hz, 2H), 7.36 (t, J = 6.8 Hz, 1H), 7.29 (d, J = 6.8 Hz, 2H), 4.36–4.30 (m, 1H), 3.85–3.79 (m, 1H), 3.73–3.65 (m, 3H), 3.61–3.54 (m, 1H), 3.45 (dd, J = 10.4, 4.8 Hz, 1H), 3.30–3.25 (m, 3H), 3.06–3.02 (m, 5H), 2.94 (dd, J = 14.4, 4.0 Hz, 1H), 1.86 (d, J = 2.4 Hz, 1H), 1.80 (d, J = 7.6 Hz, 2H), 1.75 (d, J = 12.8 Hz, 1H), 1.69– 1.65 (m, 1H), 1.60–1.57 (m, 1H), 1.47–1.39 (m, 1H), 1.37–1.23 (m, 2H), 1.10–0.96 (m, 7H), 0.87 (s, 9H). 13C NMR (100 MHz, D2O): δ 158.23, 156.14, 148.37, 146.78, 138.85, 135.69, 134.87, 129.71, 128.84, 127.23, 124.73, 58.87, 56.84, 49.54, 47.88, 45.68, 44.10, 43.60, 42.76, 36.63, 31.72, 30.37, 29.92, 29.90, 27.56, 26.84, 25.71, 20.34. MS (ESI) m/z C35H54N7 [M + H]+: 572.2.
(S)-4-Benzyl-3-((4-tert-butylcyclohexyl)methyl)-1-(4-((S)-2-imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)butyl)imidazolidin-2-imine (302)
Using general scheme for the synthesis of F, compound 302 was synthesized using the following reagents: 3-pyridinylacetic acid (R1), Boc-L-phenylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.62 (s, 1H), 8.53 (s, 1H), 8.40 (d, J = 3.6 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 7.2 Hz, 2H), 7.28–7.21 (m, 2H), 7.14 (d, J = 6.8 Hz, 2H), 4.10–4.03 (m, 1H), 4.01–3.96 (m, 1H), 3.76 (dd, J = 15.2, 8.4 Hz, 1H), 3.65–3.55 (m, 2H), 3.48 (t, J = 9.6 Hz, 2H), 3.43–3.39 (m, 1H), 3.32–3.19 (m, 3H), 3.09–3.06 (m, 1H), 3.05–3.03 (m, 1H), 2.92–2.86 (m, 2H), 2.72 (dd, J = 13.6, 8.4 Hz, 1H), 1.79–1.77 (m, 2H), 1.72 (d, J = 11.6 Hz, 2H), 1.60 (t, J = 7.6 Hz, 2H), 1.51–1.47 (m, 1H), 1.42 (t, J = 7.2 Hz, 2H), 1.21–1.18 (m, 2H), 1.10–1.02 (m, 2H), 0.97–0.96 (m, 3H), 0.82 (s, 9H). 13C NMR (100 MHz, D2O): δ 158.22, 156.49, 148.49, 146.86, 138.60, 135.65, 134.72, 129.60, 128.80, 127.19, 124.60, 58.93, 57.44, 49.69, 48.59, 47.58, 45.77, 44.17, 42.80, 36.54, 35.80, 31.67, 30.59, 30.52, 29.97, 27.05, 26.47, 25.74, 20.48. MS (ESI) m/z calcd for [M + H]+: 572.2.
(S)-1-((4-tert-Butylcyclohexyl)methyl)-3-(4-(2-imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)butyl)imidazolidin-2-imine (303)
Using general scheme for the synthesis of F, compound 303 was synthesized using the following reagents: 3-pyridinylacetic acid (R1), Boc-glycine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, D2O): δ 8.54–8.50 (m, 2H), 7.93 (d, J = 7.6 Hz, 1H), 7.55 (dd, J = 7.6, 5.2 Hz, 1H), 3.96–3.89 (m, 1H), 3.73 (d, J = 9.2 Hz, 1H), 3.71–3.67 (m, 4H), 3.64–3.62 (m, 1H), 3.37– 3.31 (m, 4H), 3.13 (d, J = 7.2 Hz, 2H), 3.07–3.03 (m, 2H), 1.85–1.82 (m, 2H), 1.76 (d, J = 10.0 Hz, 2H), 1.69–1.51 (m, 6H), 1.31–1.25 (m, 2H), 1.01 (d, J = 4.4 Hz, 3H), 0.97–0.94 (m, 1H), 0.86 (s, 9H). 13C NMR (100 MHz, D2O): δ 158.24, 157.19, 148.17, 146.57, 138.99, 134.90, 124.73, 58.89, 50.95, 47.93, 47.62, 46.14, 45.73, 45.38, 42.65, 35.52, 31.68, 31.53, 30.43, 30.40, 26.89, 26.80, 26.31, 20.45. MS (ESI) m/z [M + H]+: 482.1.
