

Abstract
Context
Mutations in X-linked lysosome-associated membrane protein gene (LAMP2; Danon disease) produce a cardiomyopathy in young patients that clinically mimics hypertrophic cardiomyopathy (HCM) due to sarcomere protein mutations. However, the natural history and phenotypic expression of this newly recognized disease is incompletely resolved and its identification may have important clinical implications.
Objectives
To determine the clinical consequences of LAMP2 cardiomyopathy and the efficacy of diagnostic and management strategies.
Setting and Design
Clinical course and outcome were assessed prospectively in 7 young LAMP2 patients (6 males) previously identified in our laboratory from the time of diagnosis (ages 7–17; median 14) to October 2008. Phenotypic expression of this disease was assessed both clinically and at autopsy.
Interventions
Implantable defibrillators, cardioactive medications, and heart transplantation.
Outcome measure
Progressive heart failure/cardiac death, and transplant.
Results
Over 7.3 ± 3 (SD) years of follow-up, and by 12 to 24 years of age, the study patients developed LV systolic dysfunction (ejection fraction 25 ± 7[SD]%) and cavity enlargement, as well as particularly adverse clinical consequences including: progressive refractory heart failure and death (n = 4), sudden death (n = 1), aborted cardiac arrest (n = 1), or heart transplantation (n = 1). LV hypertrophy was particularly marked (maximum ventricular septum, 29–65 mm; mean 44±15[SD]) including 2 patients with massive thickness of 60 mm and 65 mm at ages 23 and 15 years, respectively. In 6 patients, at study entry a ventricular pre-excitation pattern was associated with markedly increased voltages for maximum R- or S-wave (40–145 mm; mean 74±38mm), and deeply inverted T-waves. Autopsy findings included a combination of histopathologic features consistent with a storage disease (i.e., clusters of vacuolated myocytes), but also typical of HCM due to sarcomere protein mutations (i.e., myocyte disarray, small vessel disease, myocardial scarring).
Conclusions
LAMP2 cardiomyopathy is a profound disease process characterized by rapid clinical deterioration leading to cardiac death in young patients < 25 years. These observations underscore the importance of timely molecular diagnosis for predicting prognosis, and early consideration of heart transplantation.
Metabolic myocardial storage diseases which mimic the clinical and phenotypic expression of hypertrophic cardiomyopathy (HCM) have recently been reported in young patients (1), including those due to mutations in the X-linked lysosome-associated membrane protein gene (LAMP2; Danon disease) (1–7). The morphologic expression as well as the clinical course experienced by patients with this newly identified cardiomyopathy (2–7), is incompletely resolved at present. Therefore, it is informative at this time to report our early experience with an assessment of the natural history associated with LAMP2 cardiomyopathy.
METHODS
The most current clinical status of 7 previously identified LAMP2 patients (1) was reexamined, as of October 2008. Two-dimensional and Doppler echocardiographic studies were performed according to standard methodology, with commercially available instruments. As previously described in detail (1), all mutations found in probands reported here were identified using dideoxy sequencing and confirmed by restriction enzyme digestion (8). Tissue sections of LV myocardium were obtained from formalin-fixed hearts, embedded in paraffin, sectioned at 6μ thickness, and stained with hematoxylin and eosin and Masson’s trichrome. All participating centers received approval from their Institutional Review Boards.
RESULTS
Clinical profile
Clinical, demographic and outcome data were assembled for the 7 affected proband study patients, and are summarized in Table 1. At cardiac diagnosis, the one female and 6 male patients were 7 to 15 years old (mean 12). Clinical recognition in 6 patients occurred by virtue of heart murmur, family screening, and routine ECG, or by symptoms (chest pain or syncope), and in one due to atrial fibrillation. All had predominant or isolated cardiac manifestations without mental retardation, neurological or musculoskeletal deficits associated with Danon disease (2–7). On ECG at diagnosis, 6 patients had ventricular pre-excitation patterns with short PR interval (Figure 1). These patients also showed markedly increase standard and/or precordial lead voltages with maximum R- or S-wave of 40–145 mm, usually with deep negative T-waves (Table 1).
Table 1.
