

Abstract
Introduction
Reintroduction of Variola major as an agent of bioterrorism remains a concern. Time to seroconversion and plaque reduction neutralizing antibody titers (PRNT) of 1 or 2 standard doses (SD) were compared to a single high dose (HD) of modified vaccinia Ankara (MVA).
Methods
Ninety subjects were randomized 1:1 to receive 1 HD or 2 SD of MVA subcutaneously on Days 0 and 28 in a placebo-controlled trial. Serum was collected for PRNT and ELISA. Subjects were followed for safety for the entire study.
Results
The HD was well-tolerated. Using Bavarian Nordic’s ELISA, subjects in both groups achieved seroconversion by Study Day 15 (HD) and Day 28 (SD). Before second vaccination, the hazard rate of seroconverting for the HD group was 1.7 times the SD group with a median time for seroconversion of 14 days for both groups. The peak titer of one HD vaccine was superior to one dose of SD vaccine but inferior to the peak titer after the second dose of the SD vaccination regimen.
Using Saint Louis University’s PRNT, peak titers were 95.8 and 65.2 for the HD and SD groups, respectively, prior to second vaccination. Non-inferiority of the SD group was not established. The proportions of positives were 93.3% (42/45) and 82.2% (37/45) for the HD and SD groups, respectively. The peak titer after two standard doses was superior to that of the HD.
Conclusions
HD MVA was safe and well-tolerated. While the hazard rate for seroconverting was significantly higher in the HD group before second dose, the effect was small as the median time to seroconversion was identical. When comparing PRNT, non-inferiority of one SD was not established and the peak titers were low for both groups. The HD peak response was inferior to the standard two-dose regimen response based on ELISA and PRNT.
Keywords: MVA, IMVAMUNE, vaccine, ELISA, plaque reduction neutralizing antibody, smallpox, Variola
INTRODUCTION
Although smallpox was declared officially eradicated by the World Health Assembly in 1980[1], governments remain concerned about the potential use of Variola virus as an agent for bio-terrorism. ACAM2000™, a recently licensed clonal derivation of Dryvax®, has a similar safety profile as Dryvax® including reported cases of myo/pericarditis [2].
A potential alternative vaccine, Modified vaccinia Ankara (MVA), was used during the smallpox eradication campaign during the 1970s in Germany to mitigate the reactogenicity of vaccinia Elstree [3,4]. This replication-incompetent virus [5–7] is host-range restricted [8–12] due to multiple mutations and significant deletions of replicating vaccinia [2]. While MVA genes are efficiently transcribed, there is a block in viral replication at the level of virus assembly and egress in human cells [8,9].
Bavarian Nordic's proprietary MVA, derived from a purified clone of MVA-597, is produced under serum-free conditions in chicken embryo fibroblast (CEF) cells and does not fully replicate in human cells [13]. The vaccine is safe and well-tolerated and has been administered to immunocompromised subjects [14,15] and subjects with atopic dermatitis.
The standard MVA two dose vaccination regimen is 1×108 TCID50 of MVA administered on Days 0 and 28. However, in the event of a confirmed release of smallpox, a more rapid vaccination regimen is desirable. The main goal of this study was to determine whether a single, high dose (HD) of MVA provided a more rapid and/or stronger immune response relative to a single, standard dose (SD). Additional analysis included comparing a single HD with a standard two-dose regimen. This study (ClinicalTrials.gov Identifier:NCT00879762) seeks to inform policy makers about different options for post-event utilization of available smallpox vaccines.
METHODS
Study Design
This was a randomized, double-blinded study of a single HD versus two SD of MVA administered 28 days apart (Table 1). Subjects (N=45) in the HD group received two 0.5 mL injections in different arms of MVA vaccine at 5×108/mL TCID50 on Day 0 followed by a single saline placebo dose on Day 28. Subjects (N=45) in the SD group received MVA at 1×108 TCID50 in one 0.5 mL injection on Day 0 and 28 and saline placebo (0.5 mL) on Day 0. All injections were administered subcutaneously over the deltoid muscle.
Table 1.
Study Design
 |
ECGs, hematology and chemistry evaluations were obtained at screening and 14 days after each vaccination. Troponin I was evaluated at screening, 14 days after first vaccination and if clinically indicated.
Reactogenicity was followed for 14 days after each vaccination, unsolicited adverse events (AE) for 28 days after the last vaccination, and serious adverse events for the entire study.
