

Abstract
A principal mechanism by which tumors evade immune-mediated elimination is through immunosuppression. Previous approaches to tumor immunotherapy have focused on modifying the immunosuppressive environment with immune checkpoint inhibitors, cytokine therapy, and other modalities with the intent to generate T-cell based anti-tumor immunity. We hypothesized that transformation of the suppressive ovarian cancer microenvironment could be achieved by introduction of the attenuated ΔactA/ΔinlB strain of Listeria monocytogenes. ΔactA/ΔinlB introduced into the microenvironment of the aggressive ID8-Defb29/Vegf-A murine ovarian carcinoma is preferentially phagocytosed by tumor-associated macrophages (TAMs) and reprograms that population from one of suppression to immunostimulation. TAMs in the peritoneum upregulated their co-stimulatory molecules CD80 and CD86, increased transcription of inflammatory cytokines, and downregulated transcription of suppressive effector molecules. Surprisingly, therapeutic benefit was not mediated by T- or NK-cell activity. ΔactA/ΔinlB-induced repolarization of TAMs activated direct tumor cell lysis via Nos2 production of nitric oxide. Modulation of the immunosuppressive nature of the ID8-Defb29/Vegf-A microenvironment, specifically by reprogramming of the TAM suppressive population from M2 to M1 polarization, is critical for our observed immune-mediated survival benefit.
Keywords: Listeria, tumor-associated macrophages, ovarian cancer, immunotherapy, immunosuppression
Introduction
Standard of care for epithelial ovarian cancer patients includes surgical resection and chemotherapy with non-specific cytotoxic drugs such as cisplatin and paclitaxol.1 Targeted chemotherapy with the anti-angiogenic drug bevacizumab has shown limited potential with accompanying adverse side effects.2,3 These multiple approaches have not improved the five-year survival rate for ovarian cancer over the past 30 y, which remains below 50% overall and significantly lower for late stage disease.4 New therapeutic approaches are badly needed and a promising potential approach is found in immunotherapy. However, the immunosuppressive ovarian cancer microenvironment must be overcome for successful immunotherapy.
An abundant cell population in ovarian cancer ascites is tumor-associated macrophages (TAMs).5-8 This cell type is M2-polarized and is strongly immunosuppressive. TAMs are characterized by high expression of immunosuppressive and pro-angiogenic molecules such as IL-10 and VEGF-A.8 They are positive for CD11b and express low levels of MHC-II and co-stimulatory molecules such as CD86.5,9 These cells secrete high levels of immunosuppressive effector molecules arginase-1 and IDO.10,11 TAMs are deficient in antigen presentation, suppress tumor antigen-specific cytotoxic CD8+ T cell proliferation and function, and recruit CD25+Foxp3+ T regulatory cells (T-regs) to the tumor microenvironment.5-7 TAMs also recruit naïve T cells and drive their differentiation into T-regs. High TAM frequency is correlated with poor patient prognosis.5,6
Attenuated pathogens and pathogen-derived constituents are strong immunostimulants that have the potential to impact tumor-mediated immunosuppression. Listeria monocytogenes is a Gram-positive, facultative intracellular bacteria. Listeria has shown promise as an anti-tumor immunotherapeutic platform due to its potent induction of both innate and adaptive immune responses and its capacity to express human tumor-associated antigens (TAA). Listeria vaccines are currently in clinical trials for a variety of cancers including lung, pancreatic, cervical, liver, mesothelioma, and ovarian.12,13 Listeria cancer vaccines are rendered safe by attenuation through deletion of virulence factors such as ActA and Internalin B (ΔactA/ΔinlB).14 The ΔactA/ΔinlB strain has reduced capacity to invade non-phagocytic cells, essentially targeting it to antigen-presenting cells. Once taken up by phagocytic cells, this strain can escape the phagosome into the cytoplasm where it can effectively secrete antigen to be presented on MHC class-I to prime CD8+ T cell responses.15
Listeria strains engineered to express tumor-associated antigens have shown efficacy in pre-clinical tumor models.14,16-19 Treatment with Listeria induces production of inflammatory cytokines that activate natural killer (NK) cells and increase their anti-tumor cytolytic capacity.12,18Listeria can effectively induce TAA-specific CD4+ and CD8+ T cells that cause regression of primary and metastatic disease and protect mice from re-challenge, demonstrating that TAA-expressing Listeria can establish systemic anti-tumor immunity.18 Initial NK cell- and CD8+ T cell-mediated tumor cell death following treatment releases tumor antigen and can lead to epitope spreading, which has been shown to generate CD8+ T cells that are specific for tumor antigen not expressed by the Listeria vaccine.17,18 Such epitope spreading is crucial for long-lived tumor immunity.17
While the bulk of the Listeria cancer immunotherapy literature has focused on eliciting anti-tumor adaptive immunity, little focus has been directed to the modulation of the innate, immunosuppressive populations present in the tumor microenvironment. Previous work has shown that “re-educating” TAMs in the ID8 ovarian cancer model via adenoviral-mediated inhibition of NF-κB can re-polarize these cells to more of a classical M1 macrophage phenotype expressing high levels of Il-12 and MHC-II, and low levels of Il-10 and arginase-1. This manipulation of TAMs in the tumor microenvironment led to an in vivo anti-tumor immune response mediated by recruitment and cytotoxic activity of NK cells, and by macrophage direct killing of tumor cells.9 While adenoviral-mediated inhibition of NF-κB that is specifically targeted to ovarian cancer TAMs is unfeasible as a clinical application, the ΔactA/ΔinlB strain of Listeria monocytogenes may be uniquely adaptable for this purpose as its vastly reduced capacity to invade non-phagocytic cells may restrict it to this population whereby it can potently stimulate innate immunity.14 It is our hypothesis that treatment of ovarian cancer with attenuated ΔactA/ΔinlB Listeria monocytogenes (Lm) will target the immunosuppressive tumor-associated macrophage population in the microenvironment and re-polarize these cells to a pro-inflammatory phenotype. We tested this hypothesis in the aggressive ID8-Defb29/Vegf-A model.20,21
Results
Treatment of ID8-Defb29/Vegf-A ovarian tumors with Lm improves survival
Weekly intraperitoneal (IP) treatment (Fig. 1A) of ID8-Defb29/Vegf-A tumor-bearing mice with 10 million CFU of the live-attenuated Lm improved survival of C57BL/6 mice (Fig. 1B). This treatment was dependent on the presence of live Lm in the tumor microenvironment, as intravenous (IV) treatment with live or intraperitoneal (IP) treatment with heat-killed Lm did not improve survival as compared with untreated mice (Fig. 1B). Survival of mice treated intraperitoneally with live Lm differed significantly from untreated, IV-treated, and heat-killed treated groups to a similar degree (Fig. 1B; P value < 0.001 for all three). Although heat killing of Lm may potentially denature immunogenic proteins, rendering them inert, the process does not affect Lipoteichoic acid (LTA), CpG DNA, and many other bacterial products, indicating that treatment with toll-like receptor (TLR) agonists alone does not confer survival.