(R)-4-Benzyl-3-((4-tert-butylcyclohexyl)methyl)-1-(4-((R)-2-imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)butyl)imidazolidin-2-imine (304)
Using general scheme for the synthesis of F, compound 304 was synthesized using the following reagents: 3-pyridinylacetic acid (R1), Boc-d-phenylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). The only variation of the general compound synthesis for compound 304 was utilizing Fmoc-d-Lys(Boc)-OH instead of Boc-L-Lys(Fmoc)-OH. Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.93 (s, 2H), 8.61 (s, 1H), 8.52 (s, 1H), 8.41 (s, 1H), 7.76 (d, J = 6.8 Hz, 1H), 7.33 (t, J = 7.2 Hz, 2H), 7.27 (d, J = 6.4 Hz, 1H), 7.24–7.20 (m, 1H), 7.14 (d, J = 7.2 Hz, 2H), 4.10–4.04 (m, 1H), 4.01–3.96 (m, 1H), 3.76 (dd, J = 14.4, 8.4 Hz, 1H), 3.65–3.57 (m, 2H), 3.47 (t, J = 9.6 Hz, 2H), 3.43–3.39 (m, 1H), 3.29–3.23 (m, 3H), 3.09–3.06 (m, 1H), 3.05–3.02 (m, 1H), 2.94–2.84 (m, 2H), 2.72 (dd, J = 13.2, 8.4 Hz, 1H), 1.79–1.77 (m, 2H), 1.72 (d, J = 11.2 Hz, 2H), 1.65–1.55 (m, 2H), 1.51–1.37 (m, 3H), 1.24–1.16 (m, 2H), 1.08–1.04 (m, 2H), 0.99–0.94 (m, 3H), 0.82 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 159.35, 157.28, 150.20, 147.99, 137.26, 135.16, 133.97, 129.30, 129.21, 127.58, 123.90, 58.63, 57.78, 50.34, 48.82, 47.87, 46.27, 45.19, 43.03, 37.76, 36.18, 32.54, 31.20, 30.71, 27.66, 26.85, 26.50, 20.61. MS (ESI) m/z [M + H]+: 572.2.
(R)-1-((4-tert-Butylcyclohexyl)methyl)-3-(4-(2-imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)butyl)imidazolidin-2-imine (305)
Using general scheme for the synthesis of F, compound 305 was synthesized using the following reagents: 3-pyridinylacetic acid (R1), Boc-glycine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). The only variation of the general compound synthesis for compound 305 was utilizing Fmoc-d-Lys(Boc)-OH instead of Boc-L-Lys(Fmoc)-OH. Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, D2O): δ 8.54–8.46 (m, 2H), 7.98 (d, J = 8.0 Hz, 1H), 7.59 (t, J = 6.4 Hz, 1H), 3.95–3.91 (m, 1H), 3.74 (d, J = 9.2 Hz, 1H), 3.73–3.67 (m, 5H), 3.65–3.59 (m, 1H), 3.37–3.31 (m, 3H), 3.13 (d, J = 7.2 Hz, 2H), 3.08–3.05 (m, 2H), 1.87–1.80 (m, 2H), 1.76 (d, J = 10.8 Hz, 3H), 1.69–1.62 (m, 4H), 1.59–1.51 (m, 1H), 1.34–1.25 (m, 2H), 1.01 (d, J = 4.4 Hz, 3H), 0.96 (d, J = 12.4 Hz, 1H), 0.87 (s, 9H). 13C NMR (100 MHz, CCl3D): δ 159.41, 158.06, 150.16, 147.98, 137.22, 134.05, 123.91, 58.64, 51.72, 47.91, 46.51, 46.20, 45.70, 45.41, 42.91, 36.26, 32.54, 31.36, 30.89, 30.78, 27.67, 26.78, 26.70, 20.73. MS (ESI) m/z [M + H]+: 482.1.