Clinical, Demographic and Pathologic Findings in 7 Patients with LAMP2 Cardiomyopathy
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| Age: cardiac diagnosis (y) | 8 | 14 | 11 | 15 | 17 | 7 | 15 |
| Age: last evaluation/death (y)Δ | 12 | 23 | 22 | 21 | 24 | 20 | 23 |
| Gender | M | M | F | M | M | M | M |
| Presentation | heart murmur (sports exam) | syncope | heart murmur | family history | abnormal ECG | chest pain | AF |
| NYHA functional class | |||||||
| Initial | I | I | I | I | I | I | I |
| Most recent | I | IV | I | III | IV | III | III |
| Paroxysmal AF/flutter | + | 0 | + | + (x3) | + | + | + |
| Medical treatment | atenolol; verapamil; amiodarone; warfarin | sotalol; amiodarone; warfarin; spirono-lactone | Toprol | atenolol | spirono-lactone; toprol; lisinopril; digoxin; diuretics; warfarin | Sotaolol; atenolol; diurectics | atenolol; sotalol; warfarin; diuretics; amiodarone |
| Family history of cardiomyopathy | 0 | Brother: WPW/LVH Aunt: WPW |
0 | Mother: dilated CM/transplant | 0 | 0 | 0 |
| Electrocardiogram | |||||||
| WPW | +* | + | + | + | 0 | + | +* |
| Maximum voltage (mm) | 145 | 80 | 75 | 55 | 10 | 55 | 56 |
| PR interval (ms) | 105 | 80 | 125 | 80 | 154 | 80 | 110 |
| Other | T-inversion (11 mm); inferior Qs | T-inversion (30 mm); IVCD | T-inversion (25 mm) | T-inversion (22 mm) | LAD; absent R (V1–V3) | T-inversion (15 mm) | T-inversion (10 mm); LBBB |
| LV outflow gradient (rest); mm Hg | 65 | 0 (mild SAM) | 0 | 0 | 0 | 65 | 0 |
| Maximum LV wall thickness (mm) | 65† | 60 | 30 | 37‡ | 35 | 52** | 29 |
| Ejection fraction (%) | |||||||
| Initial | 70 | nl | 64 | 70 | 75 | 66 | 68 |
| Most recent | 36 | 25 | 35 | 20 | 22 | 15 | 23 |
| LV cavity end- diastole (mm) | |||||||
| Initial | 25 | 42 | 37 | 40 | 37 | 54 | 55 |
| Most recent | 43 | 70 | 53 | 60 | 49 | n/a | 68 |
| Left atrium (mm) - - Initial | 35 | 39 | 32 | 38 | 41 | 36 | 30 |
| Mitral regurgitation | moderate | mild | Mild | mild | mild | 0 | 0 |
| 24-Hour Ambulatory Holter ECG | 633 PVBs; 8 couplets | n/a | 3 PVBs; 1 couplet | Sinus bradycardia | NSVT | NSVT | 127 PVBs; 1 couplet |
| ICD | + | +α | + | + (&CRT) | + | + | + (& CRT) |
| Complications | end-stage | end-stage; embolic stroke | end-stage | end-stage | end-stage; acute cardiac/renal failure; syncope | end-stage; pulmonary hypertension; ICD shock for VT | end-stage |
| Clinical status | sudden death (found dead in bed) | acute HF death | alive (ICD shock for VT [222 bpm] | acute HF death | sudden/HF death | progressive HF death; liver/multi-system failure; pneumonia | alive; transplant |
| Serum enzymes elevatedβ | + | + | 0 | + | + | + | 0 |
| Genetic transmission | Sporadic | Maternal | Sporadic | Maternal | Maternal | Sporadic | Sporadic |
| Mutation | Y109 | IVS6+1_4 del GTGA | IVSG-2A→G | K289FS | IVS1+ 1G→T | IVS1-2A→G | V310I (mosaic) |
Symbols:
radio frequency ablation of bypass tract at age 14 years in patient #1 and at age 15 in patient #7
anomalous anterolateral papillary muscle insertion into anterior mitral leaflet; marked LV wall thinning to 23 mm over clinical course
LV wall thickness regression to 14 mm at time of death (by echocardiography)
predominant LV hypertrophy of posterior LV free wall (septum = 40 mm)
creatine kinase and alanine aminotransferase levels elevated by factor of ≥2 and organ specific enzyme isoforms indicated cardiac, as well as musculoskeletal and liver, involvement
secondary prevention ICD; all other patients with primary prevention implants
+ = present; 0=absent
Abbreviations:
AF = atrial fibrillation; CM = cardiomyopathy; CRT = cardiac resynchronization therapy; ECG = electrocardiogram; F = female; HF = heart failure; ICD = implantable cardioverter-defibrillator; IVCD = intraventricular conduction defect; LAD = left axis deviation; LBBB = left bundle branch block; LV = left ventricular; LVH = left ventricular hypertrophy; M = male; nl = normal; NSVT = nonsustained ventricular tachycardia; NYHA = New York Heart Association; PVB = premature ventricular beats; SAM = systolic anterior motion (of mitral valve); SCD = sudden cardiac death; VT = ventricular tachycardia; VF = ventricular fibrillation; WPW = Wolff-Parkinson-White (pre-excitation pattern); y = year
Figure 1.