Figure 1.Lm improves survival in ovarian cancer-bearing mice. (A) schematic of treatment regimen. After challenge with 2 x 106 ID8-Defb29/Vegf-A, mice were injected IP with 107 CFU of Lm weekly until death beginning on day 3 post-challenge. (B) C57BL/6J mice (n = 8-12 per group) were tumor-challenged and left untreated (solid black), treated with 107Lm injected IP (dashed black), treated with 107Lm injected IV (solid gray), or treated with 107 heat-killed Lm injected IP (dashed gray). P value between untreated, Lm IV, and heat-killed Lm > 0.05, n.s. P value between Lm IP and other three groups all < 0.001.
Lm preferentially accumulates in CD11b+ tumor-associated macrophages in ascites
As previously described, the predominant myeloid cell population in ID8 tumor ascites are tumor-associated macrophages (TAMs) that can be identified by the CD11b surface marker and immunosuppressive phenotype.9,22 Therefore, CD11b was used for phenotyping and cell-sorting purposes. In the hyper-aggressive ID8-Defb29/Vegf-A model, CD11b+ TAMs cells comprise approximately 20% of the total cells in the ascites (Fig. 2A). By labeling Lm with CFSE prior to IP injection, we were able to determine which cell populations in the peritoneum of tumor-bearing mice were targeted for Lm uptake by our treatment (Fig. 2B). While gating on CFSE-positive cells revealed that approximated 50% of these cells were CD11b+, it was this population that exhibited a significantly higher fluorescence intensity for CFSE labeling, leading us to conclude that Lm preferentially accumulated in this population (Fig. 2C).

Figure 2.Lm injected peritoneally preferentially accumulates in CD11b+ tumor-associated macrophages. (A) cells from ascites were harvested from mice (n = 4) bearing ID8-Defb29/Vegf-A tumors on day 35 and assessed for percentage of CD45+ (left panel) and percentage of CD11b+ out of CD45+ pre-gated (right panel). ~20% of total cells in peritoneum of tumor-bearing mice are CD11b+. (B) ascites cells of day 35 tumor-bearing mice injected with 1 × 107 CFSE-labeled Lm were harvested 16 h post-injection and assayed by FACs for in vivo cell tracking. Bottom left panel are gated on CFSE+ cells. (C) CFSE-labeled Lm accumulate in both CD11b+ and CD11b- cells in peritoneum, but mean fluorescence in CD11b+ subset is significantly higher (P value < 0.001).
The CD11b- population that was positive for Lm uptake was CD45- (Fig. S1A–C). A significant percentage of ascites cells in this model are tumor cells, which can be phagocytic, and are likely the cells comprising this sizable CFSE+CD11b-CD45- population. To investigate any direct growth inhibitory effects of Lm on the tumor cell population, we performed growth assays with increasing MOI of Lm and observed no adverse effects on ID8-Defb29/Vegf-A cell growth with Lm infection up to a MOI of 10 (Fig. S1D).
Lm invasion upregulates co-stimulatory markers on CD11b+ TAMs and reprograms their transcription of immune effector molecules
Exposure of bulk ascites cells to Lm ex vivo allows for observation of any phenotypic changes on those cells while controlling for migration of any new cells. The CD11b+ cells, in which we previously showed that Lm preferentially accumulate, upregulate their surface expression of co-stimulatory molecules CD80 and C86 in response to Lm exposure (Fig. 3A). Again utilizing CFSE-labeling of Lm before treatment, we were able to distinguish CD11b+ cells that had phagocytosed Lm (CFSE+) and those that had not (CFSE-). This enabled us to observe a pronounced bystander effect in CD11b+ cells within the same cultures whereby CD11b+CFSE+ cells had markedly higher expression of CD80 and CD86, but CD11b+CFSE- cells still had expression significantly elevated above that seen in untreated controls, likely in response to paracrine signaling. As expected, CD80 and CD86 expression were low on these cells from untreated cultures (Fig. 3A). We observed similar results in vivo where CD11b+ cells that had phagocytosed CFSE-labeled Lm displayed high levels of CD80. Again, there was a bystander effect where CD11b+CFSE- cells in the ascites of Lm-treated mice exhibited elevated CD80 expression when compared with untreated mice (Fig. 3B). Additionally, CD11b+ TAMs exhibited drastically increased MHC class II presentation following Lm treatment (Fig. S1E).