(R)-3-((4-tert-Butylcyclohexyl)methyl)-4-(cyclohexylmethyl)-1-(4-((S)-2-imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)butyl)-imidazolidin-2-imine (306)
Using general scheme for the synthesis of F, compound 306 was synthesized using the following reagents: 3-pyridinylacetic acid (R1), Boc-d-cyclohexylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, D2O): δ 8.55–8.45 (m, 2H), 7.88 (d, J = 7.6 Hz, 1H), 7.51–7.49 (m, 1H), 4.00–3.90 (m, 2H), 3.82 (t, J = 9.2 Hz, 1H), 3.77–3.72 (m, 1H), 3.68 (t, J = 7.2 Hz, 1H), 3.62–3.55 (m, 1H), 3.37–3.33 (m, 4H), 3.24–3.11 (m, 2H), 3.06–2.97 (m, 2H), 1.92–1.75 (m, 5H), 1.74–1.60 (m, 8H), 1.58– 1.50 (m, 2H), 1.43–1.36 (m, 2H), 1.32–1.25 (m, 4H), 1.17 (d, J = 10.8 Hz, 1H), 1.02–0.92 (m, 7H), 0.87 (s, 9H). 13C NMR (100 MHz, D2O): δ 158.27, 156.96, 148.71, 146.99, 138.36, 124.56, 59.01, 55.68, 51.42, 48.32, 47.54, 45.81, 44.50, 42.87, 39.08, 36.01, 34.02, 33.63, 32.17, 31.89, 30.71, 30.12, 27.28, 26.68, 26.09, 25.85, 20.59. MS (ESI) m/z [M + H]+: 578.2.
(S)-3-((4-tert-Butylcyclohexyl)methyl)-4-(cyclohexylmethyl)-1-(4-((S)-2-imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)butyl)-imidazolidin-2-imine (307)
Using general scheme for the synthesis of F, compound 306 was synthesized using the following reagents: 3-pyridinylacetic acid (R1), Boc-L-cyclohexylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CCl3D): δ 8.97 (s, 1H), 8.61 (s, 1H), 8.52 (m, 1H), 8.42 (s, 1H), 7.75 (d, J = 6.8 Hz, 1H), 7.25–7.22 (m, 1H), 4.01–3.93 (m, 1H), 3.82 (d, J = 7.2 Hz, 2H), 3.65 (t, J = 9.2 Hz, 2H), 3.58–3.52 (m, 3H), 3.44–3.37 (m, 2H), 3.25–3.21 (m, 1H), 3.16 (t, J = 8.0 Hz, 1H), 2.97 (dd, J = 14.8, 5.6 Hz, 1H), 2.90–2.86 (m, 1H), 1.81–1.78 (m, 2H), 1.72 (d, J = 10.8 Hz, 5H),1.65–1.63 (m, 4H), 1.56–1.48 (m, 3H), 1.33–1.14 (m, 8H), 1.03–0.93 (m, 7H), 0.81 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 159.41, 157.33, 150.19, 147.99, 137.24, 134.03, 58.63, 55.21, 51.81, 48.40, 47.87, 46.27, 45.31, 43.00, 39.55, 36.09, 34.43, 34.38, 32.53, 31.38, 31.21, 30.72, 27.66, 26.87, 26.59, 26.26, 26.06, 20.69. MS (ESI) m/z [M + H]+: 578.2.
