

Abstract
Aims/hypothesis
Resistin was originally identified as an adipocyte-derived factor upregulated during obesity and as a contributor to obesity-associated insulin resistance. Clinically, resistin has also been implicated in cardiovascular disease in a number of different patient populations. Our aim was to simultaneously address these phenomena.
Methods
We generated mice with modest adipocyte-specific resistin overexpression. These mice were crossed with mice deficient in the LDL receptor (Ldlr−/−) to probe the physiological role of resistin. Both metabolic and atherosclerotic assessments were performed.
Results
Resistin overexpression led to increased atherosclerotic progression in Ldlr−/− mice. This was in part related to elevated serum triacylglycerol levels and a reduced ability to clear triacylglycerol upon a challenge. Additional phenotypic changes, such as increased body weight and reduced glucose clearance, independent of the Ldlr−/− background, confirmed increased adiposity associated with a more pronounced insulin resistance. A hallmark of elevated resistin was the disproportionate increase in circulating leptin levels. These mice thus recapitulated both the proposed negative cardiovascular correlation and the insulin resistance. A unifying mechanism for this complex phenotype was a resistin-mediated central leptin resistance, which we demonstrate directly both in vivo and in organotypic brain slices. In line with reduced sympathetic nervous system outflow, we found decreased brown adipose tissue (BAT) activity. The resulting elevated triacylglycerol levels provide a likely explanation for accelerated atherosclerosis.
Conclusions/interpretation
Resistin overexpression leads to a complex metabolic phenotype driven by resistin-mediated central leptin resistance and reduced BAT activity. Hypothalamic leptin resistance thus provides a unifying mechanism for both resistin-mediated insulin resistance and enhanced atherosclerosis.
Keywords: Adipose tissue, Atherosclerosis, Brown adipose tissue, Insulin resistance, Leptin, Leptin resistance, Obesity, Resistin, Triacylglycerol, Type 2 diabetes
Introduction
Resistin is a hormone that belongs to a family of small, cysteine-rich secreted proteins collectively termed resistin-like molecules (RELMs) [1]. The source of resistin differs between rodents and humans: in mice resistin is expressed primarily by adipocytes while in humans monocytes and macrophages are the main sources. Despite this species and expression profile difference, resistin derives its name from a positive correlation with obese insulin-resistant conditions in both mice [2, 3] and humans [4].
Murine studies support a direct role for resistin in insulin resistance. Retn−/− mice display reduced fasting glucose levels [5], transgenic human resistin expressed in murine adipose tissue induces insulin resistance [6] and recombinant murine resistin reduces glucose uptake in cardiac myocytes [7]. Although the mechanisms by which resistin causes insulin resistance have not yet been wholly elucidated, work by Muse and colleagues suggests that central resistin action is driving hypothalamic-mediated peripheral metabolic responses in mice [8].
The role of resistin in the pathogenesis of insulin resistance has been questioned due to conflicting data in human clinical studies, which have yielded no relationship between resistin and insulin sensitivity. However, several clinical studies have demonstrated a close correlation between resistin levels and the severity of cardiovascular disease, independent of established risk factors such as insulin resistance [9, 10]. Furthermore, experimental studies in vitro and in vivo suggest that resistin plays a direct role in the pathogenesis of atherosclerosis [10-13]. Thus, it is possible that resistin exerts its pathophysiological effects more directly through its effects on atherosclerosis rather than on systemic insulin sensitivity.
Given the association of resistin with cardiovascular disease in humans, we hypothesised that chronically elevated resistin levels would also accelerate and/or aggravate atherosclerosis in mice. To test this hypothesis, we generated an Ap2 (also known as Fabp4)-promoter-driven Retn transgenic (tg) mouse model that demonstrates elevated circulating resistin levels and crossed it into the atherosclerosis-prone Ldlr−/− mouse background. This approach enables us to potentially disentangle the metabolic and atherogenic actions of chronically elevated resistin levels without the complications of pharmacological delivery.
Methods
Animals
C57BL/6 background Ldlr−/− mice were obtained from Jackson laboratories (Bar Harbor, ME, USA) and Retn tg mice were generated as described in electronic supplementary material (ESM) Methods. Mice were maintained on a 12 h light/dark cycle and housed in groups of three to five with unlimited access to water, standard chow (No. 2016; Harlan-Teklad, Indianapolis, IN, USA) or atherogenic ‘western diet’ (TD.88137; 42% energy from fat, 0.2% cholesterol by weight; Harlan-Teklad). For cold exposure, mice were housed individually at a constant 6°C in standard mouse caging with their chow and water. The Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center at Dallas approved all animal experiments.
Hepatic steatosis measurement
Quantification of hepatic steatosis (described in further detail in ESM Methods) was performed by either computerised tomography (CT) or through tissue extraction according to the Folch method.
Lipid clearance and oral glucose tolerance test
An oral load of Intralipid at 15 μl/g or 1.4 mol/l glucose solution at 10 μl/g was given following, respectively, a 3 h and a 12 h fast (21:00–09:00 hours) fast. Serum samples were collected as indicated in the experiments.
[3H]Triolein uptake and oxidation
For tissue-specific lipid-uptake and lipid-oxidation analyses, [3H]triolein was given through an oral load of Intralipid to mice following a 3 h fast. After 2 h, tissues were harvested and lipids were extracted using a chloroform-to-methanol-based extraction method. Samples were then counted for radioactivity.
Organotypic hypothalamic culture and measurement of phosphorylation of signal transducer and activator of transcription 3
Organotypic brain slices were obtained from 16 young male wild-type mice as previously described [14] and phosphorylation of signal transducer and activator of transcription 3 (STAT3) was determined by immunofluorescence. These methods are described in further detail in ESM Methods.
Primary brown adipocyte differentiation and culture
The stromal vascular fraction of interscapular brown adipose tissue (BAT) was isolated and cultured for differentiation into mature brown adipocytes as described previously [15] and in ESM Methods.
Statistical methods
Bar graph data are expressed as mean ± SD. Time-dependent data are expressed as mean ± SEM for visual clarity in curves. Unpaired parametric t tests were used for comparisons between groups and analysis of variance for time-dependent data. A p value of <0.05 was considered as significant and is indicated by a single asterisk (double asterisk indicates 0.05<p<0.01 and triple asterisk indicates p<0.001 compared with wild-type mice; asterisks are equivalently employed for concurrent changes by experimental condition).