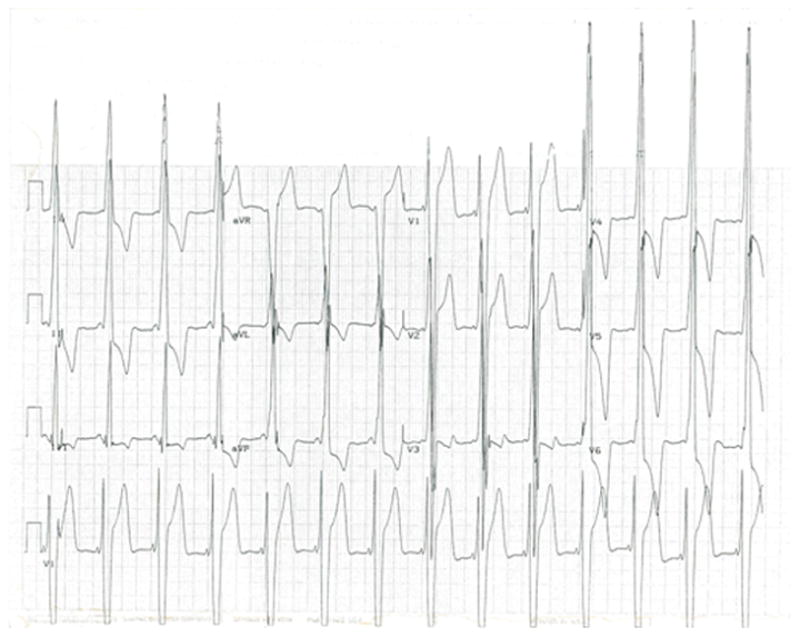
12-lead ECG (recorded at full standard) from a 16-year-old LAMP2 cardiomyopathy patient (#2 in Table 1) showing striking standard and precordial voltages and T-wave inversion.
Clinical course
At cardiac diagnosis, all patients were in New York Heart Association (NYHA) functional class I. Over the subsequent 7±3 years, each of the 7 patients experienced particularly adverse clinical consequences, by 12 to 24 years of age (mean 20). Four patients died of acute and/or progressive heart failure and one underwent heart transplantation; clinical deterioration was often rapid with the time interval from clinical stability with little or no symptoms and preserved systolic function to end-stage heart failure as brief as 6 months. Two others experienced sudden unexpected major arrhythmic events: one died suddenly (age 12) from ventricular fibrillation refractory to implantable cardioverter-defibrillator (ICD) therapy (Figure 2), and one received appropriate defibrillator shock for rapid ventricular tachycardia at age 18 (9). All 7 patients developed marked LV systolic dysfunction (ejection fraction, 20–35%; mean 25±7), associated with LV cavity dilatation in 4 and enlargement in 2 others over the follow-up period. (Table 1) (10).
Figure 2.