(R)-3-((4-tert-Butylcyclohexyl)methyl)-4-(cyclohexylmethyl)-1-(4-((R)-2-imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)butyl)-imidazolidin-2-imine (308)
Using general scheme for the synthesis of F, compound 308 was synthesized using the following reagents: 3-pyridinylacetic acid (R1), Boc-d-cyclohexylalanine (R2), 4-tert-butyl-cyclohexanecarboxylic acid, predominantly trans, 98+% (R3). The only variation of the general compound synthesis for compound 308 was utilizing Fmoc-d-Lys(Boc)-OH instead of Boc-L-Lys(Fmoc)-OH. Final crude product was purified by HPLC as described above. 1H NMR (400 MHz, CDCl3): δ 8.59 (s, 1H), 8.54–8.48 (m, 1H), 8.40 (s, 1H), 7.78 (d, J = 7.2 Hz, 1H), 7.23–7.21 (m, 1H), 4.03–3.96 (m, 1H), 3.82 (d, J = 8.4 Hz, 1H), 3.66 (t, J = 9.2 Hz, 3H), 3.61–3.58 (m, 2H), 3.54–3.52 (m, 1H), 3.46–3.40 (m, 2H), 3.26–3.23 (m, 1H), 3.17 (t, J = 8.0 Hz, 1H), 2.97 (dd, J = 15.2, 5.6 Hz, 1H), 2.91–2.85 (m. 1H), 1.78–1.61 (m, 12H), 1.56–1.51 (m, 4H), 1.29–1.24 (m, 4H), 1.23–1.13 (m, 4H), 1.05–0.99 (m, 3H), 0.96–0.93 (m, 2H), 0.80 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 159.34, 157.26, 150.23, 147.98, 137.33, 133.96, 123.87, 58.66, 55.21, 51.84, 48.43, 47.84, 46.33, 45.43, 43.10, 39.55, 36.09, 34.37, 32.53, 31.39, 31.18, 30.83, 30.69, 27.66, 26.87, 26.73, 26.25, 26.05, 20.73. MS (ESI) m/z calcd for C35H60N7 [M + H]+, 578.49; found, 578.15.
Positional Scanning Library 1169
Utilizing Scheme 1, positional scanning library 1169 was synthesized. The positional scanning library incorporates both individual and mixtures of amino acids (R2) and carboxylic acid (R1 and R3). The synthetic technique facilitates the generation of information regarding the likely activity of individual compounds from the screening of the library.26,35,36 Equimolar isokinetic ratios have been previously determined and calculated for each of the amino and carboxylic acids utilized for the respective mixtures.43,44, The bis-cyclic guanidine library 1169 has a total diversity of 45864 compounds (42 × 26 × 42 = 45864). The R1 and R3 positions as shown in Scheme 1 (F) each consist of 42 carboxylic acids and the R2 contains 26 amino acids.
Scaffold Ranking Library
The scaffold ranking library contains one sample representing each of the positional scanning libraries. This sample contains an approximate equal molar amount of each compound in that library. So, for example, the sample 1169 in the scaffold ranking library contains 45864 compounds in approximately equal molar amounts. These samples can be prepared by mixing the cleaved products of the complete positional scanning library, as was the case for sample 1169, or they can be synthesized directly as a single mixture.26,27
Calculation of xIC50s
It has been shown32 that the harmonic mean of the IC50s of constituents within a mixture accurately models the resulting IC50 of the mixture. Because percent responses are generally used as the initial screening metric, it is often necessary (as it was in this case) to extrapolate IC50s for samples using the equation
Here [x] is the concentration tested and %resp is the percent inhibition value obtained at that concentration. Obviously, these values are extrapolations and not a substitute for actual experimentally determined IC50s, but they are sufficient for the order-of-magnitude estimation used in the Harmonic Mean Analysis of position scanning libraries.
Prediction of Selectivity
For a sample with IC50 x1 against α3β4 and x2 against α4β2, we define the selectivity over α3β4 as
To predict the selectivity of an individual compound based on the values of the positional scanning mixture, we used the standard biometrical analysis approach45–47 of using the product of the mixture activity values. In particular, if a compound has functionality A1 at position R1, A2 at position R2, and A3 at position R3, then its Biometrical Analysis Score would be
Given BA values for α3β4 and α4β2, the predicted selectivity is then found using the same formula as above:
Cell Culture
KXαβ4R2 and KXα4β2R2 cells, containing rat α3β4 and α4β2 nAChR respectively (obtained from Drs. Kenneth Kellar and Yingxian Xiao, Georgetown University), were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 0.5% penicillin/streptomycin, and 0.4 mg/mL of geneticin and were maintained in an atmosphere of 7.5% CO2 in a humidified incubator at 37 °C. For binding assays, cells were passaged on 150 mm dishes and harvested when confluent.