Quantitative RT-PCR, aortic root analyses, leptin treatment, western blot and blood biochemistry methods
These are described in the ESM Methods.
Results
Retn tg mice exhibit mild insulin resistance and increased atherosclerotic progression
To test whether resistin plays a role in insulin resistance and the pathogenesis of atherosclerosis, we developed a mouse model that overexpresses murine resistin under the control of the Ap2-promoter (Fig. 1a). Retn expression in Retn tg mice was predominantly confined to adipose tissues with only marginal levels of the Ap2-driven expression found in isolated peritoneal macrophages (Fig. 1a). Male Retn tg mice fed a chow diet displayed an approximately ninefold elevation in circulating resistin levels when compared with wild-type mice; this increase was sevenfold in mice fed an atherogenic western diet (Fig. 1b). While higher than on diet-induced obesity alone [2], these transgene-driven levels are in a more physiological range than those observed in past models [16]. These mice allow us to address whether adipocyte-secreted resistin is a benign biomarker or plays an active role in the pathogenesis of cardiovascular disease.
Fig. 1.
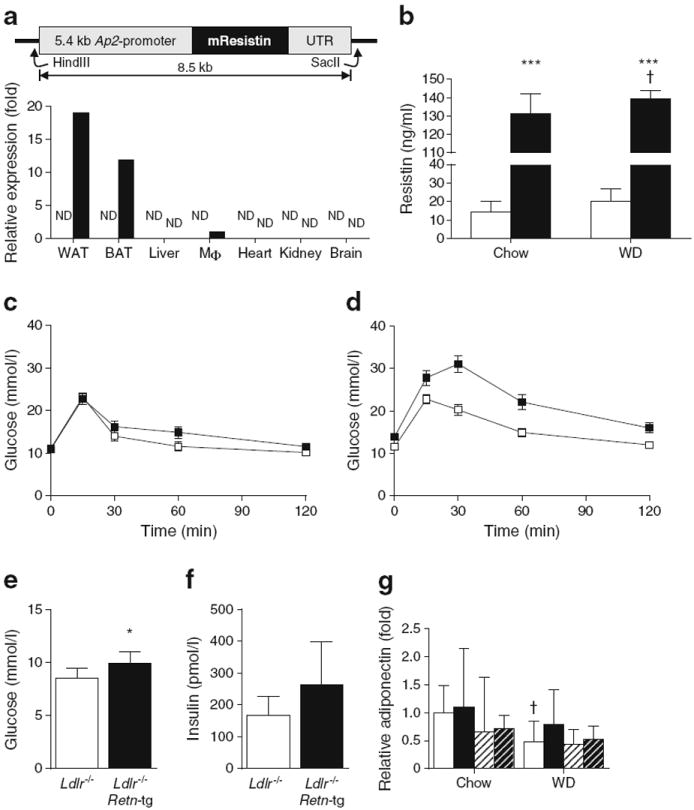
Overexpression of the resistin gene in adipocytes results in mild insulin resistance and increased atherosclerosis. (a) Full-length resistin gene expressed under the 5.4 kb Ap2 promoter in mice results in adipose tissue overexpression. Limited expression can also be detected in peritoneal macrophages (Mφ) by quantitative RT-PCR. ND indicates not detected. (b) Circulating resistin levels in Retn tg mice (black bars) on chow diet and western diet (WD) are elevated compared to wild type littermates (white bars). (c) Oral glucose tolerance test in chow-fed male mice (white squares: wild type; black squares: Retn tg mice). (d) Oral glucose tolerance test in WD-fed male mice demonstrated reduced sensitivity (p=0.023 for AUC). (e, f) Fasting glucose (e) and fasting insulin (f) levels in Ldlr−/− female mice on WD. (g) Adiponectin levels in Retn tg female mice (black bars) were no different from those in wild-type female mice (white bars); WD reduced levels in Ldlr−/− wild-type mice, but not in Ldlr−/− Retn tg mice. Male mice (hatched bars, white background: wild type; black background: Retn tg) exhibited no differences with diet or genotype. *p<0.05 and ***p<0.001 vs wild type; †p<0.05, chow vs WD
Elevated resistin levels have been implicated in insulin resistance. An oral glucose tolerance test demonstrated no significant change in the insulin sensitivity of male Retn tg mice on chow diet (Fig. 1c). Fed ad libitum western diet for 8 weeks, however, male Retn tg mice exhibited reduced glucose clearance by oral glucose tolerance (Fig. 1d). To study the impact of chronically elevated resistin levels on atherosclerosis, we crossed Retn tg mice onto the atherosclerosisprone Ldlr−/− background. Our female mice on western diet displayed significantly increased fasting blood glucose (Fig. 1e) and a trend toward increased plasma insulin (Fig. 1f). Male mice only exhibited higher basal glucose levels on western diet (ESM Fig. 1a). Adiponectin levels were unaltered by the Retn transgene (Fig. 1g). Retn tg mice therefore present only mild insulin resistance—ideal conditions in which to further examine the atherogenic and metabolic effects of increased levels of resistin.
The combination of physiological resistin overexpression, mild insulin resistance and Ldlr−/− background, and western diet feed provide a platform to determine whether resistin does indeed drive atherosclerosis. After 8 weeks on the western diet, female Retn tg Ldlr−/− mice displayed an increase in atherosclerotic plaque area in the aortic root by Oil Red O and MAC2 staining compared with littermate control mice (Fig. 2a). Plaque area quantification confirmed a small but significant increase in Oil-Red-O-positive (Fig. 2b) and MAC2-positive areas (Fig. 2c) in aortic cross-sections. Pathological examination confirmed no differences in plaque morphology, including necrotic core areas. Upon extended western diet feeding (15 weeks), the differences in atherosclerotic plaque size between genotypes were no longer present (ESM Fig. 2a), indicating that resistin enhances arteriosclerotic progression but does not lead to a more severe terminal plaque size. Combined, these findings further support observations that resistin is mechanistically involved in both insulin resistance and atherosclerosis.
Fig. 2.