The study was approved by the respective institutional review boards. All subjects signed informed consent.
Inclusion Criteria
Eligible healthy subjects were at least 18 years old and not pregnant. ECGs were acceptable if there were no clinically significant findings at screening. Subjects were excluded if they had a vaccinia scar, possible history of smallpox vaccination, active exfoliative skin conditions, or a >10% risk of developing a myocardial infarction or coronary death using the National Cholesterol Education Program’s risk assessment tool [16].
Investigational Products
Three lots of MVA vaccine, IMVAMUNE®, were provided by Bavarian Nordic (BN) A/S (Hejreskovvej 10A, 3490 Kvistgård, Denmark) in liquid-frozen aliquots of 0.65 mL as single use vials. Sterile saline for injection, supplied by BN, was used as placebo and as a diluent for IMVAMUNE® injections.
Immunogenicity Assays
The immunogenicity assays were previously described. BN used BN-MVA (non-replicating in humans) and vaccinia-Western Reserve (replicates in humans) as the assay antigens in the ELISA and PRNT assays, respectively [17] and SLU used ATCC MVA (VR-1508) as the assay antigen in the ELISA [18] and plaque reduction neutralizing antibody (PRNT) assays [19], respectively. Table 2 includes the thresholds for positivity for each assay and definitions for seroconversion and non-inferiority. Serum was obtained on Days 0, 4, 8, 14 and 28 after first vaccination and Days 14, 28 and 180 after second vaccination.
Table 2.
Summary of ELISA and PRNT
| Assay | ELISA | PRNT | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Laboratory | Bavarian Nordic (1° assay) |
Saint Louis Univ. | Saint Louis Univ. (1° assay) | Bavarian Nordic | |||||
| Antigen | MVA-BN | ATCC MVA (VR1508) | ATCC MVA (VR1508) | Vaccinia Western Reserve | |||||
| Seroconversion (SC) Threshold* |
≥50 | >50 | ≥20 | ≥15 | |||||
| Limit of Detection (LOD) |
50 | 50 | 4 | 15 | |||||
| Value Imputed when < LOD |
25 (1/2 LOD) | 50 | 2 (1/2 LOD) | 7.5 (1/2 LOD) | |||||
| Study Arm | SD | HD | SD | HD | SD | HD | SD | HD | |
| Proportion SC after One Vaccination n/N (%) P-Value |
EE: 45/45 (100) |
EE: 45/45 (100) |
EE: 15/45 (33.3) |
EE: 22/45 (48.9) |
EE: 37/45 (82.2) |
EE: 42/45 (93.3) |
EE: 21/45 (46.7) |
EE: 36/45 (80) |
|
| >0.99 | 0.20 | 0.20 | 0.002 | ||||||
| Proportion SC with Two Vaccinations n/N (%) P-Value |
TE: 42/42 (100) |
TE: 45/45 (100) |
TE: 42/42 (100) |
TE: 25/45 (55.6) |
TE: 42/42 (100) |
TE: 42/45 (93.3) |
TE: 42/42 (100) |
TE: 37/45 (82.2) |
|
| >0.99 | <0.0001 | 0.24 | 0.006 | ||||||
| Peak GMT (95% CI) with One Vaccination |
158 (116.2, 214.9) |
282.3 (220.1, 362.1) |
85.3 (65.1, 111.8) |
103.7 (81.3, 132.2) |
65.2 (42.9, 99.1) |
95.8 (67.7, 135.6) |
12.2 (9.5, 15.7) |
16.7 (13.3, 20.9) |
|
| Peak GMT (95% CI) with Two Vaccinations |
1140.6 (881.7, 1475.5) |
286.8 (223.3, 368.4) |
1120.3 (866.7, 1448.1) |
114.2 (89.8, 145.3) |
468.9 (348.3, 631.3) |
102.6 (73.1, 143.9) |
60.4 (41.0, 88.9) |
17.6 (13.9, 22.3) |
|
| SE: 0.42 (−0.17, 1.00) | EE: 0.84 (0.27, 1.40) | ||||||||
| TE: 1.66 (1.17, 2.15) | TE: 1.34 (0.85, 1.83) | ||||||||
| EE: 0.84 (0.27, 1.40) | EE: 0.28 (−0.24, 0.80) | ||||||||
| TE: 1.99 (1.48, 2.50) | TE: 3.29 (2.79, 3.79) | ||||||||
| Evaluation of Non- Inferiority (One SD versus one HD)** |
EE: Non-inferiority not established for SD; HD Superior |
EE: SD Non-inferior |
SE: Non-inferiority not established for SD |
EE: SD Non-inferior | |||||
| Evaluation of Non- Inferiority (One HD versus two SDs)** |
TE:Non-inferiority not established for HD; Two SDs Superior |
TE:Non-inferiority not established for HD; Two SDs Superior |
TE:Non-inferiority not established for HD Two SDs Superior |
TE: Non-inferiority not established for HD Two SDs Superior |
|||||
HD=high dose, SD=standard dose, SC= seroconverters, CI=confidence interval, GMT=geometric mean titer, PE=Primary Endpoint, SE=Secondary Endpoint, TE=Tertiary Endpoint, EE=Exploratory Endpoint
Threshold for seroconversion and value imputed for analysis when below the limit of detection was specific to each institution’s assay and SOP. If any subject had a baseline value at or above the threshold for seroconversion, the post vaccination titer must be 2-fold or greater increase from the baseline titer to be considered seroconversion.
