

Abstract
Purpose.
The redox function of APE1/Ref-1 is a key regulator in pathological angiogenesis, such as retinal neovascularization and tumor growth. In this study, we examined whether inhibition of APE1/Ref-1 redox function by a small molecule inhibitor E3330 suppresses experimental choroidal neovascularization (CNV) in vitro and in vivo.
Methods.
Primate choroid endothelial cells (CECs) received treatment of 0 to 100 μM E3330 alone or cotreatment of E3330 and 500 μg/mL anti-VEGF antibody bevacizumab. Choroid endothelial cell angiogenic function was examined by cell proliferation, migration, and tube formation assays. The effects of E3330 on NF-κB and STAT3 signaling pathways were determined by reporter gene assay, Western blot, and ELISA. Laser-induced CNV mouse model was used to test the effects of E3330 in vivo. Potential toxicity of E3330 was evaluated by TUNEL assay.
Results.
The E3330 of 25 to 100 μM dose-dependently suppressed CEC proliferation, migration, and tube formation, in the absence of noticeable cell toxicity. Lower doses of E3330 (10–20 μM) reduced the transcriptional activity of NF-κB and STAT3 without affecting protein phosphorylation of both molecules. At the same time, E3330 downregulated MCP-1 production in CECs. The antiangiogenic effect of E3330 was comparable and additive to bevacizumab. The E3330 effectively attenuated the progression of laser-induced CNV in mice after a single intravitreal injection.
Conclusions.
The APE1/Ref-1 redox function regulates multiple transcription factors and inflammatory molecules, and is essential for CEC angiogenesis. Specific inhibition of APE1/Ref-1 redox function with E3330 may represent a promising novel treatment for wet AMD.
Keywords: APE1/Ref-1, redox function, transcription factors, choroidal neovascularization, antiangiogenesis
Selective inhibition of APE1/Ref-1 redox function has significant anti-angiogenic effect on choroidal neovascularization both in vitro and in vivo.
Introduction
Neovascular AMD, also known as wet AMD, affects 1.2 million people in the United States.1 It is characterized by the growth of choroidal neovascularization (CNV) through Bruch's membrane and into subretinal space in the macular area, and subsequent damage of central vision. Currently, the main treatment of CNV is intravitreal injection of VEGF antagonists, such as bevacizumab and ranibizumab.2 Yet, these drugs also are known for their limited indications and insufficient efficacy.3–5 Although VEGF is a key regulator of neovascularization, solely targeting of VEGF is often not sufficient to counter the complex etiology of CNV.6,7 In addition to VEGF's role in endothelial mitogenesis and vascular hyperpermeability, other molecules, such as nuclear factor-κB (NF-κB)8–10 and signal transducer and activator of transcription 3 (STAT3),11,12 as well as their downstream inflammatory cytokines, also are known to be profoundly involved in pathological neovascularization. Therefore, research efforts on developing novel agents targeting different molecules is ongoing to improve antiangiogenic effect for CNV treatment.
Studies in the past decade have revealed that the reduction-oxidation (redox) regulatory activity of apurinic endonuclease 1/redox factor-1 (APE1/Ref-1, here referred to as APE1) is a potential target for anti-angiogenesis therapy, including retinal neovascularization.13–17 APE1/Ref-1 is named for its dual function as an AP-endonuclease in DNA base excision repair pathway and a reductive activator in transcriptional regulation.17–22 It has been shown that APE1 redox function regulates a number of transcription factors, notably activator protein 1 (AP-1),23–25 NF-κB,23,25–27 hypoxia-inducible factor 1 (HIF-1),15,17,27,28 and p53.27,29 By reducing certain conserved cysteine residues in these proteins,14 APE1 stimulates DNA-binding activity of these transcription factors and participates in a wide spectrum of cellular function, including inflammation and neovascularization. Specific inhibition of APE1 redox function can be achieved by using a small molecule inhibitor, E3330.15,17,30–34 We have reported that E3330 significantly suppressed in vitro angiogenesis of retinal vascular endothelial cells (RVEC) and reduced subretinal neovascularization (SNV) in a very-low-density lipoprotein receptor knockout (vldlr–/–) mutant mouse.13,35,36 However, it is not known whether APE1 redox function also regulates choroidal endothelial cell (CEC) angiogenesis.
In this study, we demonstrated for the first time that specific inhibition of APE1 redox activity with E3330 significantly suppressed CEC angiogenesis in vitro, and laser-induced CNV development in mouse eyes without causing any tissue toxicity. These data indicate that specific inhibition of APE1 redox function may serve as a promising novel strategy for alternative or complementary treatment for CNV.
Materials and Methods
Cell Culture and Reagents
Choroid-retinal endothelial cell line (RF/6A) from rhesus macaque and human adult retinal pigment epithelium cell line (ARPE-19) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), and cultured according to ATCC instructions. Primary human RVECs were purchased from Cell Systems (ACBRI 181; Cell Systems, Kirkland, WA, USA) and grown in an endothelial cell growth medium (EGM-2 Bulletkit; Lonza, Inc., Allendale, NJ, USA). The E3330 ([2E]-3-[5-2,3-dimethoxy-6-methyl-1,4-benzoquinoyl]-2-nonyl-2-propenoic acid) was first dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich Co., St. Louis, MO, USA) at a concentration of 20 mM before adding to the culture medium at final concentrations of 10 to 120 μM. The final concentration of DMSO in medium of all treated groups did not exceed 0.01% (vol/vol). A vehicle control with matched concentration of DMSO without E3330 was routinely included in each experiment.