Figure 3. Treatment with Lm increases presentation of costimulatory molecules on CD11b+ TAMs. (A) ascites from day 35 ID8-Defb29/Vegf-A-bearing mice (n = 4) were harvested and treated with CFSE-labeled Lm at a MOI of 1 or left untreated. CD80 (left panel) and CD86 (right panel) increase significantly in CD45+CD11b+ TAMs that have phagocytosed Lm when compared with CD45+CD11b+ TAMs from the same ex vivo cultures that were negative for CFSE or were left untreated. (B) Similar results to (A) were observed in vivo with CD80 utilizing CFSE-Lm tracking. Bottom left panel: untreated CD45+CD11b+ TAMs (dashed line), treated CD45+CD11b+CFSE- TAMs (solid black), and treated CD45+CD11b+CFSE+ TAMs (dotted black) representative histograms are shown. Bottom right panel: CD80 mean fluorescence is quantified from multiple in vivo replicates (n = 5).
Interestingly, the majority of untreated CD11b+ TAMs also stain positive for CD11c expression (Fig. S2A). However, these cells downregulate CD11c in response to Lm to the degree that the majority of TAMs no longer express any CD11c (Fig. S2B–D). Recently reported data indicate that CD11c is downregulated in response to activation via TLR signaling.23 We believe that the loss of CD11c that we observe in CD11b+ TAMs is another indicator of their activation, consistent with the reported literature and other markers enumerated above.
CD11b+ TAMs alter their transcription of immunosuppressive effector molecules in response to exposure to Lm. These cells downregulate transcription of transforming growth factor-β (Tgf-β), indoleamine 2,3-dioxygenase (Ido), the immune checkpoint B7 family ligand member V-domain Ig suppressor of T cell activation (VISTA), arginase-1 (Arg1), TNF ligand superfamily member 11 (RANKL), and matrix metallopeptidase 7 (Mmp7) (Fig. 4A).24

Figure 4. Phagocytosis of Lm by CD11b+ TAMs induces transcriptional changes indicative of M2 to M1 repolarization. (A, B) ascites of tumor-bearing mice (n = 4) were harvested and plated in culture overnight and either left untreated or exposed to Lm at a MOI of 1. 16 h later, CD11b+ cells were sorted and RNA was isolated for qRT-PCR analysis of immunosuppressive immune effector molecules (A) and immunostimulatory cytokines and Cxcl10 (B). (C) Quantification of cytokine concentrations in supernatant of ex vivo ascites cultures exposed to Lm overnight (n = 4). (D) quantification of cytokine concentrations in serum of ascites of day 35 tumor-bearing mice 16 h following treatment with 107 CFU Lm (n = 4).
TAMs exposed to Lm increase their transcription of inflammatory cytokines tumor-necrosis factor α (Tnf-α), interleukin-1 β (Il-1β), interleukin-6 (Il-6), and interleukin-12 (Il-12p35), and the chemokine Cxcl10, all by many fold (Fig. 4B). Secretion of these effector molecules was confirmed by Luminex array in supernatants of ex vivo cultures and in the ascites serum of mice 16 h post-treatment (Fig. 4C and D). With the exception of Il-1β levels in vivo (P value = 0.10), all cytokines reached statistical significance both ex vivo and in vivo. In addition to CD11b+ TAM-produced cytokines, we also observed significant increases in interferon gamma (Ifn-γ) levels, indicating activation of peritoneal-resident CD4+ T cells, CD8+ T cells, and NK cells (Fig. 4C and D). Exposure of CD11b+ TAMs to Lm caused these cells to increase cell surface expression of co-stimulatory molecules, downregulate suppressive immune effector molecule transcription, and upregulate pro-inflammatory cytokine and chemokine transcription.
Lm exposure reverses suppressive phenotype of CD11b+ TAMs
In addition to the increases we observed in the expression of cell surface co-stimulatory molecules and immunomodulatory cytokines and chemokines in CD11b+ TAMs, we sought to determine any change in their function after Lm treatment in a T cell suppression assay. It is well established that TAMs isolated from the ascites of tumor-bearing mice suppress the production of Ifn-γ from bulk splenocyte cultures that were stimulated with anti-CD3 (Fig. 5).25
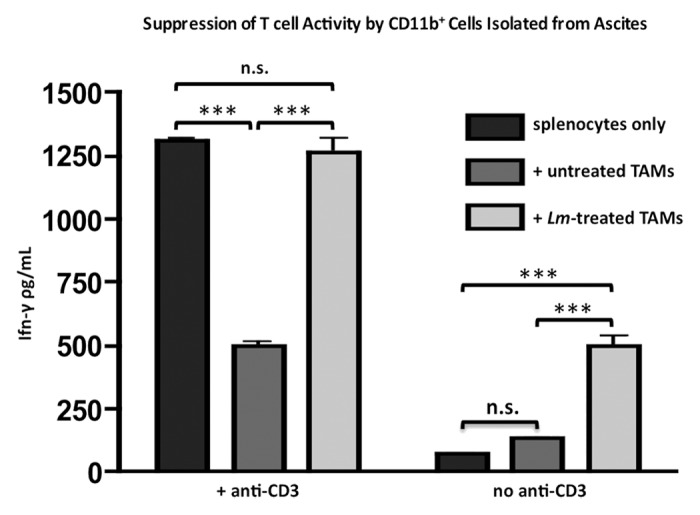
Figure 5. Exposure to Lm reverses suppressive phenotype of CD11b+ TAMs in T cell suppression assay. Ascites of tumor-bearing mice (n = 4) were harvested and plated in culture overnight and either left untreated or exposed to Lm at a MOI of 1. CD11b+ cells were sorted out and 105 were plated with 106 bulk splenocytes from naïve C57BL/6J mice with or without anti-CD3ε. Supernatants were harvested for Ifn-γ ELISA after 72 h.