The non-inferiority margin for the PRNT assay was specified as a 2.5-fold difference between the group means which translates to a mean difference of 1 (unitless) on a log2.5 scale. Non inferiority was established if the upper bound of the CI of the mean difference was below 1. If non inferiority was not established and the lower bound was greater than zero, the reference group was considered superior compared to the comparison group. For ELISA, the margin was specified as a 2-fold difference. Non inferiority for ELISA was evaluated in the same way except that it was based on the CI of the log2 mean difference.
Statistical Analysis
Sample Size
A sample size of 45 subjects per group provided 95% power to detect a significant difference of 2.5 fold or more change in median time to seroconversion using the BN ELISA assay at a 5% significance level and obtained 87% power to evaluate non-inferiority of a SD versus a HD based on SLU-PRNT geometric mean of the peak titers GMT) assuming a non-inferiority margin of 2.5 fold, SD=4 for both groups, and a significance level of 2.5 percent.
Safety
The number of each AE or reactogenicity event was summarized using the most severe response (see legend for Figure 1). The number of AEs, rates and exact 95% confidence intervals (CI) for each group were calculated. Differences in event rates were evaluated with Chi-square or Fisher’s exact test (Fisher).
Figure 1.



Maximum severity grade of any reactogenicity event collected per subject in the HD and SD groups for 15 days (Days 0–14) after each vaccination. Erythema and Induration were measured and graded as mild (<15 mm), moderate (15–30 mm) or severe (>30 mm). Other AEs including pain at the injection site, redness, swelling (induration was assessed by the clinical staff), muscle aches, chills, headache, nausea, feeling tired, underarm pain, underarm swelling, itchiness at vaccination site, change in appetite, and joint pain were graded using a functional scale of mild (present but easily tolerated), moderate (able to tolerate routine activity with effort), and severe (unable to continue routine activity). Fever grading scale for oral temperature (°C) was mild 38.0 – 39.0, moderate >39.0 – 40.0, severe >40.0; fever is captured under systemic reactogenicity. Figure 1.a. a) Systemic (functional grade) reactogenicity; b) Local (functional grade) reactogenicity; c) Local (measurement grade) reactogenicity.
Immunogenicity
The per protocol immunogenicity analysis was used and included subjects who received one dose of vaccine and contributed both pre- and post-vaccination serum samples. One subject was excluded due to a dosing error at the first vaccination. Subjects were excluded at the post dose 2 timepoints if they did not receive the second standard vaccine, but were not excluded if they did not receive placebo in the HD group. No imputations were performed to account for the missing data.
The primary endpoint, time-to-seroconversion after the first vaccination using BN-ELISA, was examined for a dose effect using interval-censored survival analysis (R package “intcox” [20]) and confidence intervals for the hazard ratio were estimated using bootstrapping [21]. Antibody titers were dichotomized into responders or non-responders, and summarized using response rates and their exact 95% CI. Fisher’s exact test was used to compare response rates. For ELISA, the geometric mean of the peak titer (GMT) among subjects receiving one SD of IMVAMUNE® was considered non-inferior if there was less than 2 fold difference in the GMT to that among subjects receiving one high dose of IMVAMUNE®.