Cell Proliferation Assay
CellTiter 96 AQueous One Solution Reagent (Promega Corp., Madison, WI, USA) was applied according to the manufacturer's instructions. Choroidal endothelial cells were treated with 25 to approximately 100 μM E3330 alone or in combination with 500 μg/mL bevacizumab (Avastin; Genentech, San Francisco, CA, USA) for 24 hours. The treated CECs were then immediately incubated with the reagent for 2 hours. The absorbance value at 490 nm was measured by a microplate reader (SpectraMax M2e; Molecular Devices LLC, Sunnyvale, CA, USA).
In Vitro TUNEL Assay
Choroidal endothelial cells treated with 25, 50, and 100 μM E3330 for 24 hours were stained using an In Situ Cell Death Detection Kit (Roche, Indianapolis, IN, USA) following the manufacture's protocol. A DNase I–treated group was included as a positive control. The positively stained cells were counted under a fluorescence microscope (Leica DM IRB; Leica Microsystem, Inc., Bannockburn, IL, USA). The percentage of positively stained cells over the total cell number in each group was calculated.
Cell Migration Assay
Choroidal endothelial cell migration was assessed using a modified Boyden chamber method according to the Klemke et al. report.37 Briefly, CECs at 5000 cells per well were seeded into the upper chamber of Matrigel (BD Bioscience, Bedford, MA, USA) precoated Transwell inserts (Transwell, 8.0 μm pore size, 6.5-mm membrane diameter; Corning, Acton, MA, USA). Basic FGF (Genentech, San Francisco, CA, USA) at 10 ng/mL was added to the lower chamber medium as a chemoattractant to induce baseline migration. After being treated with 25 to 100 μM E3330 alone or cotreated with E3330 and 500 μg/mL bevacizumab for 16 hours, CECs were labeled with cell tracker (CellTracker Green BODIPY; Life Technologies, Grand Island, NY, USA) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Un-migrated cells that remained on the upper surface of the Transwell membrane were scraped off with a cotton swab. Migrated cells attached to the lower surface of the Transwell membrane were counted under an inverted fluorescence microscopy.
In Vitro Matrigel Tube Formation Assay
After administration of 25 to 100 μM E3330 alone or in combination with 500 μg/mL bevacizumab for 24 hours, CECs were re-suspended and transferred to Matrigel precoated 96-well plates at 5000 cells per well. After 6 hours of incubation to allow tube formation, the capillary-like tube structure formed by CECs in each well was captured with a digital camera (Spot RT-KE; Diagnostic Instruments, Inc., Sterling Heights, MI, USA) and quantified with ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA) by measuring the total tube length.
Luciferase Reporter Gene Assay
The DNA-binding activity of NF-κB and STAT3 was analyzed by Cignal luciferase reporter kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Briefly, CECs were first treated with 0, 10, and 20 μM of E3330 for 24 hours and then transfected with inducible firefly luciferase (Fluc) reporter vector and constitutive Renilla luciferase (Rluc) construct for 24 hours. Fluc and Rluc expression in each sample was detected using the Dual-Luciferase Reporter Assay System (Promega Corporation, Madison, WI, USA) on a luminometer (Molecular Devices, LLC, Sunnyvale, CA, USA). The transcription activities, as indicated by Fluc reading, were normalized to Rluc. The ratio of Fluc to Rluc reading in each group was divided by the ratio of promoterless positive control luciferase to determine the fold change.
Western Blot
Whole-cell protein extracts of CECs with or without E3330 treatment were separated by 10% SDS-PAGE electrophoresis, and transferred to a nitrocellulose membrane. Blots were probed with different primary antibodies, including anti-APE1/Ref-1 (Cell Signaling, Danvers, MA, USA), anti–NF-κB p65 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-pSTAT3 (Cell Signaling). Blots were then incubated with horseradish peroxidase–conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) at room temperature for 1 hour and visualized by an enhanced chemiluminescence solution (Thermo Scientific, Rockford, IL, USA). Membranes were stripped and reprobed with β-actin antibody (Abcam, Cambridge, MA, USA) as a loading control. Quantitative analysis of all blots was performed using densitometry software (Image Lab; Bio-Rad, Hercules, CA, USA).
ELISA Assay
The protein concentration of MCP-1 in the conditioned media of CECs was determined using an ELISA kit (R&D Systems, Minneapolis, MN, USA) following the manufacturer's instructions.
Animals
All animals used in this study were conducted and treated strictly in accordance with an animal protocol reviewed and approved by Henry Ford Institutional Animal Care and Use Committee, and in adherence to the ARVO Statement for the Use of Animals in Ophthalmologic and Vision Research. Male wild-type C57BL/6 mice of 6 to 8 weeks were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA), and housed on a 12-hour light-dark cycle with free access to food and water. For all experimental procedures, mice were anesthetized by 50 mg/kg ketamine HCl and 10 mg/kg xylazine, and pupils were dilated with topical 1% tropicamide and 2.5% phenylephrine.