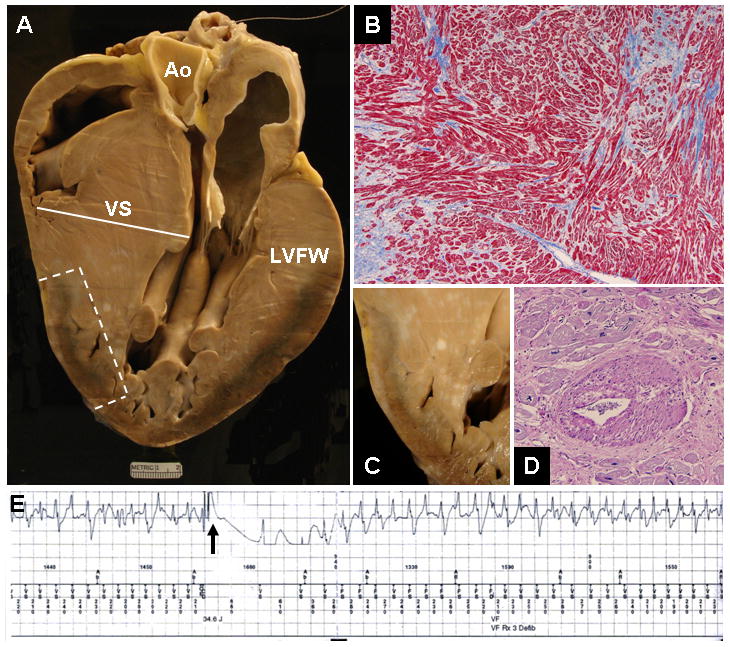
LAMP2 cardiomyopathy in a 12-year-old boy with sudden death (Table 1; patient #1). A. At autopsy, massive asymmetric LV hypertrophy. Ventricular septal (VS) thickness is 65 mm (heart weight, 1425 grams), exceeding all hearts reported to date; LV cavity is small. Ao = aorta; LVFW = left ventricular free wall. B. Disorganized LV architecture. Adjacent cardiac muscle cells (myocytes), or groups of cells, are arranged at perpendicular or oblique angles. Masson’s trichrome stain x100. C. Area of LV wall, demarcated by the broken line in A, showing subepicardial necrosis and scarring. D. Abnormal intramural coronary artery with thickened wall and narrowed lumen. PAS stain x20. E. Intracardiac ventricular electrogram. The ICD elicited a defibrillation shock (arrow) which failed to interrupt ventricular fibrillation (280 beats/min). This event was repeated 5 times until ICD capacity was extinguished and death occurred.
Phenotype
Echocardiography
Most recent echocardiographic studies demonstrated diffuse and marked LV hypertrophy in each patient. Maximum wall thickness (ventricular septum) was 29 to 65 mm (mean 44±15[SD]), including 2 patients with particularly massive thickening of 60 mm and 65 mm at age 23 and 15 years, respectively. (Table 1; patients #1 and #2). LV end-diastolic cavity dimension was documented to have dilated and/or enlarged over the follow-up period. LV outflow obstruction due to mitral valve systolic anterior motion was present at rest in 2 patients (gradient, 65 mm Hg).
Autopsy
Post-mortem examination of 2 hearts showed massive cardiac hypertrophy; heart weights were 1265 grams and 1425 grams with asymmetric LV wall thickening (Figures 2 and 3). Patient #1 showed, in addition, substantial myocyte disarray, abnormal intramural coronary arteries (with thickened walls and narrowed lumen), and replacement fibrosis including subepicardial distribution (Figure 2). Both patients showed prominent clusters of numerous myocytes with distinctive and extensive sarcoplasmic vacuolation, and inclusions of amorphous granular material in some cells within areas of scarring (Figure 3).
Figure 3.

LV myocardium from the same patient shown in Figure 1, with findings consistent with a metabolic storage disease. A. Clusters of myocytes with vacuolated sarcoplasm (stained red) embedded in an area of scar (stained blue) x100. B. At lower power, similar area of myocardium shows subepicardial distribution of scarring and vacuolated myocytes x40. C. High power photomicrograph showing a large empty myocyte surrounded by smaller vesicles in an area of replacement fibrosis x40. D. Small focal scars (stained blue) surrounded by viable myocardium x40. All photomicrographs stained with Masson’s trichrome.
DISCUSSION
The clinical course of patients reported here with LAMP2 mutations provides insights to the cardiovascular community regarding molecular diagnosis as well as the natural history, pathophysiology and clinical implications of this recently recognized genetic cardiomyopathy (1–7). Indeed, as previously suggested (1), LAMP2 mutations cause a particularly profound and accelerated cardiac disease process characterized by clinical deterioration and early death, perhaps representing one of the most lethal cardiomyopathies in the young. Such an outcome occurred in our patients despite application of the most contemporary treatment strategies, including the ICD (9) which failed to convert 5 patients to normal rhythm.