The secondary endpoint was the geometric mean of the peak SLU-PRNT post first vaccination. The SD was considered non-inferior if there was less than 2.5-fold difference in the GMT. When the PRNT were analyzed as a continuous variable, the mean difference in log2.5 transformed peak titer between groups together with its associated two-sided 95% CI was computed. If the upper limit of the 95% CI was less than 1, the SD group was considered non-inferior to the HD group. The peak subject-specific PRNT was dichotomized as positive or negative. The difference of proportion of positives by dose and the associated 95% CI were calculated. Fisher’s exact test (Fisher) was used for the association of dose and titer positivity.
RESULTS
Study Subjects
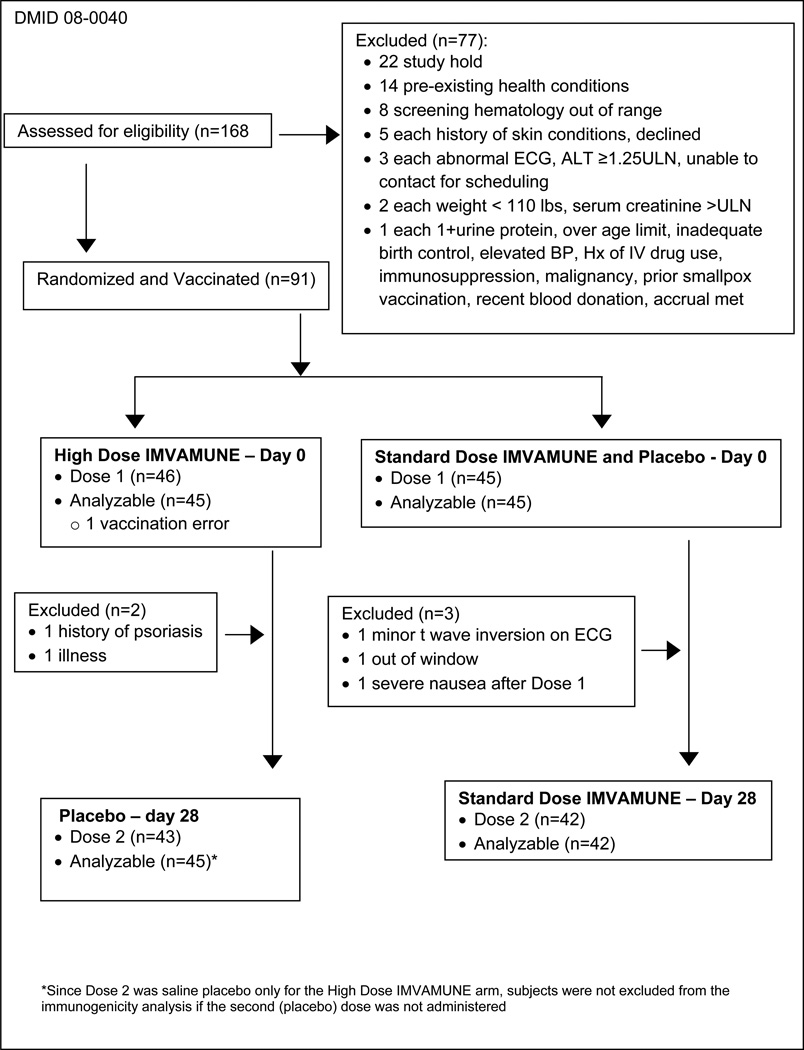
Ninety-one subjects were enrolled at 2 sites from February 9, 2009 through August 16, 2010. The majority of subjects was female (53.3%), non-Hispanic (93.3%), and white 88.9%). The distributions of gender and ethnicity were the same for both groups. There were no significant differences in race (Fisher, P=0.71) and age (one-way ANOVA, P=0.99). The median age was 26.5 years (range: 18 to 37 years).
The study was placed on FDA clinical hold on June 15 through July 10, 2009 to add an additional ECG 14 days after each vaccination, and a troponin level on 14 days post first vaccination. During September 2009 through April 2010, enrollment was interrupted because the HD vaccine titer fell below 3×108 TCID50/mL. Enrollment resumed with a new lot of “high dose” (4.6×108 TCID50/mL) vaccine.
Safety Results
Systemic Reactogenicity (Figure 1.a.)