Laser Photocoagulation, E3330 Intravitreal Injection, and Quantification of CNV Lesions
Laser photocoagulation (532-nm wavelength, 100-mW power, 50-μm spot size, and 0.1-second duration) was performed bilaterally in each mouse. A series of four laser spots were concentrically placed at approximately 3, 6, 9, and 12-o'clock positions and 75 to 100 μm around the optic disk in each eye. All laser burns had the appearance of a cavitation bubble. Right after laser photocoagulation, each animal received a single intravitreal injection of 1 μL E3330 in one eye and the same volume of balanced saline solution (BSS) as a vehicle control in the fellow eye. Target concentration of E3330 in retinochoroid tissue was approximately 20 μM. Two weeks after laser photocoagulation, mice were killed and the eyes were processed for fluorescent-labeled isolectin stain of CNV lesions according to previous reports.13,38 Briefly, after fixation with 4% paraformaldehyde and incubation with a blocking solution containing 3% normal donkey serum and 0.2% Triton x-100 in PBS, the eyecups were stained with 1:100 diluted isolectin in the blocking buffer at 4°C overnight. With four to approximately six relaxing radial cuts, RPE-choroid-sclera complex was flattened and mounted with the RPE side upward. The area of CNV lesion in each sample was measured using imaging software (SPOT Advanced; Diagnostic Instruments, Inc., Sterling Heights, MI, USA).
In Vivo TUNEL Assay
After intravitreal administration of 10 to 100 μM E3330 for 24 hours, eyeballs of adult wild-type C57BL/6 mice were harvested and fixed. The eyecups, including retina-choroidal tissue, were embedded within optimal cutting temperature compound and frozen to cut slices. Apoptotic cells in the retina-choroidal tissue were detected using an in situ cell death detection kit (Roche, Indianapolis, IN, USA) and counted in 10 random views at ×20 magnification under fluorescence microscope. Slices were also counterstained with DAPI.
Statistics
All data were presented as a percentage of the control group and calculated for mean ± SD. Differences were assessed by ANOVA or Student's t-test using IBM SPSS Statistics 19 (Armonk, New York, NY, USA). A value of P less than 0.05 is considered statistically significant.
Results
APE1 Expression in Retinal and Choroidal Cells
We and others have previously reported APE1 expression in mouse RVECs13 and in developing rat retina.39 To verify the presence of APE1 in retinochoroidal tissue in primates and humans, we examined APE1 protein level in several commonly used cell lines originally isolated from retina of human and nonhuman primates. We found highly abundant APE1 in all three cell lines, including CECs, ARPE19, and human RVECs (hRVECs) (Fig. 1), indicating potential roles of APE1 in choroidal vascular pathologies.
Figure 1.
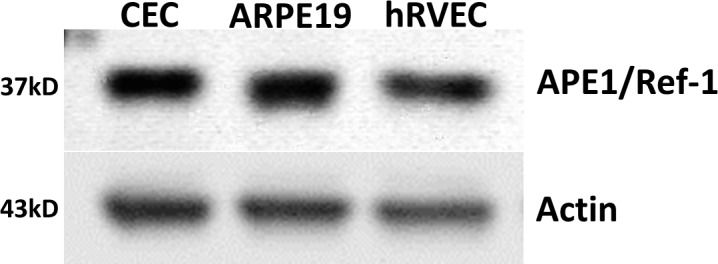
Western blot of APE1 expression in CECs, ARPE-19, and hRVECs. Apurinic endonuclease 1 was abundantly expressed in CECs, ARPE-19, and hRVECs. The result is representative of three replicates. The molecular weight is indicated to the left in kilodaltons.
APE1 Redox Inhibition Suppresses CEC Angiogenesis In Vitro
A series of in vitro assays were conducted to determine the effects of APE1 redox activity on specific angiogenic functions of CECs, including proliferation, migration, and tube formation. Choroidal endothelial cell proliferation was significantly suppressed by 50, 75, and 100 μM E3330 in a dose-dependent manner (Fig. 2A; P < 0.05), and reduced to 86.39%, 54.22%, and 5.36% of the control level, respectively. For CEC migration, a notable inhibitory effect of E3330 started from the dose of 25 μM, which depressed the number of migrated cells to 66.87% of the control group (Fig. 2B; P < 0.05). Choroidal endothelial cell migration plunged to 25.77%, 19.02%, and 12.27% of the control group when treated with 50, 75, and 100 μM E3330, respectively. The tube formation function of CECs also was suppressed dose-dependently by administration of 50, 75, and 100 μM E3330 (Fig. 2C; P < 0.05), with the total tube length decreased to 46.32%, 35.12%, and 12.79% of the control group, respectively.
Figure 2.

Effects of E3330 on CEC proliferation, migration, tube formation, and apoptosis in vitro. (A) The MTS assay indicated that 50 to 100 μM E3330 markedly reduced the CEC proliferation in a dose-dependent manner. (B) The Transwell assay showed that 25 to 100 μM E3330 significantly inhibited CEC migration dose-dependently. (C) Matrigel tube formation assay revealed a remarkable dose-dependent reduction of total tube length by 50 to 100 μM E3330. All the data points were normalized to the vehicle control. Bars represent mean ± SEM of three independent experiments.*P < 0.05 compared with the vehicle control group. (D) In vitro TUNEL assay showed up to 100 μM E3330 did not induce notable CEC apoptosis compared with the negative control. A positive control administrated with DNase I was included and had 100% apoptosis.