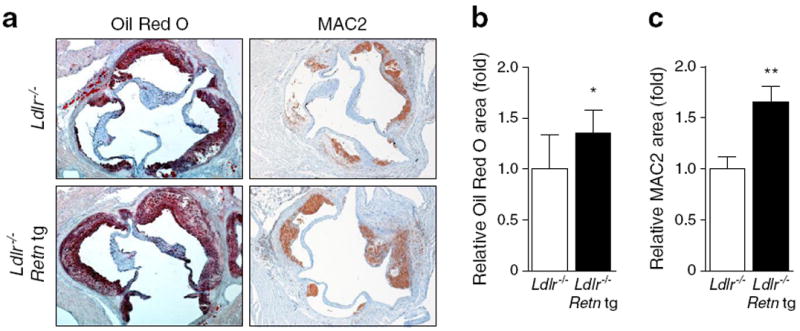
Overexpression of the resistin gene in adipocytes leads to increased atherosclerotic progression. (a) Atherosclerotic plaque areas in aorta of female Ldlr−/− mice, as imaged by Oil Red O for lipid deposition and MAC2 for macrophage accumulation, show increased plaque progression in Retn tg mice. (b) Quantified areas of Oil-Red-O-positive areas demonstrate significant increases in Retn tg mice (black bars) (areas relative to wild-type plaques on Ldlr−/− background: white bars). (c) MAC2-positive area quantification demonstrated even greater differences between genotypes. *p<0.05 and **p<0.01 vs wild type
Resistin overexpression causes delayed triacylglycerol clearance
To elucidate the underlying mechanism for the accelerated atherosclerosis in Retn tg Ldlr−/− female mice, we measured various known risk factors for cardiovascular disease in western-diet-fed animals. Fed circulating fatty acid (FA) levels were significantly elevated (Fig. 3a) but neither total cholesterol (Fig. 3b) nor the cholesterol content across lipoprotein fractions (Fig. 3c) was altered. Circulating triacylglycerol levels were nearly double those in non-transgenic Ldlr−/− mice on western diet (Fig. 3d); there was also a striking difference between genotypes in VLDL-triacylglycerol content (Fig. 3e). A challenge with an oral lipid load revealed that western-diet-fed Retn tg Ldlr−/− mice had a reduced capacity for lipid clearance (Fig. 3f). While circulating triacylglycerol levels were normal in both female and male Retn tg Ldlr−/−mice fed a chow diet (ESM Figs 1f, 2b), functionally, triacylglycerol clearance was still significantly impaired (ESM Figs 1i, 2c). Increased and extended triacylglycerol presence in the circulation of Retn tg Ldlr−/− mice may therefore explain the measured increase in atherosclerotic progression upon resistin overexpression.
Fig. 3.
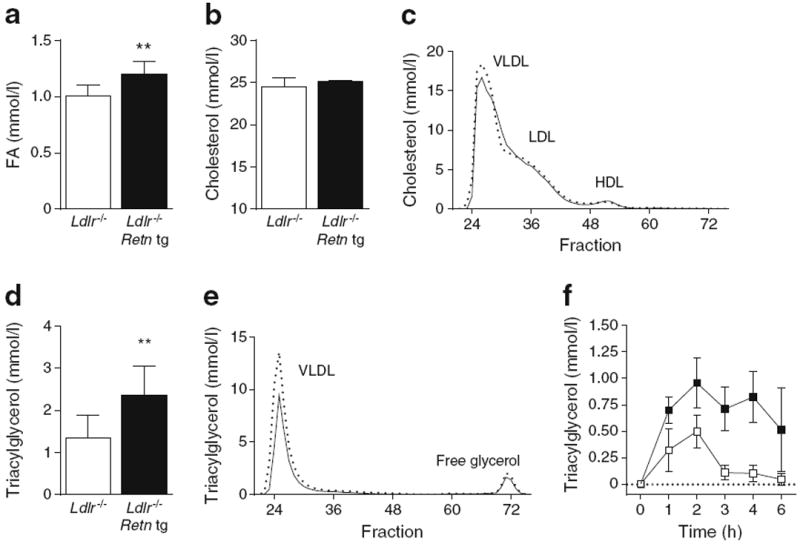
Retn tg Ldlr−/− female mice exhibit increased circulating lipids and reduced triacylglycerol clearance. (a, b) Fasting FA (a) and cholesterol levels (b) in western-diet-fed Ldlr−/− Retn tg (black bars) and Ldlr−/−(white bars) female mice. (c) Cholesterol content of VLDL, HDL and LDL particles; solid line: wild type; dashed line: Retn tg. (d, e) Serum triacylglycerol was elevated in Retn tg mice (d) and the triacylglycerol content of VLDL particles in western-diet-fed Retn tg female mice was enriched (e). (f) Lipid clearance in western-diet-fed female mice demonstrated delayed clearance in Ldlr−/− Retn tg mice (black squares) compared with Ldlr−/− (white squares) mice. **p<0.01 vs wild type
Retn tg mice present with increased adiposity and heightened leptin levels
Beyond the increased triacylglycerol phenotype, Retn tg Ldlr−/− mice also exhibited an increased weight gain (Fig. 4a). Though subtle, this effect was consistently noted in all cohorts. Measurements of cohorts containing 25 mice in each group achieved significance after 8 weeks on western diet (ESM Fig. 2d). What was more apparent, however, was an increase in adiposity in the Retn tg Ldlr−/− mice. Inguinal (subcutaneous) and gonadal white adipose tissue (IWAT and GWAT) were significantly increased in mass and in relation to body weight (Fig. 4b); interscapular BAT, however, was not. Consistent with increased adipose tissue mass, plasma leptin levels were significantly elevated in Retn tg Ldlr−/− mice (Fig. 4c, ESM 1d). An increase in leptin level is to be expected with an increase in body weight. This is apparent when leptin levels were plotted against individual body weights (Fig. 4e). Upon doing that, it was clear that leptin levels in Retn tg Ldlr−/− mice were disproportionately higher given their body weight, consistent with a potentially impaired leptin action in target tissues.
Fig. 4.