No SAEs were reported. One subject from each group experienced severe AEs after the first vaccination. Following any vaccination (2nd vaccination for HD was placebo), there was no statistically significant difference between the proportion of subjects with any solicited systemic reaction for the HD (62.2%) and the SD (48.9%) groups (Chi-square P=0.20), nor between the proportion of subjects with any moderate plus severe systemic reactions for the HD (31.1%) and the SD (22.2%) groups (Chi-square P=0.34). Headache was the most prevalent systemic reaction reported across both groups, 35.6% and 17.8% of subjects in the HD and the SD groups, respectively, after vaccination 1. In the SD group, 17.8% also reported feeling tired. Following vaccination 2, headache was the most commonly reported symptom, with 11.6% in the HD group (11.6% also reported feeling tired) and 14.3% in the SD group. Two subjects receiving the HD experienced fever, one graded as moderate and one mild, the latter of which had a concurrent illness of gastroenteritis unrelated to vaccine. There were no troponin elevations, cardiac events or clinically significant ECG abnormalities noted.
Local Reactogenicity (Figure 1.b. and c.)
Following any vaccination (2nd vaccination for HD was placebo), there were no significant differences between the proportion of subjects with any solicited local reactions (functional grade) for the HD (91.1%) and the SD (86.4%) groups (Fisher P=0.52) and between the proportion of subjects with any moderate plus severe reactions for the HD (22.2%) and the SD (29.5%) groups (Chi-square P=0.48). One subject in the HD group experienced a severe local reaction of underarm swelling. Pain at the injection site was the most prevalent local reaction, reported as mild or moderate by 88.9% and 84.1% of subjects in the HD and SD groups, respectively, after either vaccination.
Following first vaccination, there is no significant difference between the proportions of subjects with any grade of measured erythema or induration size for the HD (60.0%) and the SD (65.9%) groups (Chi-square P=0.56), and between the proportions of subjects with any moderate plus severe reaction for the HD (40.0%) and the SD (45.5%) groups (Chi-square P=0.60). Post second dose, the SD group had a significantly higher proportion of subjects with any local reactions (Chi-square P<0.001). In the HD group, 13 subjects experienced severe/large local reactions, all after the first dose. In the SD group, 7 and 10 subjects experienced severe/large local reactions, after the first dose and second dose, respectively; 4 subjects had reactions after both doses.
Unsolicited AEs (Days 0–56 after First Vaccination)
Of the 73 unsolicited non-serious AEs reported, 41 events (32 mild, 9 moderate) were reported by 22 HD recipients and 32 events (21 mild, 11 moderate) were reported by 22 SD subjects (48.9% of subjects in each group). Thirty-two events (43.8%) were considered associated with the vaccination. Eleven subjects (24.4%) in the HD and nine subjects (20.0%) in the SD group experienced an associated adverse event. There was no significant difference between the two groups (Chi-square P=0.61).
Immunogenicity Results (per protocol analysis)
Seroconverion (Primary Objective) (Table 2, Figure 2, Figure 3)
Figure 2.


Estimated survival functions describing seronconversion as a function of time based on per protocol analysis and interval-censored data. If 1 was not included in the interval, the hazard rates were considered to be significantly different. a) Estimated survival functions - BN ELISA titers, per protocol population. The corresponding 95% CI of the hazard-ratio was 0.58 (0.38, 0.85). b) Estimated survival functions -SLU ELISA titers, per protocol population. The corresponding 95% CI of the hazard-ratio was 0.61 (0.30, 1.18).
Figure 3.


a). Non inferiority of a Single High Dose Versus a Single Standard Dose of MVA. Confidence intervals are shown for mean peak titer differences (PRNT log2.5 scale, ELISA log2 scale) between one HD and one SD prior to 2nd vaccination using per protocol population. Dashed vertical line indicates non-inferiority margin.
b). Non inferiority of a Single High Dose Versus Two Single Standard Doses of MVA. Confidence intervals are shown for mean peak titer differences (PRNT log2.5 scale, ELISA log2 scale) between two SDs and one HD all timepoints using per protocol population. Dashed vertical line indicates non-inferiority margin.
BN-ELISA (Primary Assay)
Prior to the second vaccination, all (N=90) subjects in both dose groups seroconverted by 15 and 28 days post first vaccination for the HD and SD, respectively. The interval-censoring based hazard ratio (HR) for time to seroconversion for the SD group versus the HD group was 0.583 (95% CI: 0.384,0.853). The hazard rate of seroconverting for the HD group was significantly higher implying that, at any time, 1.7 times as many subjects in the HD group had a seroconversion event compared with the SD group. However, the median time to seroconversion was 14 days for both groups indicating that the overall effect size was small. The peak GMTs were 282.3 (220.1, 362.1) and 158 (116.2, 214.9) for the HD and SD groups, respectively. Exploratory analyses indicated that non-inferiority of the SD was not established and that the HD GMT was superior to that of the SD.