In addition to an overall dose-dependent inhibitory effect of E3330 on CEC angiogenesis, it was noted that CEC migration was more susceptible to E3330 than the cell proliferation or tube formation (Figs. 2A–C). At the dose of 25 μM, E3330 had no evident effect on CEC proliferation (Fig. 2A) or tube formation (Fig. 2C), but significantly decreased CEC migration (Fig. 2B; P < 0.05). The decline range of CEC migration also was more prominent than that of proliferation and tube formation when 50 and 75 μM E3330 was administrated. In other words, E3330 has differentiated inhibitory effects regarding specific angiogenic functions of CECs with a preference for CEC migration. TUNEL assay revealed that 0 to 100 μM E3330 did not induce CEC apoptosis comparing to the negative control (Fig. 2D; P > 0.10), indicating a sublethal dose range of E3330 for CECs.
APE1 Redox Inhibition Reduces Transcriptional Activity of NF-κB and STAT3, and Production of MCP-1
Dual-luciferase reporter gene assay revealed an evident decline of NF-κB and STAT3-mediated DNA binding induced by E3330 at low doses (10–20 μM). Of the two transcription factors, STAT3 appears to be more susceptible to APE1 redox inhibition than NF-κB does, because 10 μM E3330 depressed STAT3 activity to 70.27% of the control group (Fig. 3A; P < 0.01) while having no significant effect on NF-κB activity (Fig. 3A). A dose of 20 μM E3330 reduced the transcription activity of NF-κB and STAT3 to 39.92% and 27.92% of the control group, respectively (Fig. 3A; P < 0.001). In addition, the production of a key downstream effector of NF-κB and STAT3, MCP-1, was also significantly reduced by 10, 20, and 50 μM E3330 in a dose-dependent manner (Fig. 3B; P < 0.001).
Figure 3.

Effects of E3330 on the DNA binding and phosphorylated protein levels of NF-κB and STAT3, and on MCP-1 production in CECs. (A) Dual-luciferase reporter gene assay indicated that E3330 at low doses (10–20 μM) suppressed the DNA binding activity of NF-κB and STAT3. NF-κB–directed DNA binding was significantly reduced by 20 μM E3330, whereas STAT3 activity was remarkably reduced by 10 and 20 μM E3330. (B) The ELISA assay of MCP-1 concentration in CECs culture revealed that E3330 significantly suppressed MCP-1 production in a dose-dependent manner starting from 10 μM. (C, D) Representative gel images of NF-κB p65 (C) and pSTAT3 (D) expression in CECs treated with E3330. The DNA-binding inhibitory dose of E3330 (10–20 μM) did not affect the expression of both proteins. The E3330 doses of 80 and 120 μM significantly reduced p65 expression in CECs. The expression of pSTAT3 was not altered by up to 80 μM E3330. All the data points were normalized to the vehicle control. Bars represent mean ± SEM of three independent experiments. *P < 0.05 compared with the vehicle control group.
Western blot analyses showed E3330 at 10 to 20 μM had minimal effects on the protein level of phosphorylated NF-κB (p65) and STAT3 (pSTAT3) (Figs. 3C, 3D). Significant decrease of p65 was observed by E3330 only at doses greater than 80 μM (Fig. 3C; P < 0.05). No significant change of pSTAT3 was induced by up to 80 μM E3330 (Fig. 3D; P > 0.05). These results are consistent with the previous established mechanism that APE1 redox function regulates transcription activities by interfering with DNA-binding activities of transcription factors rather than affects the expression of specific transcription factors, especially at a low-dose range.
APE1 Redox Inhibition Is Comparable and Additive With the Antiangiogenic Effect of Bevacizumab
According to the data shown, a dose range of 0 to 50 μM E3330 was adapted to test if E3330 cotreatment with bevacizumab, a widely used VEGF-A antagonist in the treatment of AMD, results in promoted antiangiogenic effects compared with the exclusive use of either drug. We discovered an additive interaction between E3330 and bevacizumab regarding inhibition of CEC proliferation, migration, and tube formation. A decline of 22.87% in CEC proliferation was caused by 25 μM E3330 cotreatment with 500 μg/mL bevacizumab, whereas the reduction caused by 25 μM E3330 and 500 μg/mL bevacizumab, respectively, was 7.15% and 14.55%. Administration of 50 μM E3330 and 500 μg/mL bevacizumab led to comparable (13.61% and 14.55% reduction by the exclusive use of the two drugs, respectively) and significantly additive (33.24% by cotreatment of the two drugs) (Fig. 4A; P < 0.05 compared with the exclusive treatment of either compound at corresponding dose) antiproliferation effect. The additive effect of E3330 and bevacizumab in CEC migration inhibition was dramatic. Treatment with 500 μg/mL bevacizumab suppressed CEC migration by 26.38%, cotreatment with 25 and 50 μM E3330 reduced CEC migration by 82.21% (Fig. 4B; P < 0.05 compared with the exclusive treatment of either compound at corresponding dose) and 90.18% (Fig. 4B; P < 0.05 compared with the exclusive treatment of 500 μg/mL bevacizumab), respectively. For CEC tube formation, a decline of 19.52% in the total tube length was induced by 500 μg/mL bevacizumab. This extent of decline reached 56.63% and 69.72%, respectively, when 25 and 50 μM E3330 was added (Figs. 4C, 4D; both P < 0.05 compared with the exclusive treatment of either compound at corresponding dose).
Figure 4.