Binding Assays
Cells were harvested by scraping the plates with a rubber policeman, suspended in 50 mM Tris buffer pH 7.4, homogenized using a Polytron homogenizer, and the centrifugation was repeated twice at 20000g (13500 rpm) for 20 min. For binding, the cell membranes were incubated with the test compounds or mixtures in the presence of 0.3 nM of [3H]epibatidine. After 2 h of incubation, at room temperature, samples were filtered, using a Tomtec cell harvester, through glass fiber filters that had been presoaked in 0.05% polyethyleneimine. Filters were counted on a betaplate reader (Wallac). Nonspecific binding was determined by using 1 µM of AT-1001 or 0.1 µM of unlabeled epibatidine. IC50 values were determined by using the program Graphpad/PRISM. Ki values were calculated using the Cheng-Prusoff transformation: Ki = IC50/(1 + L/ Kd)48 where, L is radioligand concentration and Kd is the binding affinity of the radioligand, as determined previously by saturation analysis.
nAChR Functional Assays
nAChR functional activity was determined by measuring nAChR-induced membrane potential change, which can be directly read by Molecular Devices Membrane Potential Assay Kit (Blue Dye) (Molecular Devices), using the FlexStation 3 microplate reader (Molecular Devices). The HEK cells transfected with α3β4 or α4β2 nAChR were seeded in a 96-well plate (4000 cells per well) one day prior to the experiments. For agonist assays, after brief washing, the cells were loaded with 225 µL of HBSS assay buffer (Hank’s Balanced Salt Solution with 20 mM of HEPES, pH 7.4), containing the blue dye, and incubated at 370C. After 30 min, 25 µL of the appropriate compounds were dispensed into the wells by the FlexStation and nAChR stimulation-mediated membrane potential change was recorded every 3 s for 120 s by reading 550–580 nM fluorescence excited at 530 nM wavelength. For the antagonist assay, the cells were loaded with 200 µL of HBSS buffer containing the blue dye and incubated at 37 °C After 20 min, 25 µL of test compounds were added, and after another 10 min, 25 µL of epibatidine (or nicotine) was added by the FlexStation to a final concentration of 100 nM, with fluorescence measured as described above. The change in fluorescence represents the maximum response minus the minimum response for each well. Graphpad PRISM was used to determine the EC50 and IC50 values.
ACKNOWLEDGMENTS
This work was supported by a NIH-NIDA grant R01-DA031370 and the State of Florida, Executive Office of the Governor’s Department of Economic Opportunity. We also thank Clemencia Pinilla and Jon Appel for their critical reading of the manuscript and insightful suggestions.
ABBREVIATIONS USED
- nAChR
nicotinic acetylcholine receptors
- DHβE
dihydro-β-erythroidine hydrobromide
- 18-MC
18-methoxycoronaridine
- GWAS
Genome Wide Association Studies
- MHb
medial habenula
- KO
knockout
- NAc
nucleus accumben
- ADAM 17
ADAM metallopeptidase domain 17
- MBHA
p-methylbenzdryl-amine resin
- DIEA
disopropylethylamide
- DCM
dichloromethane
- Fmoc
g-fluoromethyloxycarbonyl
- Boc
tert-butyloxycarbonyl
- DMF
dimethylformamide
- DIC
diisopropylcarbodiimide
- HOBt
hydroxybenzotriazole hydrate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- LCMS
liquid chromatograph/mass spectrometer
- BA
biometric analysis
- DMEM
Dulbecco’s Modified Eagle’s Medium
- FBS
fetal bovine serum
- HBSS
Hank’s Balanced Salt Solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
Footnotes
ASSOCIATED CONTENT
Supporting Information
Detailed description of the scaffold ranking library samples and their corresponding dose response data against α4β2 and α3β4 nAChR as well as 1H and 13C NMR spectra for key compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
REFERENCES
- 1.Gotti C, Fornasari D, Clementi F. Human neuronal nicotinic receptors. Prog. Neurobiol. 1997;53:199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- 2.McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 3.McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu. Rev. Physiol. 1995;57:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- 4.Perry DC, Xiao Y, Nguyen HN, Musachio JL, Davila-Garcia MI, Kellar KJ. Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J. Neurochem. 2002;82:468–481. doi: 10.1046/j.1471-4159.2002.00951.x. [DOI] [PubMed] [Google Scholar]