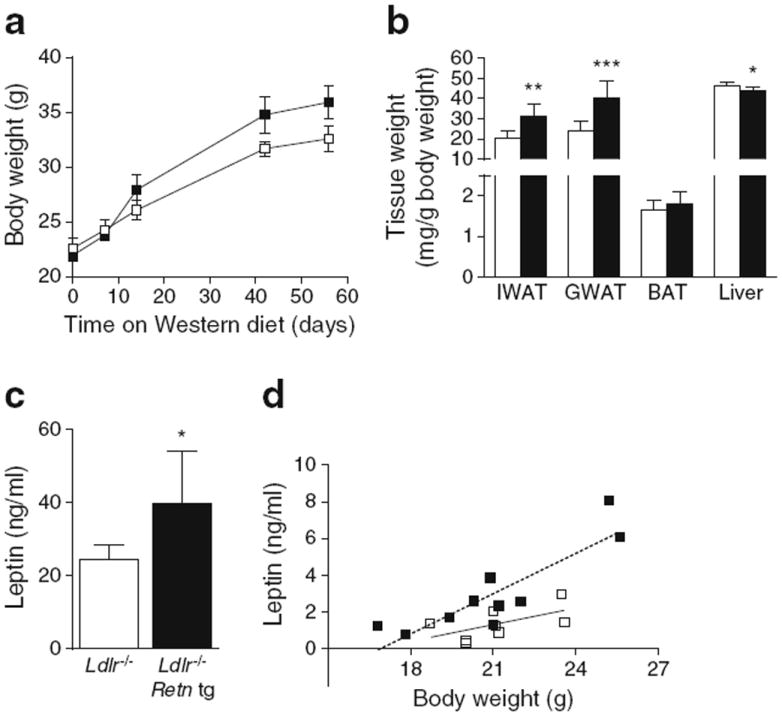
Overexpression of resistin results in increased weight and adiposity and heightened leptin levels. (a) Body weight over time in western-diet-fed Ldlr−/− (white squares) and Ldlr−/− Retn tg (black squares) female mice. (b) IWAT, GWAT and BAT adipose tissue and liver weights in Ldlr−/− Retn tg (black bars) and Ldlr−/− (white bars) female mice on western diet. (c) Leptin levels in western-diet-fed Ldlr−/− female mice. (d) Leptin levels in relation to body weight in chow-fed female mice demonstrate higher leptin:body weight correlations in Ldlr−/− Retn tg mice (R2=0.764) than in Ldlr−/− female mice (R2=0.347). *p<0.05, **p<0.01 and ***p<0.001 vs wild type
Liver weights were reduced in the female Retn tg Ldlr−/−mice (Fig. 4b). Using our previously established CT-based method [17], we found that hepatic steatosis was lower than expected in the Retn tg Ldlr−/− female mice (ESM Fig. 3a). After 15 weeks of western diet, the liver triacylglycerol and cholesterol content were significantly reduced in the Retn tg Ldlr−/− female mice compared with littermate controls (ESM Fig. 3b, c). Hepatic VLDL secretion was comparable between genotypes (ESM Fig. 3d). Sphingolipidomic analysis revealed no differences in the 28 sphingolipid species measured in the liver by mass spectroscopy (data not shown). Furthermore, quantitative RT-PCR analysis did not show any differences in expression levels of inflammatory cytokine genes, such as Tnf and Il1b, in the liver (data not shown). Liver function in Retn tg Ldlr−/− female mice was, therefore, normal and increased circulating triacylglycerol levels were not due to decreased liver uptake or prevailing inflammation in the liver—a potential peripheral target tissue for resistin [8].
All weight and metabolic measurements were repeated in Retn tg male mice in a wild-type background fed both chow and western diets to eliminate potential confounding effects due to sex or LDL receptor deficiency. Trends and significant increases in body weight, adiposity and leptin levels and reduced liver lipids were still apparent under those conditions (ESM Fig. 1a–h). Most notably, triacylglycerol clearance was significantly reduced in Retn tg male wild-type mice on chow diet (ESM Fig. 1i), an effect lost once the mice were placed on western diet (ESM Fig. 1j). Energy balance assessment using indirect calorimetry and food intake measurements did not show any measurable differences between chow-fed male genotypes (data not shown). This lack of difference is not surprising given the very mild weight gain phenotype in the Retn tg mice. Western diet exacerbated weight and metabolic differences in male mice (ESM Fig. 1b), but also induced a metabolic dysregulation potentially greater than that caused by elevated resistin alone.
Reduced BAT activity in Retn tg Ldlr−/− mice
Reduced lipid clearance and increased leptin levels were the most prominent metabolic readouts measured with chronically elevated resistin levels. To explore these differences further, we first repeated the oral lipid clearance experiments with the addition of a tritium-labelled lipid tracer to identify the responsible tissue(s) with reduced lipid clearance. We detected a substantially reduced lipid uptake in BAT in the Retn tg Ldlr−/− mice compared with their littermate controls (Fig. 5a). Lipid uptake in liver, muscle, heart and WAT was similar between genotypes. This supports a model in which the increased adipose tissue size in the Retn tg mice is due to a reduction in lipolysis rather than an increase in lipid uptake. At room temperature, BAT in Retn tg mice was visually less ‘brown’ and exhibited more unilocular cells than wild-type littermates upon histological examination (Fig. 5b; ESM Fig. 4a). Quantitative RT-PCR measurements of an array of BAT genes demonstrated a trend in reduced BAT-defining genes in Retn tg mice, with a significant reduction in Ucp1 (Fig. 5c). Lower levels of uncoupling protein-1 (UCP-1) were confirmed in BAT isolated from Retn tg Ldlr−/− mice (ESM Fig. 4b). To eliminate any potentially confounding systemic metabolic effects in intact mice, brown pre-adipocytes were isolated from wild-type and Retn tg mice and induced to differentiate into mature brown adipocytes in vitro. The cells isolated from the transgenic mice differentiated equally well (ESM Fig. 4c) but exhibited a reduced ‘brown’ gene expression profile (ESM Fig. 4d), suggesting a potential cell-autonomous component to this BAT phenotype as well. The adrenergic response to isoprenaline was still present, but muted by overall reduced gene levels of Ppargc1a and Ucp1 in newly differentiated brown adipocytes (ESM Fig. 4e).
Fig. 5.