The additive interaction between E3330 and 500 μg/mL bevacizumab on CEC proliferation, migration, and tube formation. (A) The MTS assay showed that cotreatment with 0, 25, and 50 μM E3330 plus 500 μg/mL bevacizumab significantly reduced CEC viability compared with the exclusive treatment of E3330 at corresponding doses. Cotreatment of 50 μM E3330 and 500 μg/mL bevacizumab significantly reduced CEC proliferation compared with the use of either drug alone at corresponding dose. (B) The Transwell assay indicated 25 μM E3330 cotreatment with 500 μg/mL bevacizumab remarkably reduced CEC migration compared with the exclusive application of either drug. Coadministration of 50 μM E3330 significantly enhanced the suppressive effect of 500 μg/mL bevacizumab on CEC migration. (C, D) Representative images (C) and quantitative analysis (D) of matrigel tube formation assay revealed an additive effect between E3330 at 25, 50, and 75 μM and bevacizumab at 500 μg/mL, by which CEC tube formation was significantly reduced compared with the exclusive use of either drug. Scale bar: 100 μm. All the data points were normalized to the vehicle control. Two-way ANOVA showed P less than 0.01 between the combined treatment and mono-treatment in all above experiments. Bars represent mean ± SEM of three independent experiments. *P < 0.05 compared with the exclusive use of E3330 at corresponding dose; #P < 0.05 compared with the exclusive use of 500 μg/mL bevacizumab.
Intravitreal E3330 Suppresses Experimental CNV Lesion Without Causing Tissue Toxicity
A classic model of wet AMD, laser-induced CNV, was applied to determine whether in vitro antiangiogenic effects of E3330 also presents in vivo. A single intravitreal injection of 1 μL E3330 at 200 μM, which is approximately equal to a final concentration of 20 μM intravitreally, remarkably inhibited CNV development caused by laser photocoagulation in mouse eyes (Figs. 5A–D). Fluorescein angiography revealed E3330 markedly reduced CNV leakage compared with vehicle control (Figs. 5A, 5B). The size of CNV lesion was also much smaller in the E3330-treated group than that in the vehicle control–treated group (Figs. 5C, 5D). The mean value of CNV size was significantly reduced (9779.29 μm2) to nearly half of the value in the vehicle control–treated group (17,429.98 μm2) (Fig. 5E; n = 14; P < 0.01) by E3330. Furthermore, we calculated the percentage of large (>10,000 μm2), medium (5000–10,000 μm2), and small (<5000 μm2) CNV area according to the size of CNV. As shown in the Table, whereas most (63%) CNV lesions in vehicle control–treated eyes was more than 10,000 μm2 in the large group, the number of CNV lesions in the same category in the E3330-treated group was much less (41%). This reduction of more than 20% in the large-sized group was shifted into the small-sized category of less than 5000 μm2. These data indicate that APE1 redox inhibition by E3330 significantly attenuates the growth of CNV lesion.
Figure 5.

Effect of E3330 on laser-induced CNV in mice. Male adult wild-type C57BL/6 mice received a single intravitreal injection of 1 μL E3330 at 200 μM (equivalent to an intravitreal centration of approximately 20 μM) in one eye and 1 μL vehicle (BSS) in the fellow eye immediately after laser photocoagulation of RPE-Bruch's membrane. (A, B) Representative fluorescein angiography of central retina 2 weeks after laser photocoagulation and intravitreal injection of BSS (A) or E3330 (B). All four CNV lesion sites were clearly visible with significant fluorescein dye leaked out of the vessels in the control eyes. In contrast, fluorescein leakage was much less in the E3330-treated eyes with two small leaky spots and two nonleaking lesion sites. (C, D) Representative images of isolectin stain of RPE-choroid-sclera whole mount that performed 2 weeks after laser. The area of CNV lesion in E3330-treated eyes (D) was smaller than that of the BSS-treated eye (C). Scale bar: 100 μm. (E) Quantitative measurement of CNV area showed that the mean value of the CNV area treated by E3330 was significantly lower than that by vehicle control (n = 14, P = 0.007129).
Table.
The Distribution of CNV Lesion Size in Single Intravitreal E3330 or Vehicle (BSS)-Administrated Eyes After Laser Photocoagulation
|
Treatment |
E3330 |
BSS |
|
| CNV lesion area, μm2 | Sum | 537,861 | 958,649 |
| Mean | 9,779.29 | 17,429.98 | |
| Distribution of CNV size, % | >10,000 μm2 | 41 | 63 |
| 5000–10,000 μm2 | 21 | 21 | |
| <5000 μm2 | 38 | 16 | |
The isolectin-stained CNV lesion was quantified by three grades: large (>10000 μm2), medium (5000–10000 μm2), and small (<5000 μm2). The size range was chosen arbitrarily; 56 lesions of 14 choroidal samples were graded for each treatment group. The percent incidence of each category revealed that large-sized CNV proportion was significantly lower in the E3330-treated group than that in the BSS-treated group.
For evaluation of potential retinal toxicity, intravitreal concentration of 0 to 100 μM E3330 was achieved and retinal sections were examined for apoptotic cell death 3 days later. TUNEL stain revealed that intravitreal doses of 10, 20, and 30 μM E3330 did not induce notable cell apoptosis compared with vehicle control. The stain of retinal cell apoptosis was significantly increased only when the intravitreal dose of E3330 reached 100 μM (Fig. 6; P < 0.05).
Figure 6.

In vivo TUNEL assay of E3330 or vehicle-treated retina. Normal eyes of young adult wild-type C57BL/6 mice received intravitreal injection of various doses of E3330. Statistical analysis of the TUNEL stain revealed that the equivalent intravitreal concentration of 10, 20, or 30 μM E3330 did not elicit apoptosis in mouse retina, whereas 100 μM E3330 induced significant retinal neuron cell apoptosis. Bars represent mean ± SEM of three independent experiments. *P < 0.05 compared with the vehicle control group.