- 5.Xiao Y, Kellar KJ. The comparative pharmacology and up-regulation of rat neuronal nicotinic receptor subtype binding sites stably expressed in transfected mammalian cells. J. Pharmacol. Exp. Ther. 2004;310:98–107. doi: 10.1124/jpet.104.066787. [DOI] [PubMed] [Google Scholar]
- 6.Picciotto MR. Common aspects of the action of nicotine and other drugs of abuse. Drug Alcohol Depend. 1998;51:165–172. doi: 10.1016/s0376-8716(98)00074-x. [DOI] [PubMed] [Google Scholar]
- 7.Stolerman IP, Shoaib M. The neurobiology of tobacco addiction. Trends Pharmacol. Sci. 1991;12:467–473. doi: 10.1016/0165-6147(91)90638-9. [DOI] [PubMed] [Google Scholar]
- 8.Perry DC, Mao D, Gold AB, McIntosh JM, Pezzullo JC, Kellar KJ. Chronic nicotine differentially regulates alpha6- and beta3-containing nicotinic cholinergic receptors in rat brain. J. Pharmacol. Exp. Ther. 2007;322:306–315. doi: 10.1124/jpet.107.121228. [DOI] [PubMed] [Google Scholar]
- 9.Maisonneuve IM, Glick SD. Anti-addictive actions of an iboga alkaloid congener: a novel mechanism for a novel treatment. Pharmacol., Biochem. Behav. 2003;75:607–618. doi: 10.1016/s0091-3057(03)00119-9. [DOI] [PubMed] [Google Scholar]
- 10.Toll L, Zaveri NT, Polgar WE, Jiang F, Khroyan TV, Zhou W, Xie XS, Stauber GB, Costello MR, Leslie FM. AT-1001: A High Affinity and Selective alpha3beta4 Nicotinic Acetylcho-line Receptor Antagonist Blocks Nicotine Self-Administration in Rats. Neuropsychopharmacology. 2012;37:1367–1376. doi: 10.1038/npp.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, Waterworth D, Muglia P, Mooser V. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol. Psychiatry. 2008;13:368–373. doi: 10.1038/sj.mp.4002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saccone SF, Saccone NL, Swan GE, Madden PA, Goate AM, Rice JP, Bierut LJ. Systematic biological prioritization after a genome-wide association study: an application to nicotine dependence. Bioinformatics. 2008;24:1805–1811. doi: 10.1093/bioinformatics/btn315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson KJ, Marks MJ, Vann RE, Chen X, Gamage TF, Warner JA, Damaj MI. Role of alpha5 nicotinic acetylcholine receptors in pharmacological and behavioral effects of nicotine in mice. J. Pharmacol. Exp. Ther. 2010;334:137–146. doi: 10.1124/jpet.110.165738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, Changeux JP, Maskos U, Fratta W. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J. Neurosci. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epping-Jordan MP, Picciotto MR, Changeux JP, Pich EM. Assessment of nicotinic acetylcholine receptor subunit contributions to nicotine self-administration in mutant mice. Psychopharmacology (Berlin) 1999;147:25–26. doi: 10.1007/s002130051135. [DOI] [PubMed] [Google Scholar]
- 17.Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 18.Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- 19.Watkins SS, Epping-Jordan MP, Koob GF, Markou A. Blockade of nicotine self-administration with nicotinic antagonists in rats. Pharmacol., Biochem. Behav. 1999;62:743–751. doi: 10.1016/s0091-3057(98)00226-3. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee S, Steensland P, Simms JA, Holgate J, Coe JW, Hurst RS, Shaffer CL, Lowe J, Rollema H, Bartlett SE. Partial agonists of the alpha3beta4* neuronal nicotinic acetylcholine receptor reduce ethanol consumption and seeking in rats. Neuro-psychopharmacology. 2011;36:603–615. doi: 10.1038/npp.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA, Lu Y, Mansbach RS, Mather RJ, Rovetti CC, Sands SB, Schaeffer E, Schulz DW, Tingley FD, III; Williams KE. Pharmacological profile of the alpha4beta2 nicotinic acetylcho-line receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology. 2007;52:985–994. doi: 10.1016/j.neuropharm.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 22.Ahmed AI, Ali AN, Kramers C, Harmark LV, Burger DM, Verhoeven WM. Neuropsychiatric adverse events of varenicline: a systematic review of published reports. J. Clin. Psychopharmacol. 2013;33(1):55–62. doi: 10.1097/JCP.0b013e31827c0117. [DOI] [PubMed] [Google Scholar]