In contrast, CD11b+ TAMs that had been exposed to Lm prior to co-culture did not suppress Ifn-γ secretion (P value < 0.001) and, in fact, Ifn-γ levels reached the same level as control wells containing only splenocytes and anti-CD3, with no significant difference between the two groups. Additionally, Lm-treated CD11b+ TAMs on their own were able to induce the production of Ifn-γ from splenocyte cultures even in the absence of activating anti-CD3, likely as a result of their inflammatory cytokine production. This assay demonstrates that Lm exposure abrogates the functional suppressive phenotype of CD11b+ TAMs.
Lm treatment induces an increase in anti-tumorigenic chemokine abundance in mouse ascites and subsequent recruitment of effector cell types
Due to the increased production of inflammatory cytokines from CD11b+ TAMs in the tumor microenvironment as a response to Lm, we sought to determine changes in chemokines that might recruit new effector cells into the peritoneum that could augment the anti-tumor immune response we had observed (Fig. 1B). In addition to the Cxcl10 that is produced by the Lm-treated TAMs (Fig. 4B), we also assessed levels of the T cell chemoattractant Ccl5 (RANTES) and myeloid cell chemoattractants Ccl2 (MCP-1), Ccl3 (MIP-1α), and Ccl4 (MIP-1β). All five chemokines were increased in the ascites of mice that received Lm when compared with untreated mice (Fig. 6A). Next, we assessed whether there was a concomitant increase in abundance of effector cell subsets in the ascites post-Lm administration. Indeed, we observed significant increases in CD8+ T cells (Fig. 6B), CD4+ T cells (Fig. 6C), CD11b+Gr-1hi neutrophils, and CD11b+Gr-1lo monocyte/macrophages (Fig. 6D) in ascites following Lm treatment. Given that CD11b+Gr-1+ cells migrate to the peritoneum after Lm treatment in the context of a massive inflammatory response, we believe these cells to be newly activated neutrophils and macrophages/monocytes and not myeloid-derived suppressor cells (MDSCs), although this may be a possibility.

Figure 6.Lm treatment of tumor-bearing mice induces accumulation of chemoattractants in peritoneum and subsequent recruitment of immune effector cells. (A) Mice bearing ID8-Defb29/Vegf-A tumors for 35 d were injected with 107 CFU Lm. 16 h later ascites serum was harvested for quantification of chemokine levels. (B-D) ascites cells were harvested from day 35 ID8-Defb29/Vegf-A-bearing mice 16 h post-Lm treatment and assessed for changes in numbers of CD45+CD3ε+CD8+ T cells (B), CD45+CD3ε+CD4+ T cells (C), CD45+CD11b+Gr-1hi neutrophils, and CD45+CD11b+Gr-1lo monocytes/macrophages (D). Data for treated mice appear as light gray bars and untreated mice as dark gray bars for all four panels.
We hypothesized that this increased trafficking of effector cell subsets into the tumor microenvironment induced by Lm treatment might be responsible for the increased survival in these treated mice. However, tumor-challenged mice depleted of CD8+ or CD4+ T cells and treated with Lm still exhibited significantly improved survival when compared with CD8- or CD4-depleted mice that were not treated with Lm (Fig. 7A and B). While Lm treatment drastically alters the milieu of immunomodulatory molecules and recruitment of effector cell subsets into the tumor microenvironment, it does not appear that the function of these effector cell subsets is required for the overall survival increase in response to treatment.

Figure 7.Lm treatment improves survival in immunocompromised mice that is dependent on direct macrophage tumoricidal action. (A–C) survival of mice challenged with ID8-Defb29/Vegf-A and treated with weekly Lm treatments is not dependent on CD8+ T cells (A), CD4+ T cells (B), or NK cells (C). (D) NSG mice treated with Lm have improved survival: P value < 0.001. (E) ascites of tumor-bearing mice (n = 4) were harvested and plated in culture and either left untreated or exposed to Lm at a MOI of 1. 16 h later, CD11b+ cells were sorted and RNA was isolated for qRT-PCR analysis of Nos2 expression. Supernatants from same cultures were assayed with Griess reagent for functional validation of Nos2 activity by CD11b+ TAMs. (F) CD11b+ cells as harvested in (E) were co-cultured with 51Cr-pulsed ID8-Defb29/Vegf-A cells at an effector-target ratio of 10:1 overnight with or without the selective iNOS inhibitor L-nil and assayed after 16 h for tumor target cell Chromium release.