Discussion
Clinical treatment of CNV remains a challenge despite the most current application of anti-VEGF agents.3–5,40,41 One major reason is that targeting one single angiogenic molecule in a complex multifactorial pathogenic process may not be sufficient to halt CNV progression in all circumstances.42 With our growing understanding of cellular and molecular events involved in CNV development,7,43,44 the redox activity of APE1 has emerged as an appealing therapeutic target due to its regulation of transcription factors.
APE1 reduces the redox-sensitive cysteine residues of various transcription factors, facilitates their DNA-binding and transcriptional activities, and thereby controls multiple cell functions, including proliferation, migration, and stress responses.16,17,19,21 A number of transcription factors are directly regulated by APE1 redox function, including NF-κB,23,25–27 HIF-1α,15,17,27,28 AP-1,45 and p53.27,29 As these transcription factors participate in multiple aspects of endothelial cell behavior, APE1 redox function exerts its biological effects on endothelial activation and angiogenesis. In fact, the redox activity of APE1 was found required for RVEC proliferation and tube formation in vitro,13,17 as well as for retinal neovascularization in vldlr–/– mutant mouse.13,35
This current study demonstrated that an APE1 redox inhibitor, E3330, significantly inhibited CEC proliferation, migration, and capillary tube formation at a wide and safe dose range (25–100 μM), without causing cell apoptosis. The effects of E3330 in suppressing CEC activation and angiogenesis also could be interpreted as an ability in maintaining a mature or differentiated state of CECs, which allows the cell to perform physiological functions, such as transendothelial exchange and vascular permeability control.46 This notion is supported by previous reports that APE1 redox function participates in development of CD31(+) endothelial cells,15 differentiation of hemangioblasts,32 and endothelial functions, including nitric oxide production and vascular tone modulation.47 When this evidence is taken into account, APE1 redox activity is supposed to have a profound impact in regulating endothelial gene expression rather than merely modulating angiogenesis, which merits further investigation.
We previously reported a synergistic effect between E3330 and bevacizumab in inhibiting human endothelial colony-forming cell angiogenesis,13 here a significant additive action between these two compounds also was observed in suppressing CEC proliferation, migration, and tube formation. As is known, E3330 and bevacizumab have different targets in endothelial cells. The E3330 affects a range of transcription factors, such as NF-κB15,17,27 and HIF-1α,15,17,27,28 through specific inhibition of APE1 redox function. Bevacizumab is a humanized anti–VEGF-A antibody. The E3330 coadministration with bevacizumab compromises more than just VEGF signaling, and thereby produces a more prominent antiangiogenesis effect than the exclusive application of either compound. These findings implied the possibility of using supplementary APE1 redox inhibitor for improvement of the current regiment of anti-VEGF treatment for CNV, including enhancement of effectiveness and reduction of readministration frequency.
The impacts of E3330 on NF-κB and STAT3 were investigated due to their major roles in chronic inflammation in angiogenesis.48–50 At low doses (10–20 μM), E3330 significantly reduced the DNA-binding activity of NF-κB and STAT3, without affecting phosphorylated NF-κB and STAT3 level. These results agree well with previous reports in Jurkat cells,26 cancer cells,51 and RPEs.27 The DNA binding mediated by NF-κB and STAT3 is highly redox-sensitive, as evidenced by the fact that oxidation or chemical modification of conserved redox-reactive cysteine(s) within their DNA binding domain eliminated their DNA binding activities.52–54 On the contrary, reduction of disulfide bond involving the conserved cysteine residue in NF-κB stimulated its DNA binding.53 It is believed that APE1 regulates transcriptional activities through maintaining a reduced state of certain conserved cysteine residues.14 Thereby, we speculatively attribute the analogous inhibition of NF-κB and STAT3 by E3330 to the block of disulfide bond reduction mediated by specific cysteine residues. Monocyte chemoattractant protein-1 is regulated by NF-κB and STAT3,50,55 and dominates attraction of monocytes to promote chronic inflammation during CNV initiation and progression.56–58 Here we found a significant drop in MCP-1 level following the inhibition of NF-κB and STAT3 activities by E3330. In light of existing data suggesting MCP-1 as a potential target for CNV treatment in addition to VEGF,59,60 reduction of MCP-1 by E3330 may represent an important mechanism of its antiangiogenesis activity.
In vivo study confirmed the therapeutic potency of E3330 for CNV treatment in a laser-induced CNV model. However, it is noteworthy that laser-induced CNV is basically a wound-healing response in which inflammation is a major pathological component. Our findings that E3330 significantly reduced CNV lesion size in this model again suggested an anti-inflammatory role of E3330. In conclusion, the therapeutic potential of targeting APE1 redox function for CNV treatment is considerable and merits further investigation. Suppression of CEC proliferation, migration, and tube formation represents important cellular basis of E3330-mediated antiangiogenesis. Inhibition of NF-κB and STAT3 activity, as well as MCP-1 production, revealed a critical role of E3330 in resolution of inflammatory response in neovascularization. The comparable and additive effects with the VEGF antagonist bevacizumab indicated E3330 could serve as an alternative or complementary medication to achieve better visual prognosis and fewer side effects. In vivo experiments validated the effectiveness of E3330 for CNV treatment. These data corroborate our previous findings in RVECs and RPEs, together they provided solid proof that E3330 attenuates multiple aspects of retinochoroidal angiogenesis and inflammation through transcriptional regulation. Although many questions remain open, the potential of APE1 redox function as a novel target for CNV treatment should be considered.