The clinical presentation of LAMP2 cardiomyopathy mimics HCM caused by mutations in genes encoding cardiac sarcomere proteins (1,2), as both are associated with marked LV hypertrophy. However, while LAMP2 cardiomyopathy is a phenocopy of HCM, it represents a fundamentally different pathologic process which results from a metabolic storage disease. In 2005, we reported genetic diagnosis in the present 7 patients with LAMP2 mutations (1), and in the ensuing and relatively brief 3-year period prospectively recognized that these patients had all experienced adverse and lethal disease consequences. Specifically, each patient evolved into an end-stage phase characterized by LV systolic dysfunction with dilated and/or enlarging cavity size (10), and experienced severe outcome: sudden death, heart failure death, heart transplantation or an appropriate ICD intervention triggered by rapid ventricular tachyarrhythmia (9) at youthful ages of 12 to 24 years. Of note, the patients presented here with LAMP2 cardiomyopathy demonstrated a clinical profile dominated by cardiac manifestations, largely without evidence of multisystem disease (e.g., mental retardation, hepatic involvement, and overt skeletal myopathy) reported in other patients with Danon disease (2–7). These observations underscore the heterogeneous clinical expression of LAMP2 mutations.
Reliably predicting future clinical events and prognosis in HCM by genetic testing for sarcomeric mutations has proved challenging in HCM (11–13). LAMP2 cardiomyopathy appears to represent an exception. Although the clinical outcome in our relatively small cohort was uniformly adverse, we should emphasize that this is not necessarily characteristic of all patients affected by LAMP2 mutations given the heterogeneity in disease expression and clinical course which has been reported in some affected relatives experiencing a generally more benign course (2,3). Indeed, 7 female LAMP2 obligate carriers (age range 19–51 years) in 2 of the families remain asymptomatic, and at present have not developed LV hypertrophy or systolic dysfunction, underscoring the striking differences in clinical phenotypes and outcomes between female carriers and affected males with LAMP2 mutations (2,4,5,7).
The early experience with the distinctive natural history and prognosis of patients with LAMP2 mutations establishes the importance of molecular diagnosis and underscores the utility of genetic testing. In this regard, a high index of suspicion should be raised for LAMP2 cardiomyopathy (and genetic testing) in young patients demonstrating massive LV hypertrophy and distinctive ECG patterns with greatly increased voltages. These observations also raise legitimate consideration for early intervention with heart transplantation (4,15) as a treatment for LAMP2 cardiomyopathy, probably when LV systolic dysfunction intervenes, despite the possibility of extra-cardiac organ involvement in this disease (3,4).
The present observations also provide insight into the phenotypic and pathologic expression of LAMP2 cardiomyopathy. Despite youthful age, most of our patients with this metabolic storage disease showed particularly massive LV hypertrophy, and two had the most substantial hypertrophy reported in man (16–18). Histopathology of the hypertrophied LV showed a hybrid architecture with features of traditional HCM due to sarcomere protein mutations (i.e., myocyte disarray, intramural small vessel disease, and myocardial scarring including subepicardial distribution) (17,19–21), but also distinctive evidence of a storage process in which clusters of numerous vacuolated and vesicle-like myocytes were embedded in areas of replacement fibrosis. The material contents of these myocytes could not be defined on the available postmortem tissue. However, a murine model of LAMP2 cardiomyopathy has demonstrated the accumulation of non-specific, partially degraded biologic material (presumably lysosomal cellular debris), in large vacuoles indicative of impaired autophagy and associated with only a modest elevation in cardiac glycogen (22).
In conclusion, LAMP2 cardiomyopathy in young patients appears to be a lethal genetic disease. The clinical resemblance of LAMP2 to sarcomeric HCM underscores the necessity and power of timely genetic testing in young patients with substantial LV hypertrophy for early molecular identification of this myocardial storage disease characterized by adverse clinical course.
Acknowledgments
Grant study support provided by the Howard Hughes Medical Institute had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; and preparation, review, or approval of the manuscript.