Survival efficacy depends on the presence of reprogrammed CD11b+ TAMs in tumor microenvironment and induction of direct tumor cell lysis by CD11b+ cells
Given that Lm treatment still conferred a significant survival benefit in the absence of CD8+ T cells (Fig. 7A), CD4+ T cells (Fig. 7B), and even NK cells (Fig. 7C), we next investigated treatment efficacy in NOD/scid/Il2R-γ−/− (NSG) mice lacking adaptive immunity, but that still have TAMs that polarize to an activated M1 phenotype in response to Lm treatment (Fig. S3). Surprisingly, Lm treatment still improved survival in NSG mice (Fig. 7D; P value < 0.001). Due to the absence of any direct growth-inhibitory effect on ID8-Defb29/Vegf-A cells by Lm (Fig. S1D) and the observation that treated NSG mice lacking T, B, and NK cells still exhibited improved survival, we next investigated whether the anti-tumor response elicited by Lm-based treatment in this model is due to activated CD11b+ TAMs and a direct lytic effect on tumor cells. CD11b+ TAMs, the predominant innate cell population in the ascites of tumor-bearing mice, are known to perform anti-tumor effector function via nitric oxide (NO) secretion upon repolarization from M2 to M1.9,22 We isolated this population from tumor ascites and found that, in addition to changes in expression of co-stimulatory markers and immune effector molecules indicative of a switch from M2 to M1 phenotype, these cells also upregulated their transcription of inducible nitric oxide synthase (Nos2) upwards of 2500-fold in response to Lm uptake (Fig. 7E). This was associated with a significant increase in measurable nitric oxide in the media of ascites cells cultured ex vivo (Fig. 7E). We next determined whether exposure to Lm and subsequent production of NO could have a direct anti-tumor lytic effect on tumor cells and if this was the mechanism for the improved survival in Lm-treated NSG and WT mice. Indeed, CD11b+ TAMs exposed to Lm were able to directly kill ID8-Defb29/Vegf-A tumor cells whereas untreated CD11b+ TAMs displayed minimal killing ability (Fig. 7F). This tumor cell lysis was abrogated when the selective iNOS inhibitor L-nil was added to culture medium, leading us to conclude that direct anti-tumor effector function of reprogrammed CD11b+ TAMs was dependent on nitric oxide production via Nos2.22
Discussion
Ovarian cancer currently has dismal five-year survival and is one of the most commonly occurring epithelial cancers.4 The most abundant myeloid population in the ovarian tumor ascites is the CD11b+ tumor-associated macrophage that exhibits immunosuppressive activity.5-8 Here, we demonstrate that treatment with the attenuated ΔactA/ΔinlB strain of Listeria monocytogenes preferentially targets this cell population and repolarizes them from an immunosuppressive M2 phenotype to an immunostimulatory, classical M1 phenotype that triggers therapeutic antitumor immunity. CD11b+ TAMs that phagocytose Lm upregulate co-stimulatory molecules CD80 and CD86, as well as MHC class II. They downregulate transcription of immunosuppressive effector molecules including Tgf-β, arginase-1, and Ido. They upregulate transcription of pro-inflammatory cytokines Tnf-α, Il-1β, Il-6, and Il-12. In addition to changes in expression of immune effector molecules on a transcriptional level, we have shown that Lm can reverse the ability of this CD11b+ TAM population to suppress T cell function and, in fact, to become immunostimulatory. Lm-treated, repolarized TAMs also become directly tumoricidal through iNOS-mediated production of nitric oxide. We believe this demonstrates that Lm treatment is a viable method to repolarize suppressive tumor-associated macrophages in the ovarian cancer microenvironment.
There is a body of literature describing a tumor necrosis factor and inducible nitric oxide synthase-producing dendritic cell population in mice derived from Gr-1+ monocytes following infection with L. monocytogenes that mediates host innate immune defense.26 So-called TIP-DCs bear a resemblance to the repolarized tumor-associated macrophages described above. However, as noted by others, these TIP-DCs have no obvious distinction from classically activated M1 macrophages.27 Initial work identifying TIP-DCs was performed on splenocytes 48 h following L. monocytogenes infection, outside of a cancer context. Given that our work presents data on macrophage polarization markers 16 h post-Lm treatment on cells harvested from peritoneal ascites of an ovarian serous carcinoma, and that CD11b+ TAMs in our model lose CD11c expression following Lm treatment (Fig. S2), we feel comfortable in classifying our studies as pertaining to tumor-associated macrophages and not TIP-DCs. We do, however, acknowledge that the CD11b marker used in our phenotyping may be expressed by multiple subsets of immune cells that expand during tumor growth, including TAMs, granulocytic and monocytic myeloid-derived suppressor cells (MDSCs), and dendritic cells. CD11b was used in lieu of the classical macrophage marker F4/80, as F4/80 has been shown to be downregulated on macrophages in response to Ifn-γ, which is induced in our model following Lm treatment (Fig. 4C and D).28 In short, there is currently no cell surface marker that can definitively distinguish between the myriad suppressive innate immune cell populations occurring in cancer. We feel, however, that the context of our model—ovarian ascites—and the T cell suppression and tumor cell lysis functional assays presented here support our assertion of CD11b+ tumor-associated macrophages that alter their polarization from M2 to M1.
Our results show that Lm has a profound immunomodulatory effect on the tumor microenvironment in increasing the abundance of pro-inflammatory cytokines and chemokines and the recruitment of immune effector cell subsets that have known positive influences on anti-tumor immunity.9,14,16,18,19,21,22 However, it is surprising that survival studies depleting CD4+ T cells, CD8+ T cells, and NK cells have shown that no effector cell population alone is critical for our observed treatment efficacy. In fact, it appears that reprogramming of tumor-associated macrophages and induction of their direct anti-tumor lytic function via nitric oxide is the mechanism underlying the survival benefit of the treatment. This is not the first evidence in which tumor regression has been shown to be mediated exclusively by innate immunity. In the KPS model of murine pancreatic adenocarcinoma, one group demonstrated that CD-40 agonist-mediated ‘reeducation’ of tumor-associated macrophages induced their direct tumoricidal activity, resulting in tumor regression that was not dependent upon CD8+ or CD4+ T cells.29 We believe that our data reports the first instance that this phenomenon—anti-tumor immunity mediated solely by innate immune mechanisms—may be present in ovarian cancer, as well. While the pancreatic study did not define a mechanism underlying TAM effector function, it is tantalizing to speculate that it requires the same Nos2-mediated production of nitric oxide described here. This study and ours support the pursuit of therapeutic strategies that target the innate suppressive populations within the tumor microenvironment.