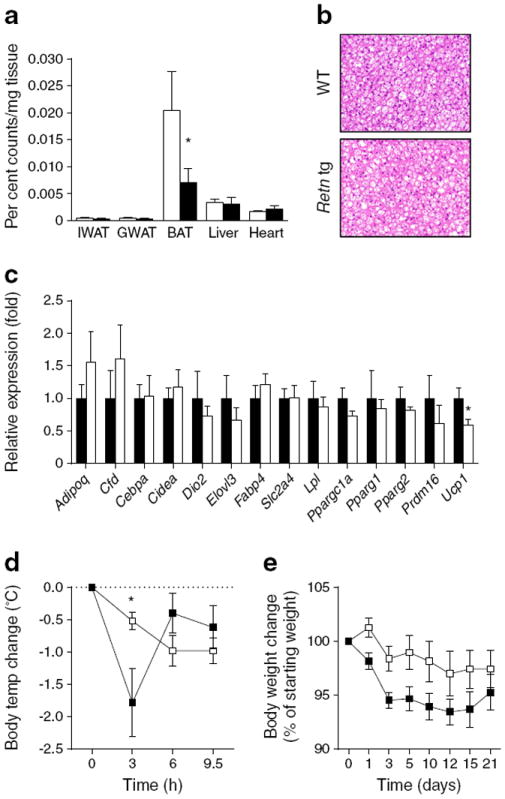
BAT is metabolically less responsive, resulting in reduced thermogenic capacity upon cold exposure in Retn tg mice. (a) Tritiated triolein uptake in Ldlr−/− Retn tg mouse tissues (black bars) compared with tissues of Ldlr−/− (white bars) female mice fed western diet. (b) BAT appearance in chow-fed Retn tg and wild-type (WT) male mice. (c) mRNA expression of adipocyte genes in BAT taken from chow-fed male wild type (white bars) and Retn tg (black bars) mice. (d, e) Body temperature (d) and weight change (e) upon exposure to cold (6°C) in chow-fed Ldlr−/− female mice. *p<0.05 vs wild type
To assess further functional consequences of reduced BAT activity, mice were challenged by housing them at a cooler temperature (6°C). This led to a more pronounced initial drop in body temperature in the Retn tg Ldlr−/− mice compared with their littermate controls (Fig. 5d). Thereafter, both genotypes were able to maintain normal body temperature despite the cool ambient temperature. However, the Retn tg Ldlr−/− mice lost consistently more body weight, at least during the first few days, indicating a higher energy cost for the maintenance of body temperature in these mice (Fig. 5e). These data suggest that BAT-driven, non-shivering thermogenesis (NST) is reduced in the presence of elevated resistin levels. Reduced BAT function and lipid clearance provide an explanation for the heightened triacylglycerol levels in the Retn tg mice.
Resistin causes central leptin resistance
An increase in circulating leptin is a potential reflection of central leptin resistance. We therefore hypothesised that chronically elevated resistin levels lead to leptin resistance and consequently a reduced sympathetic nervous system (SNS) outflow. This might explain both the susceptibility to weight gain and, at least in part, to BAT function, including reduced lipid clearance. Chow- and western-diet-fed male wild-type and Retn tg mice were treated intraperitoneally with recombinant leptin and weight loss was measured over a period of 5 days. Indeed, Retn tg mice on the chow diet had a reduced leptin response with respect to acute body weight loss (Fig. 6a). Interestingly, this effect was not present in western-diet-fed mice, with an upregulation of the endogenous leptin and resistin secretion in both strains (Fig. 1b; ESM Fig. 4d). To bypass the complex systemic metabolic perturbations, we tested the effect of resistin directly on hypothalamic leptin signalling in two ways. First, leptin was delivered into the third cerebral ventricle via intracerebroventricular (ICV) injection. Retn tg mice exhibited a blunted response to ICV injection of leptin, as judged by STAT3 phosphorylation in the hypothalamic arcuate nucleus (Fig. 6b, c), compared with wild-type littermates. These acute observations confirm the effects of chronic resistin overexpression on leptin resistance that also manifested in a trend towards reduced gene expression of both Ldlr and inhibitors of leptin signalling in the hypothalamus of Retn tg mice (ESM Fig. 5a). Second, to assess this signalling free from the systemic metabolic differences, organotypic hypothalamic sections from wild-type mice were pre-treated with or without resistin and then activated by leptin. Acute resistin treatment inhibited leptin-mediated STAT3 phosphorylation (Fig. 6d) in hypothalamic sections (Fig. 6e); variability in surviving tissue volumes likely increased pSTAT3 variation (ESM Fig. 5b). It should be noted that resistin alone acutely increased pSTAT3 in our in vitro experiments, supporting previous findings [18]. These combined in vivo and in vitro measures of reduced hypothalamic leptin responsiveness in the presence of resistin thus support a novel unifying mechanism (Fig. 7) of central resistin-mediated metabolic effects.
Fig. 6.
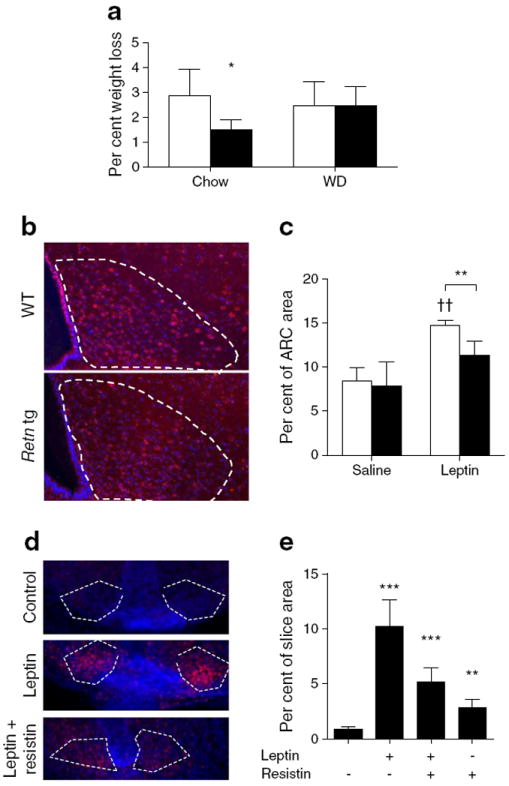
Retn tg mice exhibit reduced hypothalamic leptin responsiveness. (a) Leptin-induced weight loss in chow- and western diet-fed male wild-type (white bars) and Retn tg (black bars) male mice. (b) Phosphorylated STAT3 (pSTAT3, red; DAPI, blue) in response to administration of leptin into the third cerebral ventricle and (c) quantified pSTAT3-positive hypothalamic arcuate nucleus (ARC) area. (d) pSTAT3 labelling upon acute resistin exposure before leptin treatment. (e) Quantified pSTAT3-positive area in the ARC of hypothalamic slices. Resistin mutes leptin signalling (p=0.068). White dashed lines indicate the ARC. *p<0.05, **p<0.01 and ***p<0.001 vs wild type or no treatment. ††p<0.01 vs saline
Fig. 7.