- 23.Carroll FI, Lee JR, Navarro HA, Ma W, Brieaddy LE, Abraham P, Damaj MI, Martin BR. Synthesis, nicotinic acetylcholine receptor binding, and antinociceptive properties of 2-exo-2-(2′,3′-disubstituted 5′-pyridinyl)-7-azabicyclo[2.2.1]heptanes: epibatidine analogues. J. Med. Chem. 2002;45:4755–4761. doi: 10.1021/jm0202268. [DOI] [PubMed] [Google Scholar]
- 24.Carroll FI, Ware R, Brieaddy LE, Navarro HA, Damaj MI, Martin BR. Synthesis, nicotinic acetylcholine receptor binding, and antinociceptive properties of 2′-fluoro-3′-(substituted phenyl)-deschloroepibatidine analogues. Novel nicotinic antagonist. J. Med. Chem. 2004;47:4588–4594. doi: 10.1021/jm040078g. [DOI] [PubMed] [Google Scholar]
- 25.Ondachi P, Castro A, Luetje CW, Damaj MI, Mascarella SW, Navarro HA, Carroll FI. Synthesis and nicotinic acetylcholine receptor in vitro and in vivo pharmacological properties of 2′-fluoro-3′-(substituted phenyl)deschloroepibatidine analogues of 2′-fluoro-3′-(4-nitrophenyl)deschloroepibatidine. J. Med. Chem. 2012;55:6512–6522. doi: 10.1021/jm300575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houghten RA, Pinilla C, Giulianotti MA, Appel JR, Dooley CT, Nefzi A, Ostresh JM, Yu Y, Maggiora GM, Medina-Franco JL, Brunner D, Schneider J. Strategies for the use of mixture-based synthetic combinatorial libraries: scaffold ranking, direct testing in vivo, and enhanced deconvolution by computational methods. J. Comb. Chem. 2008;10:3–19. doi: 10.1021/cc7001205. [DOI] [PubMed] [Google Scholar]
- 27.Santos RG, Appel JR, Giulianotti MA, Edwards BS, Sklar LA, Houghten RA, Pinilla C. The mathematics of a successful deconvolution: a quantitative assessment of mixture-based combinatorial libraries screened against two formylpeptide receptors. Molecules. 2013;18:6408–6424. doi: 10.3390/molecules18066408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minond D, Cudic M, Bionda N, Giulianotti M, Maida L, Houghten RA, Fields GB. Discovery of novel inhibitors of a disintegrin and metalloprotease 17 (ADAM17) using glycosylated and non-glycosylated substrates. J. Biol. Chem. 2012;287:36473–36487. doi: 10.1074/jbc.M112.389114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rideout MC, Boldt JL, Vahi-Ferguson G, Salamon P, Nefzi A, Ostresh JM, Giulianotti M, Pinilla C, Segall AM. Potent antimicrobial small molecules screened as inhibitors of tyrosine recombinases and Holliday junction-resolving enzymes. Mol. Divers. 2011;15:989–1005. doi: 10.1007/s11030-011-9333-2. [DOI] [PubMed] [Google Scholar]
- 30.Ranjit DK, Rideout MC, Nefzi A, Ostresh JM, Pinilla C, Segall AM. Small molecule functional analogs of peptides that inhibit λ site-specific recombination and bind Holliday junctions. Bioorg. Med. Chem. Lett. 2010;20:4531–4534. doi: 10.1016/j.bmcl.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reilley KJ, Giulianotti M, Dooley CT, Nefzi A, McLaughlin JP, Houghten RA. Identification of two novel, potent, low-liability antinociceptive compounds from the direct in vivo screening of a large mixture-based combinatorial library. AAPS J. 2010;12:318–329. doi: 10.1208/s12248-010-9191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santos RG, Giulianotti MA, Dooley CT, Pinilla C, Appel JR, Houghten RA. Use and implications of the harmonic mean model on mixtures for basic research and drug discovery. ACS Comb. Sci. 2011;13:337–344. doi: 10.1021/co100065a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Latli B, D’Amour K, Casida JE. Novel and potent 6-chloro-3-pyridinyl ligands for the alpha4beta2 neuronal nicotinic acetylcholine receptor. J. Med. Chem. 1999;42:2227–2234. doi: 10.1021/jm980721x. [DOI] [PubMed] [Google Scholar]