Others have shown that repolarization of tumor-associated macrophages from M2 to M1 confers a survival advantage in the ID8 ovarian model that is predicated on a dual mechanism: induction of direct macrophage tumoricidal activity and increased NK cell activity that is dependent on repolarized macrophage-produced Il-12.9 Such mechanisms depended on Stat1 activation in TAMs. Macrophages are known to be the dominant innate immune responder and to activate Stat1 in response to L. monocytogenes infection.30 Stat1 is a potent transcriptional activator of Il-12, MHC class II, and iNOS. Indeed, we see induction of all three in CD11b+ TAMs after exposure to Lm in what is a feasible approach to targeting this population for immunotherapy.9 It is plausible to hypothesize that repolarization of TAMs by Lm may depend in part on activation of the Stat1 signaling pathway.
While other groups have shown a causal relationship between L. monocytogenes vaccines, Il-12 production, and NK cell activity in anti-tumor immunity, neither Il-12 nor NK cells are critical in the ovarian treatment model described in this study as both Il-12p35−/− and NK1.1-depleted mice exhibited significant survival increase in response to treatment (Fig. S4; Fig. 7C).12,18 While we do not discount that there is an anti-tumor contribution from other effector cell types, it appears that the iNOS-mediated tumoricidal activity of repolarized TAMs in our model is so robust that it alone can confer a survival benefit even in the absence of NK- and CD8+ T cells (Fig. 7D).
We present data here elucidating the repolarization of immunosuppressive tumor-associated macrophages from M2 to M1 phenotype and subsequent survival advantage conferred by treatment with the ΔactA/ΔinlB strain of Listeria monocytogenes in an aggressive ovarian carcinoma model. ΔactA/ΔinlB vaccines have demonstrated success as tumor immunotherapies in their ability to induce antigen-specific anti-tumor CD8+ T cell responses through expression of human tumor-associated antigens in both pre-clinical mouse models and ongoing human clinical trials. The strain of Lm currently being used clinically, human mesothelin-expressing CRS-207, is administered intravenously.12 Patients receive the Lm vaccine prior to receiving standard of care chemotherapy, and research is also in development to combine CRS-207 with GVAX, radiation, and immune checkpoint inhibitors. In lieu of our data elucidating the effect of Lm on myeloid cells and the nitric oxide response, treatment options that activate myeloid cells locally in the tumor microenvironment either to induce their direct lytic function against tumor cells or as radiosensitizers via nitric oxide might become attractive additional strategies to augment intravenous Lm vaccination focused on eliciting CD8+ T cell responses. This previously described induction of adaptive anti-tumor immunity and the under-investigated but equally significant modulation of immunosuppressive innate cell function described here present Listeria vaccines as a powerful and multifaceted anti-tumor immunotherapeutic platform in ovarian cancer.
Materials and Methods
Mice
C57BL/6J (01C55) were purchased from the National Cancer Institute. Il-12p35−/− (002692), and NOD/scid/IL2Rγ−/− (005557) mice were purchased from The Jackson Laboratory. All mouse studies were performed in accordance with the Institutional Animal Care and Use Committee of Dartmouth.
Cell lines
ID8-Defb29/Vegf-A orthotopic peritoneal tumors were established in mice as described previously.20 Briefly, ID8-Defb29/Vegf-A cells were cultured in complete media (RPMI supplemented with 10% FBS, penicillin/streptomycin, and sodium pyruvate), then harvested, washed in phosphate-buffered saline (PBS), and injected in 200 μL of PBS at 1 × 107 cells/mL to establish tumors.
Listeria treatment of tumor-bearing mice
The ΔactA/ΔinlB Listeria monocytogenes strain is based on wild-type L. monocytogenes strain 10403S and is deleted of both ActA and Internalin B virulence factors as previously described.14 Heat-killed Lm was generated by heating bacterial suspensions at 80 °C for 1 h. C57BL/6J mice were intraperitoneally (IP) injected with 2 × 106 ID8-Defb29/Vegf-A and were treated IP with 200 μL Lm at 5 × 107 CFU/mL in phosphate-buffered saline (PBS) beginning on day 3 and repeated on a weekly basis thereafter (day 10, 17, etc). For analysis of Lm-invaded cells in the peritoneal tumor ascites, ID8-Defb29/Vegf-A tumors were established for 35 d, then were treated IP with 1 × 107 CFU Lm labeled with CFSE. Lm were incubated in PBS with 10 μM CFSE (Biolegend) for 10 min at 37 °C in the dark then washed twice and resuspended in PBS prior to injection.
Antibodies and flow cytometry
Anti-mouse antibodies were specific for CD45 (30-F11), CD11c (N418), MHC-II (M5/114.15.2), CD3ε (145–2C11), CD8β (YTS156.7.7), CD80 (16–10A1), CD86 (GL-1), CD11b (M1/70), CD4 (GK1.5), and Gr-1 (RB6–8C5) from Biolegend and CD16/CD32 (93) from eBioscience. Red blood cells were removed from ascites samples using lysis buffer of 150 mM NH4Cl, 10 mM KHCO3, and 0.5 mM EDTA. Flow cytometry was performed on a FACS-Canto system (BD Biosciences) or MACSQuant analyzer (Miltenyi). Data were analyzed using FlowJo software version 8.7.
T-cell suppression assay
Suppression assay was performed as described previously.25 Briefly, ascites were harvested from day 35 tumor-bearing C57BL/6J mice and red blood cells were removed using lysis buffer. Ascites cells were plated in bulk immediately after harvest in complete media either without Lm or at a multiplicity of infection (MOI) of 1. Following overnight culture of 16 h, cells were harvested and sorted for CD11b+ cells using magnetic MicroBeads according to manufacturers protocol (Miltenyi). Lm treatment was done on bulk cells and CD11b sorting performed after in order to more closely approximate in vivo conditions.