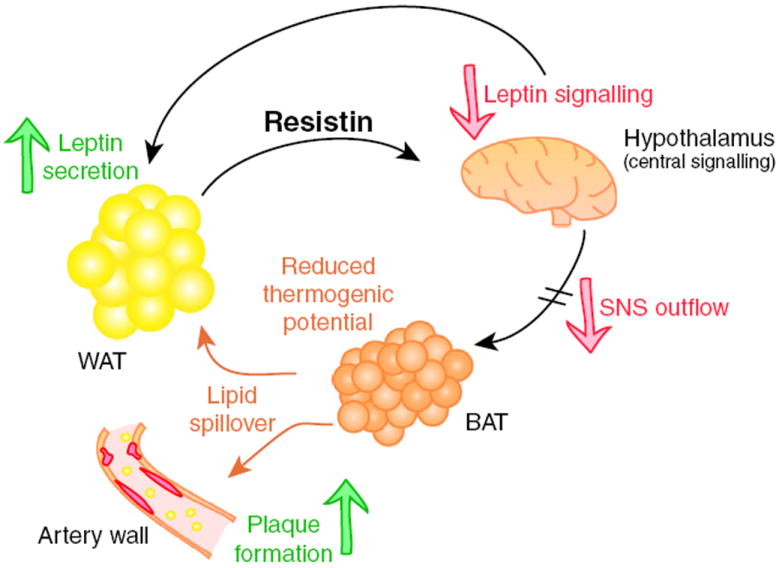
Proposed model for resistin-mediated metabolic differences. Increased resistin levels cause central leptin resistance. This reduces sympathetic outflow from the brain and particularly affects BAT, which also experiences reduced BAT marker expression in a tissue-autonomous fashion in the transgenic state. Functionally impaired BAT is responsible for the delayed triacylglycerol clearance, impaired insulin sensitivity and, in the case of the Ldlr−/− background, more rapid atherosclerotic plaque formation
Discussion
Our overexpressing Retn tg mouse model demonstrates significant atherogenic and metabolic differences in Ldlr−/− mice as a function of circulating resistin levels. These effects are more striking in female mice than in male mice, in which similar trends were observed. While in male mice many of these differences fail to reach statistical significance, male mice are in general more prone to diet-induced weight gain and insulin resistance making them metabolically more challenged even at baseline. Male Retn tg mice, regardless of background, were inherently more leptin resistant over the entire disease progression. Hence, having physiologically elevated resistin levels neither specifically accelerates the metabolic nor the atherogenic disease course in western-diet-fed male mice. However, elevated resistin quite strikingly affects female mice in which there is more room for modulation. The role of resistin as a modulator of atherogenesis is therefore critically dependent on the experimental setting. Importantly however, in the context of a wild-type background in the absence of an Ldlr−/− challenge, the effects on leptin resistance were observed in both sexes. Our Retn tg mice therefore offer an ideal setting for the careful deconvolution of multiple events that take place in different tissues, giving rise to a complex phenotype that would be nearly impossible to dissect in normal wild-type mice. The predominant unifying event with Retn overexpression appears to be accelerated weight gain through resistin-mediated central leptin resistance.
One component of our proposed mechanism for resistin’s central action is a reduction in sympathetic nervous activity to BAT resulting in reduced acute thermogenic response and lipid uptake. When recently measured by Kosari and colleagues, acute ICV administration of recombinant resistin indeed reduced BAT sympathetic nerve activity [19]. While supportive of our findings, there is a marked difference between the chronically elevated resistin levels in our model and the acute stimulation with pharmacological resistin levels in other work. Acute effects may trigger somewhat contradictory results under some circumstances. For example, central resistin infusion counteracts the effects of leptin by first elevating the phospo-STAT3/STAT3 ratio, food intake and body weight in diabetic rats; central resistin infusion also increased peripheral insulin sensitivity in this study [18]. Our ex vivo hypothalamic cultures confirmed this as a potential mechanism. The levels of peripheral resistin overexpression in our tg mice are, however, physiologically more meaningful in the context of chronic disease [2]. The model presented here thus provides a better setting in which to critically assess the effects of increased circulating resistin and distinguishes these mice from acute infusion studies or high-level overexpression [16].
Along with adaptation to low temperatures, the capacity for NST increases through recruitment of BAT. This process critically depends on an increased activation of the SNS and subsequent β-adrenergic receptor signalling in BAT [20]. Mice housed at thermoneutrality (i.e. around 30°C) have very little capacity for NST, while mice housed at room temperature display at least some degree of cold adaptation in this regard. Thus, since the overall adiposity (i.e. insulation) is similar or possibly even increased in the Retn tg mice, these data suggest that the observed lower capacity for NST reflects a lower BAT activity in the Retn tg mice, at least during the first days of cold adaptation. Acutely administered resistin has such an effect [21, 22]. BAT activity is not only important for heat generation, but has also been proven to be highly important for triacylglycerol clearance [23]. Indeed, when resistin was reduced by siRNA in adipocytes, β-oxidation was increased [9]. In our tg mice with peripheral Retn overexpression, β-oxidative activity is decreased. A reduced ability for SNS activation also leads to a reduced rate of lipolysis in both WAT and BAT. This may not only explain increased adipose depot size, but is also consistent with reduced hepatic steatosis in Retn tg mice as a consequence of reduced substrate availability for hepatic triacylglycerol synthesis. While reduced SNS outflow naturally reduces lipolysis of WAT and BAT, the emerging insulin resistance that develops over time in the tg mice will push the system increasingly towards enhanced lipolysis in the fed state. Nevertheless, the net effects in the Retn tg mice appear to be a reduced lipolytic activity and increased adiposity that persists.