SLU-ELISA (Additional Exploratory Assay)
Prior to the second vaccination, 22/45 (48.9%) and 15/45 subjects (33.3%) in the HD and SD groups seroconverted, respectively. The 95% CI for the difference of the proportion was (−0.045,0.356) (Fisher P=0.198). The hazard ratio for the SD group versus the HD group was 0.614 (95% CI: 0.302,1.181). The hazard rate for the HD group was 1.6 times higher but was not significantly different from the SD group with alpha=0.05. The median time to seroconversion was 20 days for both groups.
PRNT Assay (Secondary and Tertiary Objectives) (Table 2, Figure 3)
SLU-PRNT (Primary Assay)
Prior to the second vaccination (secondary objective), the peak PRNTs were 95.8 (67.7, 135.6) and 65.2 (42.9, 99.1) for the HD and SD groups, respectively. Non-inferiority of the SD was not established. The proportions of positive responders for the HD, 93.3% (42/45) and SD, 82.2% (37/45), were not significantly different (CI: −0.0226,0.244) (Fisher P=0.197). After two vaccinations including placebo for HD (tertiary objective), peak titers were 468.9 (348.3, 631.3) and 102.6 (73.1, 143.9) for the SD and the HD groups, respectively. Non-inferiority of the HD was not established. The two dose regimen was considered superior to a one HD regimen. The proportion of positives responders for the SD, 100% (42/42), and the HD, 93.3% (42/45), were not significantly different (CI: −0.006,0.140) (Fisher P=0.242).
BN-PRNT (Additional Exploratory Assay)
Prior to the second vaccination (exploratory objective), the peak PRNTs were 16.7 (13.3, 20.9) and 12.2 (9.5, 15.7) for the HD and SD groups, respectively. The SD group was non-inferior to the HD group. The proportion of positives responders for the HD group, 80.0% (36/45), was significantly higher than the SD, 46.7% (21/45), group (CI:0.147,0.520) (Fisher P=0.002).
After two vaccinations including placebo for HD (tertiary objective), peak PRNTs were 60.4 (41.0, 88.9) and 17.6 (13.9, 22.3) for the SD and HD groups, respectively. Non-inferiority was not established for the HD group. The two SD regimen was superior to a one HD regimen. The proportion of positive responders for the SD, 100% (42/42), was significantly higher than the HD group, 82.2% (37/45), (CI:0.066,0.289) (Fisher P=0.006).
Discussion
The objectives of this study were to evaluate the safety of a single HD MVA vaccine, assess the time to seroconversion of HD vaccine compared to a single SD vaccine, and compare the peak GMT of the neutralizing antibody (PRNT) between the HD and 1 or 2 SDs.
Regarding safety when both vaccinations are considered, there were no significant differences in the number or severity of reactogenicity events or unsolicited adverse events or the number of events associated with MVA between the HD group and the SD group. In general, the HD vaccine was safe and well-tolerated.
When comparing the time to seroconversion based on BN-ELISA titer (primary endpoint), all subjects in the HD and SD MVA groups seroconverted by Day 28. While the time to event analysis showed that at any time prior to second vaccination proportionately more subjects seroconverted in the HD group, the effect was small as no difference in median seroconversion time was observed. Interestingly, with the SLU-ELISA, only 49% and 33% of subjects in the HD and SD groups, respectively, seroconverted. Median time to seroconversion was 20 days for both groups (compared to 14 days for BN-ELISA) and the hazard rates were not significantly different. Reasons for the different results between the two laboratories’ assays are discussed below.
When comparing the peak geometric mean of the PRNT, the HD group had higher GMT than the SD group after one dose. This finding was consistent for both laboratories. Non-inferiority for the SD was established using the BN assay but not for the SLU assay, however it was close to being non-inferior (in the intent-to-treat analysis non-inferiority was established; data not shown). Likewise the proportion of positives was higher for the HD group; the difference was statistically significant only for the BN assay.
Peak titers achieved after two SDs of vaccine were superior to those after the single HD vaccination. With the BN-PRNT, the proportion of seroconverters in the HD group was significantly lower compared to the proportion of seroconverters in the SD group (in the intent-to-treat analysis, the difference was not significant; data not shown).