Co-cultures of 2 × 105 CD11b+ cells with 1 × 106 bulk responder splenocytes from naïve C57BL/6J were stimulated with 1 μg of anti-CD3 (145–2C11) in 96-well round-bottomed plates in 200 μL of culture media. Culture supernatants were collected after 72 h and analyzed for Ifn-γ production using the mouse Ifn-γ ELISA MAX™ Deluxe kit according to manufacturer’s protocol (Biolegend, 430804).
mRNA expression analysis
Ascites cells were harvested, left untreated or Lm-treated at a MOI of 1, and sorted for CD11b as in the suppression assay. RNA was extracted from CD11b+ cells using the RNeasy kit (Qiagen, 74104). cDNA was synthesized using iScriptTM cDNA synthesis kit (Bio-Rad, 170-8891). q-PCR was performed on a CFX96TM Real-Time PCR Detection System (Bio-Rad) using iQTM SYBR® Green Supermix (Bio-Rad, 170-8882) with primers at a concentration of 0.5 μM in final volume of 20 μL. Cycling parameters and primer sequences are available in Figure S5. mRNA transcript fold-change was calculated using the ΔΔCT method with all samples normalized to mouse Gapdh.31
Cytokine assay
For in vivo cytokine data, ascites were harvested from mice 16 h post-treatment with 1 × 107 CFU Lm and serum was harvested. For ex vivo cytokine results, ascites were harvested from day 35 mice, red blood cells were removed with lysis buffer, and cells were plated for 16 h in complete media with or without Lm at a MOI of 1. Cytokines were quantified using mouse 32plex Luminex assay (MPXMCYTO70KPMX32, Millipore).
Chromium-51 release macrophage killing assay
Ascites were harvested as in the suppression assay and cultured for 4 h with Lm at a MOI of 1 before sorting with CD11b magnetic beads (Miltenyi). CD11b+ cells were plated at 105 cells/well in round-bottomed 96-well plates (Costar) in 200 μL of complete media with 104 ID8-Defb29/Vegf-A target cells pulsed with 51Cr (NEZ030005MC, Perkin Elmer). Co-cultures were incubated at 37 °C for 16 h and then supernatants were harvested and read on a Wizard 1470 Automatic Gamma Counter (PerkinElmer). The selective iNOS inhibitor L-nil was used at a concentration of 500 μM in indicated samples.
Lymphocyte depletion
C57BL/6J mice were injected with mAbs depleting CD4 (clone GK1.5) and CD8 (clone 2.43) that were produced as bioreactor supernatants and administered IP in doses of 250 μg one day prior to Lm treatment and then once weekly for the duration of survival studies. Depleting anti-NK1.1 (clone PK136) was produced similarly and administered IP in doses of 200 μg for four treatments on days –2, 0, 2, and day 9 relative to tumor challenge. Greater than 95% depletion of target cell populations was confirmed by flow cytometry (Fig. S6).
Statistics
Unless noted otherwise, all experiments were repeated at least 2 times and results were similar between repeats. Experiments used between 4 and 12 mice per group. Figures denote statistical significance of P < 0.05 as *, P < 0.01 as **, and P < 0.001 as ***. A P value < 0.05 was considered to be statistically significant. Data for bar graphs was calculated using the unpaired Student t test. Error bars represent standard error of the mean from independent samples assayed within the represented experiments. Survival experiments utilized the Log-rank Mantel-Cox test for survival analysis. Statistical analysis was done with Graph Pad Prism 4 software.
Supplementary Material
Disclosure of Potential Conflicts of Interests
P.L. and D.G.B. are employees of Aduro, Inc., and hold stock options.
Financial Support
Dartmouth Immunobiology of Myeloid and Lymphoid Cells NIH Training Grant 5T32AI007363-22 (P.H.L.); Dartmouth Center of Nanotechnology Excellence NIH 1 U54 CA151662 (S.F.); Center for Molecular, Cellular, and Translational Immunological Research NIGMS 1P30RR032136-01 (S.F.); Norris Cotton Cancer Support Grant P30 CA023108 (S.F.).
Acknowledgments
We thank Paul Spear for help with iNOS macrophage assays. We thank the Immune Monitoring Lab and DartLab at the Geisel School of Medicine at Dartmouth for support of immunological assays. We would like to acknowledge the Dartmouth Transgenic and Genetic Shared Resource for support of transgenic mouse utilization. DartLab and the Transgenic shared resources are made possible through generous COBRE support.