The reduced expression of ‘brown’ genes, such as Ucp1, Cidea and Ppargc1a in cultured brown Retn tg adipocytes suggests that resistin exerts effects directly on brown adipocytes, independent of its central impact on hypothalamic leptin signalling. Previous results in Retn−/− mice demonstrated decreased UCP-1 levels, but only in the absence of leptin signalling on an ob/ob background [5]. These effects appear to be independent of the demonstrated effects of central resistin action on thermogenesis [21, 22]. While central effects and/or reduced SNS activity in the Retn tg BAT might cause epigenetic imprinting detectable even ex vivo, it seems most likely that the transgene-mediated overexpression of the characteristically white adipocyte-associated protein resistin in brown adipocytes may push these cells towards a ‘whiter’—though still responsive—functional phenotype with increased endoplasmic reticulum stress or reduced mitochondrial activity [24, 25].
Although retention of cholesterol-rich lipoproteins within the sub-endothelial matrix of the arterial wall is a key initiator of atherosclerosis, we observe here increased atherosclerotic progression without elevated cholesterol levels in the Retn tg Ldlr−/− female mice. Instead, we find an association between elevated triacylglycerol levels and accelerated atherogenesis in the context of western diet and LDL receptor deficiency. The role of elevated triacylglycerol levels in the pathogenesis of atherosclerosis is not clear, but epidemiological data confirm an independent association of triacylglycerol levels with vascular risk [26]. An increased circulating leptin level may, on its own, drive increased atherosclerosis [27], an effect that we cannot directly discount in our model and which may exacerbate disease progression.
A significant body of data suggests that the FIZZ (found in inflammatory zone) family of molecules is strongly implicated in atherogenesis in mice and humans; many of these studies suggest a macrophage immunomodulatory role for the RELM proteins [28]. At this time we cannot exclude resistin from having a direct mechanism of action, driving the atherosclerotic phenotype of Retn tg Ldlr−/− mice through the immune system or an action on endothelium—a mechanism that may well be relevant in human disease and plays a role in some animal models [28, 29]. We have, however, identified a centrally mediated aggravated metabolic state in the Retn tg mice that offers an atherosclerotic driving force of weight gain, heightened leptin activity and elevated triacylglycerol levels. Regardless of whether resistin functions directly in the plaques themselves or through altering systemic lipid metabolism as we propose, these findings demonstrate the far-reaching effects of adipokine function and highlight the role of adipokine signalling in the adipose–CNS axis of metabolic regulation.
Supplementary Material
Acknowledgments
The authors thank the UT Southwestern Transgenic Core Facility under the direction of R. Hammer for the generation of the transgenic lines, the Molecular Pathology Core and J. Shelton for assistance with histology and the Mouse Metabolic Phenotyping Core for lipid and lipoprotein measurements.
Funding sources The study was supported by the National Institutes of Health (grants R01-DK55758 and R01-DK099110 to PES and P01-DK088761 to PES and JKE). IWA was supported by the Throne-Holst Foundation, the Swedish Research Council (2006-3931 and 2012-1601), VINNOVA (2011-01336) and a NovoNordisk Excellence Project Award. JMR was supported by the American Heart Association (12SDG12050287). TF was supported by Juvenile Diabetes Research Foundation (3-2011-405). MF was supported by the AHA (09SDG2080223). RKG was supported by the NIH (grants K01-DK090120-02 and R03-DK099428) and Searle Scholars Program (Chicago, Illinois).
Abbreviations
- BAT
Brown adipose tissue
- CT
Computerised tomography
- FA
Fatty acids
- GWAT
Gonadal white adipose tissue
- ICV
Intracerebroventricular
- IWAT
Inguinal white adipose tissue
- NST
Non-shivering thermogenesis
- RELM
Resistin-like molecule
- SNS
Sympathetic nervous system
- STAT3
Signal transducer and activator of transcription 3
- tg
Transgenic mouse model
- UCP-1
Uncoupling protein-1
- WAT
White adipose tissue
Footnotes
Contribution statement IWA and JMR designed and carried out the research, interpreted the results, and co-authored the manuscript. TF, YRC, MF, CT, RKG and ZVW assisted in study design, performed research, and revised and reviewed the manuscript. JKE and PES designed the study, analysed the data, and reviewed the manuscript. PES is responsible for the integrity of this work. All authors approved the final version of the manuscript.
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material The online version of this article (doi:10.1007/s00125-014-3210-3) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Contributor Information
Ingrid W. Asterholm, Touchstone Diabetes Center, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8549, USA
Joseph M. Rutkowski, Touchstone Diabetes Center, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8549, USA
Teppei Fujikawa, Division of Hypothalamic Research, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX, USA.
You-Ree Cho, Touchstone Diabetes Center, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8549, USA.
Makoto Fukuda, Division of Hypothalamic Research, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX, USA.
Caroline Tao, Touchstone Diabetes Center, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8549, USA.
Zhao V. Wang, Touchstone Diabetes Center, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8549, USA
Rana K. Gupta, Touchstone Diabetes Center, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8549, USA
Joel K. Elmquist, Division of Hypothalamic Research, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX, USA
Philipp E. Scherer, Email: Philipp.Scherer@utsouthwestern.edu, Touchstone Diabetes Center, Department of Internal Medicine, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8549, USA; Department of Cell Biology, University of Texas Southwestern Medical Center, Dallas, TX, USA.