There are several possible reasons for the difference in the assay results between the two laboratories for numbers seroconverted and peak GMT. Among them are differences in antigens and assay reagents and controls. Importantly, differences in assay results tended to be more pronounced in those samples with lower GMTs. Therefore, it is difficult to know how much of the difference is due to variability seen as one gets closer to the lower limit of quantitation. For samples with higher GMTs, in the range where one might expect a correlation with protection, the results of the two assays were more similar.
While a single HD provided a greater percent of individuals with positive seroconversion than one SD dose as anticipated, the overall peak titers were significantly lower than those of the standard two dose regimen. A titer that correlates with protection has not been defined for any immunological readout. However, immunogenicity and efficacy data obtained to date suggest that a two-dose regimen of IMVAMUNE® provides a similar antibody response to Dryvax/Acam2000. In addition, this regimen was shown to reduce the size of replicating vaccinia takes [22,23] and was as effective as Dryvax in producing peak 60% and 90% variola virus neutralization GMTs [24]. As such, at this time, the immune response induced by the two-dose IMVAMUNE® regimen serves as a standard against which other regimens/doses are compared and should be considered when developing policy perspective until more data is available.
CONCLUSION
Multiple antibody assays using different assay antigens provided a more complete picture of the antibody immune response following vaccination. The lack of consistently superior responses of a single HD of MVA relative to one SD suggests the difference in the immune response between HD and SD is small. These results, coupled with the low mean peak titers of HD relative to the two-dose SD regimen and the limited availability of MVA, make the relatively modest increase in rate of sero-conversion and mean peak titers after the administration of a HD unlikely to provide significant benefit over a SD of MVA.
Highlights.
single dose of high titer MVA vaccine was compared to 1 or 2 standard doses.
High dose MVA was safe and well-tolerated.
Median time to seroconversion was similar between the two groups.
ELISA and PRNT peak titers after 1 high dose were higher than 1 standard dose.
ELISA and PRNT peak titers after 1 high dose were inferior to 2 standard doses.
ACKOWLEDGMENTS
The authors would like to thank Irene Graham, Edwin Anderson Sharon Irby-Moore, Mahendra Mandava and Tammy Blevins and the staff of Saint Louis University’s Vaccine and Treatment Evaluation Unit; Jack Stapleton, Jeffrey Meier, Nancy Wagner, Geraldine Dull and the staff of the University of Iowa Vaccine and Treatment Evaluation Unit and the Clinical Research Unit ; Aruna Acharyya and Lisa Davis of EMMES, and Robert Johnson, Carol Ostrye and Stephen Heyse of the Division of Microbiology and Infectious Diseases, NIAID, NIH.
Funding: HHSN272200800003C (SLU), HHSN272200800008C (IU)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. Smallpox and its Eradication. Geneva: World Health Organization; 1988. pp. 1–1460. [Google Scholar]
- 2.ACAM2000™ Smallpox Vaccine, Vaccines and Related Biological Products Advisory Committee (VRBPAC) Briefing Document. [April 18, 2007 [Internet] [cited August 1, 2013]];:70. Available from: http://www.fda.gov/ohrms/dockets/ac/07/briefing/2007-4292b2-02.pdf.