Footnotes
The principal investigator (Dr. Maron) had full access to all the data in this study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Arad M, Maron BJ, Gorham JM, et al. Glycogen storage disease presenting as hypertrophic cardiomyopathy. N Eng J Med. 2005;352:362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, McMahon CJ, Smith LR, et al. Danon disease as a frequent cause of hypertrophic cardiomyopathy in children. Circulation. 2005;12:1612–1617. doi: 10.1161/CIRCULATIONAHA.105.546481. [DOI] [PubMed] [Google Scholar]
- 3.Charron P, Villard E, Sébillon P, et al. Danon’s disease as a cause of hypertrophic cardiomyopathy: a systemic survey. Heart. 2004;90:842–846. doi: 10.1136/hrt.2003.029504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sugie K, Yamamoto A, Murayama K, et al. Clinicopathological features of genetically confirmed Danon disease. Neurology. 2002;58:1773–1778. doi: 10.1212/wnl.58.12.1773. [DOI] [PubMed] [Google Scholar]
- 5.Taylor MRG, Ku L, Slavov D, et al. Danon disease presenting with dilated cardiomyopathy and a complex phenotype. J Hum Genet. 2007;52:830–835. doi: 10.1007/s10038-007-0184-8. [DOI] [PubMed] [Google Scholar]
- 6.Nishino I, Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 7.Fanin M, Nascimbeni AC, Fulizio L, et al. Generalized lysosome-associated membrane protein-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in Danon’s disease. Am J Pathol. 2006;168:1309–1320. doi: 10.2353/ajpath.2006.050646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–1257. doi: 10.1056/NEJM199804303381802. [DOI] [PubMed] [Google Scholar]
- 9.Maron BJ, Spirito P, Shen W-K, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405–412. doi: 10.1001/jama.298.4.405. [DOI] [PubMed] [Google Scholar]
- 10.Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation. 2006;114:216–225. doi: 10.1161/CIRCULATIONAHA.105.583500. [DOI] [PubMed] [Google Scholar]
- 11.Ackerman MJ, Van Driest SL, Ommen SR, et al. Prevalence and age dependence of malignant mutations in the beta-myosin heavy chain and troponin T genes in hypertrophic cardiomyopathy: a comprehensive outpatient perspective. J Am Coll Cardiol. 2002;39:2042–2048. doi: 10.1016/s0735-1097(02)01900-9. [DOI] [PubMed] [Google Scholar]
- 12.Van Driest SL, Ackerman MJ, Ommen SR, et al. Prevalence and severity of “benign” mutations in the beta-myosin heavy chain, cardiac troponin T, and alpha-tropomyosin genes in hypertrophic cardiomyopathy. Circulation. 2002;106:3085–3090. doi: 10.1161/01.cir.0000042675.59901.14. [DOI] [PubMed] [Google Scholar]
- 13.Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2003;42:1687–1713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 14.Bertini E, Donati MA, Broda P, et al. Phenotypic heterogeneity in two unrelated Danon patients associated with the LAMP-2 gene mutation. Neuropediatrics. 2005;36:309–13. doi: 10.1055/s-2005-872844. [DOI] [PubMed] [Google Scholar]
- 15.Echaniz-Laguna A, Mohr M, Epailly E, Nishino I, et al. Novel Lamp-2 Gene Mutation and Successful Treatment with Heart Transplantation in a Large Family with Danon Disease. Muscle Nerve. 2006;33:393–397. doi: 10.1002/mus.20471. [DOI] [PubMed] [Google Scholar]
- 16.Maron BJ, Gross BW, Stark SI. Extreme left ventricular hypertrophy. Circulation. 1995;92:2748. doi: 10.1161/01.cir.92.9.2748. [DOI] [PubMed] [Google Scholar]
- 17.Roberts CS, Roberts WC. Progress in Cardiology. Vol. 2. Lea and Febinger Publishers; Philadelphia: 1989. Morphologic features; pp. 3–32. [Google Scholar]
- 18.Roberts WC, Podolak MJ. The king of hearts: analysis of 23 patients with hearts weighing 1,000 grams or more. Am J Cardiol. 1985;55:485–94. doi: 10.1016/0002-9149(85)90399-6. [DOI] [PubMed] [Google Scholar]
- 19.Varnava AM, Elliott PM, Mahon N, Davies MJ, McKenna WJ. Relation between myocyte disarray and outcomes in hypertrophic cardiomyopathy. Am J Cardiol. 2001;88:275–279. doi: 10.1016/s0002-9149(01)01640-x. [DOI] [PubMed] [Google Scholar]
- 20.Maron BJ, Roberts WC. Quantitative analysis of cardiac muscle cell disorganization in the ventricular septum of patients with hypertrophic cardiomyopathy. Circulation. 1979;59:689–706. doi: 10.1161/01.cir.59.4.689. [DOI] [PubMed] [Google Scholar]
- 21.Maron BJ, Wolfson JK, Epstein SE, Roberts WC. Intramural (“small vessel”) coronary artery disease in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1986;8:545–557. doi: 10.1016/s0735-1097(86)80181-4. [DOI] [PubMed] [Google Scholar]
- 22.Alcalai R, Arad M, Wang L, et al. Lysosomal dysfunction and impaired autophagy causes cardiomyopathy and arrhythmias in LAMP2 knock-in mouse model. Submitted. [Google Scholar]