Acknowledgments
Supported by grants from the International Retinal Research Foundation, Midwest Eye Bank, Reeves Foundation, Alliance for Vision Research, and the Henry Ford Research Foundation (XQ); National Eye Institute Grant R41 EY019784 (XQ, MRK); National Cancer Institute Grants CA121168 and CA167291 and a Riley Children's Foundation grant (MRK); and grants from the National Natural Science Foundation of China (81000390) and Alliance for Vision Research (YL).
Disclosure: Y. Li, None; X. Liu, None; T. Zhou, None; M.R. Kelley, None; P.A. Edwards, None; H. Gao, None; X. Qiao, None
References
- 1. Friedman DSOCB, Muñoz B, Tomany SC, et al. Eye Diseases Prevalence Research Group. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004; 122: 564–572 [DOI] [PubMed] [Google Scholar]
- 2. Chiang ARC. Preferred therapies for neovascular age-related macular degeneration. Curr Opin Ophthalmol. 2011; 22: 199–204 [DOI] [PubMed] [Google Scholar]
- 3. CATT Research Group. Martin DF, Maguire MG, et al. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011; 364: 1897–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosenfeld PJBD, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY; MARINA Study Group. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006; 355: 1419–1431 [DOI] [PubMed] [Google Scholar]
- 5. Tufail APP, Egan C, Hykin P, et al. ABC Trial Investigators. Bevacizumab for neovascular age related macular degeneration (ABC Trial): multicentre randomised double masked study. BMJ. 2010; 340: c2459 [DOI] [PubMed] [Google Scholar]
- 6. Campa CCC, Incorvaia C, Sheridan C, et al. Inflammatory mediators and angiogenic factors in choroidal neovascularization: pathogenetic interactions and therapeutic implications. Mediators Inflamm. 2010; 2010: 546826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Oliveira Dias JRRE, Maia M, Magalhães O Jr, Penha FM, Farah ME. Cytokines in neovascular age-related macular degeneration: fundamentals of targeted combination therapy. Br J Ophthalmol. 2011; 95: 1631–1637 [DOI] [PubMed] [Google Scholar]
- 8. Read MAWM, Williams AJ, Collins T. NF-kappa B and I kappa B alpha: an inducible regulatory system in endothelial activation. J Exp Med. 1994; 179: 503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Martin RHM, Hofer-Warbinek R, Schmid JA. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb Vasc Biol. 2000; 20: E83–E88 [DOI] [PubMed] [Google Scholar]
- 10. Dryden NHSA, Martin-Almedina S, Hannah RL, et al. The transcription factor Erg controls endothelial cell quiescence by repressing the activity of nuclear factor (NF)-κB p65. J Biol Chem. 2012; 287: 12331–12342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen ZHZ. STAT3: a critical transcription activator in angiogenesis. Med Res Rev. 2008; 28: 185–200 [DOI] [PubMed] [Google Scholar]
- 12. Chen SHMD, Lassoued W, Thurston G, Feldman MD, Lee WM. Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Biol Ther. 2008; 7: 1994–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang AGH, Kelley MR, Qiao X. Inhibition of APE1/Ref-1 redox activity with APX3330 blocks retinal angiogenesis in vitro and in vivo. Vision Res. 2011; 51: 93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelley MRGM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012; 5: 36–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou GMKC, Kabe Y, Handa H, Anders RA, Maitra A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J Cell Physiol. 2009; 219: 209–218 [DOI] [PubMed] [Google Scholar]
- 16. Tell GQF, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009; 11: 601–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luo MDS, Jiang A, Reed A, et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal. 2008; 10: 1853–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McNeill DR, Wilson DM III. A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Mol Cancer Res. 2007; 5: 61–70 [DOI] [PubMed] [Google Scholar]
- 19. Evans ARL-FM, Kelley MR. Going APE over ref-1. Mutat Res. 2000; 461: 83–108 [DOI] [PubMed] [Google Scholar]
- 20. Tell GDG, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005; 7: 367–384 [DOI] [PubMed] [Google Scholar]
- 21. Bhakat KKMA, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal. 2009; 11: 621–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xanthoudakis SMG, Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc Natl Acad Sci U S A. 1994; 91: 23–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ando KHS, Kabe Y, Ogura Y, et al. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008; 36: 4327–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xanthoudakis SCT. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992; 11: 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xanthoudakis SMG, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992; 11: 3323–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hiramoto MSN, Sugimoto K, Tang J, et al. Nuclear targeted suppression of NF-κB activity by the novel quinone derivative E3330. J Immunol. 1998; 160: 810–819 [PubMed] [Google Scholar]
- 27. Li YLX, Zhou T, Kelley MR, Edwards P, Gao H, Qiao X. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014; 2: 485–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang LEAZ, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996; 271: 32253–32259 [DOI] [PubMed] [Google Scholar]
- 29. Jayaraman LMK, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 1997; 11: 558–570 [DOI] [PubMed] [Google Scholar]