References
- 1.Steppan CM, Brown EJ, Wright CM, et al. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci U S A. 2001;98:502–506. doi: 10.1073/pnas.98.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rajala MW, Qi Y, Patel HR, et al. Regulation of resistin expression and circulating levels in obesity, diabetes, and fasting. Diabetes. 2004;53:1671–1679. doi: 10.2337/diabetes.53.7.1671. [DOI] [PubMed] [Google Scholar]
- 3.Steppan CM, Bailey ST, Bhat S, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 4.Savage DB, Sewter CP, Klenk ES, et al. Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes. 2001;50:2199–2202. doi: 10.2337/diabetes.50.10.2199. [DOI] [PubMed] [Google Scholar]
- 5.Qi Y, Nie Z, Lee YS, et al. Loss of resistin improves glucose homeostasis in leptin deficiency. Diabetes. 2006;55:3083–3090. doi: 10.2337/db05-0615. [DOI] [PubMed] [Google Scholar]
- 6.Park HK, Qatanani M, Briggs ER, Ahima RS, Lazar MA. Inflammatory induction of human resistin causes insulin resistance in endotoxemic mice. Diabetes. 2011;60:775–783. doi: 10.2337/db10-1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graveleau C, Zaha VG, Mohajer A, et al. Mouse and human resistins impair glucose transport in primary mouse cardiomyocytes, and oligomerization is required for this biological action. J Biol Chem. 2005;280:31679–31685. doi: 10.1074/jbc.M504008200. [DOI] [PubMed] [Google Scholar]
- 8.Muse ED, Lam TK, Scherer PE, Rossetti L. Hypothalamic resistin induces hepatic insulin resistance. J Clin Invest. 2007;117:1670–1678. doi: 10.1172/JCI30440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikeda Y, Tsuchiya H, Hama S, Kajimoto K, Kogure K. Resistin affects lipid metabolism during adipocyte maturation of 3T3-L1 cells. FEBS J. 2013;280:5884–5889. doi: 10.1111/febs.12514. [DOI] [PubMed] [Google Scholar]
- 10.Reilly MP, Lehrke M, Wolfe ML, Rohatgi A, Lazar MA, Rader DJ. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111:932–939. doi: 10.1161/01.CIR.0000155620.10387.43. [DOI] [PubMed] [Google Scholar]
- 11.Burnett MS, Lee CW, Kinnaird TD, et al. The potential role of resistin in atherogenesis. Atherosclerosis. 2005;182:241–248. doi: 10.1016/j.atherosclerosis.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Kawanami D, Maemura K, Takeda N, et al. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: a new insight into adipocytokine–endothelial cell interactions. Biochem Biophys Res Commun. 2004;314:415–419. doi: 10.1016/j.bbrc.2003.12.104. [DOI] [PubMed] [Google Scholar]
- 13.Verma S, Li SH, Wang CH, et al. Resistin promotes endothelial cell activation: further evidence of adipokine–endothelial interaction. Circulation. 2003;108:736–740. doi: 10.1161/01.CIR.0000084503.91330.49. [DOI] [PubMed] [Google Scholar]
- 14.Fukuda M, Jones JE, Olson D, et al. Monitoring FoxO1 localization in chemically identified neurons. J Neurosci. 2008;28:13640–13648. doi: 10.1523/JNEUROSCI.4023-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fasshauer M, Klein J, Ueki K, et al. Essential role of insulin receptor substrate-2 in insulin stimulation of Glut4 translocation and glucose uptake in brown adipocytes. J Biol Chem. 2000;275:25494–25501. doi: 10.1074/jbc.M004046200. [DOI] [PubMed] [Google Scholar]
- 16.Rangwala SM, Rich AS, Rhoades B, et al. Abnormal glucose homeostasis due to chronic hyperresistinemia. Diabetes. 2004;53:1937–1941. doi: 10.2337/diabetes.53.8.1937. [DOI] [PubMed] [Google Scholar]
- 17.Asterholm IW, Scherer PE. Enhanced metabolic flexibility associated with elevated adiponectin levels. Am J Pathol. 2010;176:1364–1376. doi: 10.2353/ajpath.2010.090647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park S, Hong SM, Sung SR, Jung HK. Long-term effects of central leptin and resistin on body weight, insulin resistance, and beta-cell function and mass by the modulation of hypothalamic leptin and insulin signaling. Endocrinology. 2008;149:445–454. doi: 10.1210/en.2007-0754. [DOI] [PubMed] [Google Scholar]
- 19.Kosari S, Rathner JA, Chen F, Kosari S, Badoer E. Centrally administered resistin enhances sympathetic nerve activity to the hindlimb but attenuates the activity to brown adipose tissue. Endocrinology. 2011;152:2626–2633. doi: 10.1210/en.2010-1492. [DOI] [PubMed] [Google Scholar]
- 20.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 21.Kosari S, Camera DM, Hawley JA, Stebbing M, Badoer E. ERK1/2 in the brain mediates the effects of central resistin on reducing thermogenesis in brown adipose tissue. Int J Physiol Pathophysiol Pharmacol. 2013;5:184–189. [PMC free article] [PubMed] [Google Scholar]
- 22.Kosari S, Rathner JA, Badoer E. Central resistin enhances renal sympathetic nerve activity via phosphatidylinositol 3-kinase but reduces the activity to brown adipose tissue via extracellular signal-regulated kinase 1/2. J Neuroendocrinol. 2012;24:1432–1439. doi: 10.1111/j.1365-2826.2012.02352.x. [DOI] [PubMed] [Google Scholar]
- 23.Bartelt A, Bruns OT, Reimer R, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 24.Lefterova MI, Mullican SE, Tomaru T, Qatanani M, Schupp M, Lazar MA. Endoplasmic reticulum stress regulates adipocyte resistin expression. Diabetes. 2009;58:1879–1886. doi: 10.2337/db08-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou L, Yu X, Meng Q, et al. Resistin reduces mitochondria and induces hepatic steatosis in mice by the protein kinase C/protein kinase G/p65/PPAR gamma coactivator 1 alpha pathway. Hepatology. 2013;57:1384–1393. doi: 10.1002/hep.26167. [DOI] [PubMed] [Google Scholar]
- 26.Tziomalos K, Athyros VG, Karagiannis A, Kolovou GD, Mikhailidis DP. Triglycerides and vascular risk: insights from epidemiological data and interventional studies. Curr Drug Targets. 2009;10:320–327. doi: 10.2174/138945009787846425. [DOI] [PubMed] [Google Scholar]
- 27.Taleb S, Herbin O, Ait-Oufella H, et al. Defective leptin/leptin receptor signaling improves regulatory T cell immune response and protects mice from atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2691–2698. doi: 10.1161/ATVBAHA.107.149567. [DOI] [PubMed] [Google Scholar]
- 28.Kushiyama A, Sakoda H, Oue N, et al. Resistin-like molecule beta is abundantly expressed in foam cells and is involved in atherosclerosis development. Arterioscler Thromb Vasc Biol. 2013;33:1986–1993. doi: 10.1161/ATVBAHA.113.301546. [DOI] [PubMed] [Google Scholar]
- 29.Cho Y, Lee SE, Lee HC, et al. Adipokine resistin is a key player to modulate monocytes, endothelial cells, and smooth muscle cells, leading to progression of atherosclerosis in rabbit carotid artery. J Am Coll Cardiol. 2011;57:99–109. doi: 10.1016/j.jacc.2010.07.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.