Glossary
Abbreviations:
- TAM
Tumor-associated macrophage
- TAA
tumor-associated antigen
- Lm
Listeria monocytogenes
- MDSC
myeloid-derived suppressor cells
- TLR
toll-like receptor
- IP
intraperitoneal
- NSG
NOD/scid/Il2R-γ−/−
References
- 1.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA, Gynecologic Oncology Group Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 2.Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, Sovak MA, Yi J, Nycum LR. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30:2039–45. doi: 10.1200/JCO.2012.42.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ranpura V, Hapani S, Wu S. Treatment-related mortality with bevacizumab in cancer patients: a meta-analysis. JAMA. 2011;305:487–94. doi: 10.1001/jama.2011.51. [DOI] [PubMed] [Google Scholar]
- 4.Lavoué V, Thédrez A, Levêque J, Foucher F, Henno S, Jauffret V, Belaud-Rotureau MA, Catros V, Cabillic F. Immunity of human epithelial ovarian carcinoma: the paradigm of immune suppression in cancer. J Transl Med. 2013;11:147. doi: 10.1186/1479-5876-11-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, Preisser L, Anegon I, Catala L, Ifrah N, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319–30. doi: 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- 6.Duluc D, Corvaisier M, Blanchard S, Catala L, Descamps P, Gamelin E, Ponsoda S, Delneste Y, Hebbar M, Jeannin P. Interferon-γ reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer. 2009;125:367–73. doi: 10.1002/ijc.24401. [DOI] [PubMed] [Google Scholar]
- 7.Kryczek I, Wei S, Zhu G, Myers L, Mottram P, Cheng P, Chen L, Coukos G, Zou W. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007;67:8900–5. doi: 10.1158/0008-5472.CAN-07-1866. [DOI] [PubMed] [Google Scholar]
- 8.Reinartz S, Schumann T, Finkernagel F, Wortmann A, Jansen JM, Meissner W, Krause M, Schwörer AM, Wagner U, Müller-Brüsselbach S, et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: correlation of CD163 expression, cytokine levels and early relapse. Int J Cancer. 2014;134:32–42. doi: 10.1002/ijc.28335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–8. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Q, Kuang DM, Wu Y, Xiao X, Li XF, Li TJ, Zheng L. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J Immunol. 2012;188:1117–24. doi: 10.4049/jimmunol.1100164. [DOI] [PubMed] [Google Scholar]
- 12.Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, Nemunaitis J, Sterman DH, Hassan R, Lutz E, Moyer B, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 2012;18:858–68. doi: 10.1158/1078-0432.CCR-11-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maciag PC, Radulovic S, Rothman J. The first clinical use of a live-attenuated Listeria monocytogenes vaccine: a Phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine. 2009;27:3975–83. doi: 10.1016/j.vaccine.2009.04.041. [DOI] [PubMed] [Google Scholar]
- 14.Brockstedt DG, Giedlin MA, Leong ML, Bahjat KS, Gao Y, Luckett W, Liu W, Cook DN, Portnoy DA, Dubensky TW., Jr. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci U S A. 2004;101:13832–7. doi: 10.1073/pnas.0406035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahjat KS, Liu W, Lemmens EE, Schoenberger SP, Portnoy DA, Dubensky TW, Jr., Brockstedt DG. Cytosolic entry controls CD8+-T-cell potency during bacterial infection. Infect Immun. 2006;74:6387–97. doi: 10.1128/IAI.01088-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SH, Castro F, Paterson Y, Gravekamp C. High efficacy of a Listeria-based vaccine against metastatic breast cancer reveals a dual mode of action. Cancer Res. 2009;69:5860–6. doi: 10.1158/0008-5472.CAN-08-4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seavey MM, Maciag PC, Al-Rawi N, Sewell D, Paterson Y. An anti-vascular endothelial growth factor receptor 2/fetal liver kinase-1 Listeria monocytogenes anti-angiogenesis cancer vaccine for the treatment of primary and metastatic Her-2/neu+ breast tumors in a mouse model. J Immunol. 2009;182:5537–46. doi: 10.4049/jimmunol.0803742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bahjat KS, Prell RA, Allen HE, Liu W, Lemmens EE, Leong ML, Portnoy DA, Dubensky TW, Jr., Brockstedt DG, Giedlin MA. Activation of immature hepatic NK cells as immunotherapy for liver metastatic disease. J Immunol. 2007;179:7376–84. doi: 10.4049/jimmunol.179.11.7376. [DOI] [PubMed] [Google Scholar]
- 19.Yoshimura K, Laird LS, Chia CY, Meckel KF, Slansky JE, Thompson JM, Jain A, Pardoll DM, Schulick RD. Live attenuated Listeria monocytogenes effectively treats hepatic colorectal cancer metastases and is strongly enhanced by depletion of regulatory T cells. Cancer Res. 2007;67:10058–66. doi: 10.1158/0008-5472.CAN-07-0573. [DOI] [PubMed] [Google Scholar]
- 20.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na H, Zhang L, et al. Tumor-infiltrating dendritic cell precursors recruited by a β-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–8. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 21.Baird JR, Fox BA, Sanders KL, Lizotte PH, Cubillos-Ruiz JR, Scarlett UK, Rutkowski MR, Conejo-Garcia JR, Fiering S, Bzik DJ. Avirulent Toxoplasma gondii generates therapeutic antitumor immunity by reversing immunosuppression in the ovarian cancer microenvironment. Cancer Res. 2013;73:3842–51. doi: 10.1158/0008-5472.CAN-12-1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J Immunol. 2012;188:6389–98. doi: 10.4049/jimmunol.1103019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh-Jasuja H, Thiolat A, Ribon M, Boissier MC, Bessis N, Rammensee HG, Decker P. The mouse dendritic cell marker CD11c is down-regulated upon cell activation through Toll-like receptor triggering. Immunobiology. 2013;218:28–39. doi: 10.1016/j.imbio.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, Lu LF, Gondek D, Wang Y, Fava RA, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–92. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hart KM, Bak SP, Alonso A, Berwin B. Phenotypic and functional delineation of murine CX(3)CR1 monocyte-derived cells in ovarian cancer. Neoplasia. 2009;11:564–73, 1, 573. doi: 10.1593/neo.09228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/S1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 27.Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol. 2010;10:453–60. doi: 10.1038/nri2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ezekowitz RA, Gordon S. Down-regulation of mannosyl receptor-mediated endocytosis and antigen F4/80 in bacillus Calmette-Guérin-activated mouse macrophages. Role of T lymphocytes and lymphokines. J Exp Med. 1982;155:1623–37. doi: 10.1084/jem.155.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kernbauer E, Maier V, Stoiber D, Strobl B, Schneckenleithner C, Sexl V, Reichart U, Reizis B, Kalinke U, Jamieson A, et al. Conditional Stat1 ablation reveals the importance of interferon signaling for immunity to Listeria monocytogenes infection. PLoS Pathog. 2012;8:e1002763. doi: 10.1371/journal.ppat.1002763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.