- 30. Nyland RLLM, Kelley MR, Borch RF. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1). J Med Chem. 2010; 53: 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zou GMMA. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther. 2008; 7: 2012–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zou GMLM, Reed A, Kelley MR, Yoder MC. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood. 2007; 109: 1917–1922 [DOI] [PubMed] [Google Scholar]
- 33. Shimizu NSK, Tang J, Nishi T, et al. High-performance affinity beads for identifying drug receptors. Nat Biotechnol. 2000; 18: 877–881 [DOI] [PubMed] [Google Scholar]
- 34. Curtis CDTD, Ziegler YS, Sarkeshik A, Yates JR, Nardulli AM. Apurinic/apyrimidinic endonuclease 1 alters estrogen receptor activity and estrogen-responsive gene expression. Mol Endocrinol. 2009; 23: 1346–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu WJA, Liang J, Meng H, Chang B, Gao H, Qiao X. Expression of VLDLR in the retina and evolution of subretinal neovascularization in the knockout mouse model's retinal angiomatous proliferation. Invest Ophthalmol Vis Sci. 2008; 49: 407–415 [DOI] [PubMed] [Google Scholar]
- 36. Jiang AHW, Meng H, Gao H, Qiao X. Loss of VLDL receptor activates retinal vascular endothelial cells and promotes angiogenesis. Invest Ophthalmol Vis Sci. 2009; 50: 844–850 [DOI] [PubMed] [Google Scholar]
- 37. Klemke RLCS, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997; 137: 481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mullins RFGM, Skeie JM. Glycoconjugates of choroidal neovascular membranes in age-related macular degeneration. Mol Vis. 2005; 509–517 [PubMed] [Google Scholar]
- 39. Chiarini LBFF, Petrs-Silva H, Linden R. Evidence that the bifunctional redox factor/AP endonuclease Ref-1 is an anti-apoptotic protein associated with differentiation in the developing retina. Cell Death Differ. 2000; 7: 272–281 [DOI] [PubMed] [Google Scholar]
- 40. Brown DMKP, Michels M, Soubrane G, et al. ANCHOR Study Group. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006; 355: 1432–1444 [DOI] [PubMed] [Google Scholar]
- 41. Brown DMMM, Kaiser PK, Heier JS, Sy JP, Ianchulev T; ANCHOR Study Group. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study. Ophthalmology. 2009; 116: 57–65.e55 [DOI] [PubMed] [Google Scholar]
- 42. Spaide RF. Rationale for combination therapies for choroidal neovascularization. Am J Ophthalmol. 2006; 141: 149–156 [DOI] [PubMed] [Google Scholar]
- 43. Sengupta NAA, Caballero S, Chang KH, et al. Paracrine modulation of CXCR4 by IGF-1 and VEGF: implications for choroidal neovascularization. Invest Ophthalmol Vis Sci. 2010; 51: 2697–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nagineni CNKV, William A, Detrick B, Hooks JJ. Regulation of VEGF expression in human retinal cells by cytokines: implications for the role of inflammation in age-related macular degeneration. J Cell Physiol. 2012; 227: 116–126 [DOI] [PubMed] [Google Scholar]
- 45. Xanthoudakis SCT. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992; 11: 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004; 5: 261–270 [DOI] [PubMed] [Google Scholar]
- 47. Jeon BHGG, Park YC, Qi B, et al. Apurinic/apyrimidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circ Res. 2004; 95: 902–910 [DOI] [PubMed] [Google Scholar]
- 48. Gupta SCSC, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010; 1799: 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nagineni CNKV, William A, Detrick B, Hooks JJ. Regulation of VEGF expression in human retinal cells by cytokines: implications for the role of inflammation in age-related macular degeneration. J Cell Physiol. 2012; 227: 116–126 [DOI] [PubMed] [Google Scholar]
- 50. Jougasaki MIT, Takenoshita Y, Setoguchi M. Statins suppress interleukin-6-induced monocyte chemo-attractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br J Pharmacol. 2010; 159: 1294–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cardoso AAJY, Luo M, Reed AM, et al. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS One. 2012; 7: e47462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Toledano MBLW. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A. 1991; 88: 4328–4332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matthews JRWN, Virelizier JL, Yodoi J, Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992; 20: 3821–3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li LCS, Evans EL, Shaw PE. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010; 70: 8222–8232 [DOI] [PubMed] [Google Scholar]
- 55. Wang XCJC, Allen JB, Roberts WL, Jaffe GJ. Suppression of NF-kappaB-dependent proinflammatory gene expression in human RPE cells by a proteasome inhibitor. Invest Ophthalmol Vis Sci. 1999; 40: 477–486 [PubMed] [Google Scholar]
- 56. Grossniklaus HELJ, Wallace TM, Dithmar S, et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. 2002; 119–126 [PubMed] [Google Scholar]
- 57. Suzuki MTM, Itabe H, Du ZJ, et al. Chronic photo-oxidative stress and subsequent MCP-1 activation as causative factors for age-related macular degeneration. J Cell Sci. 2012; 125: 2407–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Raoul WAC, Camelo S, Guillonneau X, Feumi C, Combadière C, Sennlaub F. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J Neuroinflammation. 2010; 7: 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamada KSE, Itaya M, Yamasaki S, Ogura Y. Inhibition of laser-induced choroidal neovascularization by atorvastatin by downregulation of monocyte chemotactic protein-1 synthesis in mice. Invest Ophthalmol Vis Sci. 2007; 48: 1839–1843 [DOI] [PubMed] [Google Scholar]
- 60. Tomida DNK, Kataoka K, Yasuma TR, et al. Suppression of choroidal neovascularization and quantitative and qualitative inhibition of VEGF and CCL2 by heparin. Invest Ophthalmol Vis Sci. 2011; 52: 3193–3199 [DOI] [PubMed] [Google Scholar]