- 34.Dooley CT, Houghten RA. The use of positional scanning synthetic peptide combinatorial libraries for the rapid determination of opioid receptor ligands. Life Sci. 1993;52:1509–1517. doi: 10.1016/0024-3205(93)90113-h. [DOI] [PubMed] [Google Scholar]
- 35.Houghten RA, Pinilla C, Appel JR, Blondelle SE, Dooley CT, Eichler J, Nefzi A, Ostresh JM. Mixture-based synthetic combinatorial libraries. J. Med. Chem. 1999;42:3743–3778. doi: 10.1021/jm990174v. [DOI] [PubMed] [Google Scholar]
- 36.Pinilla C, Appel JR, Blanc P, Houghten RA. Rapid identification of high affinity peptide ligands using positional scanning synthetic peptide combinatorial libraries. Biotechniques. 1992;13:901–905. [PubMed] [Google Scholar]
- 37.Pinilla C, Edwards BS, Appel JR, Yates-Gibbins T, Giulianotti MA, Medina-Franco JL, Young SM, Santos RG, Sklar LA, Houghten RA. Selective agonists and antagonists of formylpeptide receptors: duplex flow cytometry and mixture-based positional scanning libraries. Mol. Pharmacol. 2013;84:314–324. doi: 10.1124/mol.113.086595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao Y, Fan H, Musachio JL, Wei ZL, Chellappan SK, Kozikowski AP, Kellar KJ. Sazetidine-A, a novel ligand that desensitizes alpha4beta2 nicotinic acetylcholine receptors without activating them. Mol. Pharmacol. 2006;70:1454–1460. doi: 10.1124/mol.106.027318. [DOI] [PubMed] [Google Scholar]
- 39.Zwart R, Carbone AL, Moroni M, Bermudez I, Mogg AJ, Folly EA, Broad LM, Williams AC, Zhang D, Ding C, Heinz BA, Sher E. Sazetidine-A is a potent and selective agonist at native and recombinant alpha 4 beta 2 nicotinic acetylcholine receptors. Mol. Pharmacol. 2008;73:1838–1843. doi: 10.1124/mol.108.045104. [DOI] [PubMed] [Google Scholar]
- 40.Nefzi A, Giulianotti MA, Houghten RA. Solid-phase synthesis of bis-heterocyclic compounds from resin-bound orthogonally protected lysine. J. Comb. Chem. 2001;3:68–70. doi: 10.1021/cc000061t. [DOI] [PubMed] [Google Scholar]
- 41.Nefzi A, Ostresh JM, Yu Y, Houghten RA. Combinatorial chemistry: libraries from libraries, the art of the diversity-oriented transformation of resin-bound peptides and chiral polyamides to low molecular weight acyclic and heterocyclic compounds. J. Org. Chem. 2004;69:3603–3609. doi: 10.1021/jo040114j. [DOI] [PubMed] [Google Scholar]
- 42.Houghten RA. General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. U. S. A. 1985;82:5131–5135. doi: 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Acharya AN, Ostresh JM, Houghten RA. Determination of isokinetic ratios necessary for equimolar incorporation of carboxylic acids in the solid-phase synthesis of mixture-based combinatorial libraries. Biopolymers. 2002;65:32–39. doi: 10.1002/bip.10206. [DOI] [PubMed] [Google Scholar]
- 44.Ostresh JM, Winkle JH, Hamashin VT, Houghten RA. Peptide libraries: determination of relative reaction rates of protected amino acids in competitive couplings. Biopolymers. 1994;34:1681–1689. doi: 10.1002/bip.360341212. [DOI] [PubMed] [Google Scholar]
- 45.Falta MT, Pinilla C, Mack DG, Tinega AN, Crawford F, Giulianotti M, Santos R, Clayton GM, Wang Y, Zhang X, Maier LA, Marrack P, Kappler JW, Fontenot AP. Identification of beryllium-dependent peptides recognized by CD4+ T cells in chronic beryllium disease. J. Exp. Med. 2013;210:1403–1418. doi: 10.1084/jem.20122426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hemmer B, Gran B, Zhao Y, Marques A, Pascal J, Tzou A, Kondo T, Cortese I, Bielekova B, Straus SE, McFarland HF, Houghten R, Simon R, Pinilla C, Martin R. Identification of candidate T-cell epitopes and molecular mimics in chronic Lyme disease. Nature Med. 1999;5:1375–1382. doi: 10.1038/70946. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Y, Gran B, Pinilla C, Markovic-Plese S, Hemmer B, Tzou A, Whitney LW, Biddison WE, Martin R, Simon R. Combinatorial peptide libraries and biometric score matrices permit the quantitative analysis of specific and degenerate interactions between clonotypic TCR and MHC peptide ligands. J. Immunol. 2001;167:2130–2141. doi: 10.4049/jimmunol.167.4.2130. [DOI] [PubMed] [Google Scholar]
- 48.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]