- 3.Mayr A, Stickl H, Muller HK, et al. The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism (author's transl) Zentralbl Bakteriol. 1978;167:375–390. [PubMed] [Google Scholar]
- 4.Stickl H, Hochstein-Mintzel V, Mayr A, Huber H Ch, Schäfer H, Holzner A. MVA vaccination against smallpox: clinical tests with an attenuated live vaccinia virus strain (MVA) (author's transl) Dtsch Med Wochenschr. 1974;99:2386–2392. doi: 10.1055/s-0028-1108143. [DOI] [PubMed] [Google Scholar]
- 5.Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology. 1998;244:365–366. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- 6.Rosel JL, Earl PL, Weir JP, Moss B. Conserved TAAATG sequence at the transcriptional translational initiation sites of vaccinia virus late genes deduced by structural and functional analysis of the Hind III H genomic fragment. J Virology. 1986;6:436–449. doi: 10.1128/jvi.60.2.436-449.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol. 1991;72:1031–1038. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- 8.Sutter G, Moss B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc Natl Acad Sci USA. 1992;89:10847–10851. doi: 10.1073/pnas.89.22.10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carroll MW, Moss B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a non-human mammalian cell line. Virology. 1997;238:198–211. doi: 10.1006/viro.1997.8845. [DOI] [PubMed] [Google Scholar]
- 10.Blanchard TJ, Alcami A, Andrea P, Smith GL. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J Gen Virol. 1998;79:1159–1167. doi: 10.1099/0022-1317-79-5-1159. [DOI] [PubMed] [Google Scholar]
- 11.Drexler I, Heller K, Wahren B, Erfle V, Sutter G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. Gen Virol. 1998;79:347–352. doi: 10.1099/0022-1317-79-2-347. [DOI] [PubMed] [Google Scholar]
- 12.Mayr A, Hochstein M, Mintzel V.,Stickl H. Passage history, properties, and applicability of the attenuated vaccinia virus strain MVA. Infection. 1975;3:6–14. [Google Scholar]
- 13.Suter M, Meisinger-Henschel C, Tzatzaris M, Hulsemann V, Lukassen S, Wulff NH, et al. Modified vaccinia Ankara strains with identical coding sequences actually represent complex mixtures of viruses that determine the biological properties of each strain. Vaccine. 2009;27:7442–7450. doi: 10.1016/j.vaccine.2009.05.095. [DOI] [PubMed] [Google Scholar]
- 14.Walsh SR, Wilck MB, Dominguez DJ, Zablowsky E, Bajimaya S, Gagne LS, et al. Safety and Immunogenicity of Modified Vaccinia Ankara in Hematopoietic Stem Cell Transplant Recipients: A Randomized, Controlled Trial. J Infect Dis. 2013;207:1888–1897. doi: 10.1093/infdis/jit105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg RN, Overton ET, Haas DW, Frank I, Goldman M, von Krempelhuber A, et al. Safety, Immunogenicity, and Surrogate Markers of Clinical Efficacy for Modified Vaccinia Ankara as a Smallpox Vaccine in HIV-Infected Subjects. J Infect Dis. 2013;207:749–758. doi: 10.1093/infdis/jis753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Risk Assessment Tool for Estimating Your 10-year Risk of Having a Heart Attack. [[last accessed 12/19/13]]; Available from: http://cvdrisk.nhlbi.nih.gov/calculator.asp.
- 17.Von Krempelhuber A, Vollman J, Pokorny R, Rapp P, Wulff N, Petzold B, et al. A randomized, double-blind, dose-finding phase II study to evaluate immunogenicity and safety of the third generation smallpox vaccine candidate IMVAMUNE®. Vaccine. 2010;28:1209–1216. doi: 10.1016/j.vaccine.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iacono-Connors LC, Novak J, Rossi C, Mangiafico J, Ksiazek T. Enzyme-linked immunosorbent assay using a recombinant baculovirus-expressed Bacillus anthracis protective antigen (PA): measurement of human anti-PA antibodies. Clin Diagn Lab Immunol. 1994;1:78–82. doi: 10.1128/cdli.1.1.78-82.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes C, Newman F, Davidson W, Olson V, Smith S, Holman R, et al. Analysis of variola and vaccinia neutralization assays for smallpox vaccines. Clin Vacc Immunol. 2012;19:1116–1118. doi: 10.1128/CVI.00056-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan W. Extending the iterative convex minorant algorithm to the cox model for interval-censored data. J of Comput Grap Stat. 1999;8:109–120. [Google Scholar]
- 21.Efron B. Bootstrap methods: another look at the jackknife. Ann Statist. 1979;7:1–26. [Google Scholar]
- 22.Frey SE, Newman FK, Kennedy JS, Sobek V, Ennis FA, Hill H, et al. Clinical and immunologic responses to multiple doses of IMVAMUNE (R) (Modified Vaccinia Ankara) followed by Dryvax (R) challenge. Vaccine. 200;25:8562–8573. doi: 10.1016/j.vaccine.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seaman MS, Wilck MB, Baden LR, Walsh SR, Grandpre LE, Devoy C, et al. Effect of vaccination with modified vaccinia Ankara (ACAM3000) on subsequent challenge with Dryvax. JID. 2010;20:1353–1360. doi: 10.1086/651560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Damon IK, Davidson WB, Hughes CM, Olson VA, Smith SK, Holman RC, et al. Evaluation of smallpox vaccines using variola neutralization. J Gen Virol. 2009;90:1962–1966. doi: 10.1099/vir.0.010553-0. [DOI] [PMC free article] [PubMed] [Google Scholar]