

Abstract
Piperine activates TRPV1 (transient receptor potential vanilloid type 1 receptor) receptors and modulates γ-aminobutyric acid type A receptors (GABAAR). We have synthesized a library of 76 piperine analogues and analyzed their effects on GABAAR by means of a two-microelectrode voltage-clamp technique. GABAAR were expressed in Xenopus laevis oocytes. Structure–activity relationships (SARs) were established to identify structural elements essential for efficiency and potency. Efficiency of piperine derivatives was significantly increased by exchanging the piperidine moiety with either N,N-dipropyl, N,N-diisopropyl, N,N-dibutyl, p-methylpiperidine, or N,N-bis(trifluoroethyl) groups. Potency was enhanced by replacing the piperidine moiety by N,N-dibutyl, N,N-diisobutyl, or N,N-bistrifluoroethyl groups. Linker modifications did not substantially enhance the effect on GABAAR. Compound 23 [(2E,4E)-5-(1,3-benzodioxol-5-yl)-N,N-dipropyl-2,4-pentadienamide] induced the strongest modulation of GABAA (maximal GABA-induced chloride current modulation (IGABA-max = 1673% ± 146%, EC50 = 51.7 ± 9.5 μM), while 25 [(2E,4E)-5-(1,3-benzodioxol-5-yl)-N,N-dibutyl-2,4-pentadienamide] displayed the highest potency (EC50 = 13.8 ± 1.8 μM, IGABA-max = 760% ± 47%). Compound 23 induced significantly stronger anxiolysis in mice than piperine and thus may serve as a starting point for developing novel GABAAR modulators.
Introduction
γ-Aminobutyric acid type A (GABAA) receptors are the major inhibitory neurotransmitter receptors in mammalian brain.1−3 GABAA receptors belong to the superfamily of Cys loop ligand-gated ion channels. Five receptor subunits form a central chloride-conducting pore.4−6 Nineteen genes encoding different subunits have been discovered in the human genome, comprising α1–6, β1–3, γ1–3, δ, ε, θ, π, and ρ1–3.7,8 Different subunit combinations may theoretically form a vast number of receptor subtypes with different pharmacological properties (see ref (9) for review). There is consensus that the most abundantly occurring receptor subtype is formed of two α1, two β2, and one γ2 subunits (α1β2γ2 receptor).10−12
Drugs that enhance chloride currents through GABAA receptors play an important role in the treatment of general anxiety, panic disorders, sleep disturbances, and epilepsy.13−17 The most widely used benzodiazepines induce, however, a variety of side effects including dependence, unwanted sedation, and amnesia, complicating their long-term use.18−20 Hence, there is high unmet medical need for GABAA receptor modulators lacking these unwanted effects.
Besides their modulation by clinically used drugs such as benzodiazepines, barbiturates, neurosteroids, and anesthetics,3,9,15,21−27 GABAA receptors are modulated by numerous natural products that may provide lead structures for drug development.28−30
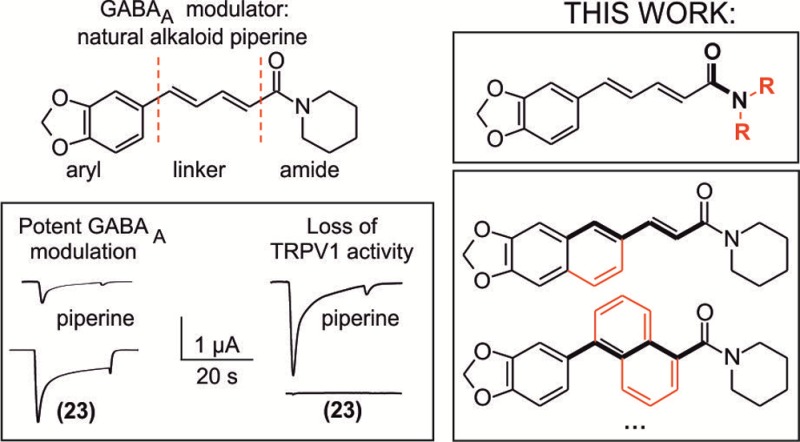
In this context, we31 and others32 have reported that piperine (1-piperoylpiperidine), the pungent component of several pepper species and activator of transient receptor potential vanilloid type 1 receptor (TRPV1),33 also modulates GABAA receptors. We could establish that replacing the piperidine ring of piperine by a N,N-diisobutyl residue, resulting in (2E,4E)-5-(1,3-benzodioxol-5-yl))-N,N-diisobutyl-2,4-pentadienamide (SCT-66;34 referred to as 24 in this work), diminishes the interaction with TRPV1 receptors. Furthermore, 24 enhanced chloride currents through GABAA receptors more potently and more efficiently than piperine and displayed, concordantly, a stronger anxiolytic action.34
Based on these findings, a library of piperine derivatives was synthesized and investigated with respect to modulation of α1β2γ2S GABAA receptors expressed in Xenopus laevis oocytes. Within this study we emphasized modifications at the amide functionality and on the diene motif within piperine in order to enhance the modulatory potential of analogue structures. Their structure–activity relationship on GABAA receptors was analyzed by establishing binary classification models.
Results and Discussion
Modification of Amide Nitrogen
Starting with piperine as lead structure from prior biological assessment, the molecule can be structurally divided into three parts: the 1,3-benzodioxole or aromatic function, the olefinic linker region comprising four carbon atoms, and the amide function natively constituted by a piperidine ring (Figure 1). Within this study, we investigated modifications at the amide group as well as in the linker region.
Figure 1.

Piperine molecule can be structurally divided into three moieties: the 1,3-benzodioxole or aromatic function, the linker region comprising four carbon atoms, and the amide function natively constituted by a piperidine ring.
Modifications at the amide function were implemented in a straightforward fashion (Scheme 1). Piperic acid amides (1–16, 20–23, and 25–43) were synthesized by treating piperic acid chloride with the corresponding amine in the presence of triethylamine in tetrahydrofuran (THF). Compounds 17 and 18 were prepared in the same way from benzodioxolyl acryloyl chloride. Treatment of piperine with Lawesson’s reagent35 gave thioamide 44. Reduction of the carbonyl group of piperine with lithium aluminum hydride afforded unsaturated amine 45 (Scheme 2).
Scheme 1. Structural Modifications of the Piperine Scaffold.

Scheme 2. Synthesis of Piperine Derivatives with Modification of the Amide Function and Truncated Alkene Spacer.

Conditions: (a) Amine (3.5 equiv), dry THF, rt. (b) Lawesson’s reagent, dry THF, rt. (c) LiAlH4, THF, rt.
First, we studied the effects of systematic modifications of the amide nitrogen on IGABA modulation through α1β2γ2S receptors. As illustrated in Figure 2A,B, 10 compounds (22, 23, 25, 28, 33, 34, 35, 38, and 43) at 100 μM induced stronger IGABA modulation than piperine (≥220%)31 and were classified as highly active. IGABA potentiation of these compounds ranged between 294% ± 66% (28) and 1091% ± 257% (23, see Table 1). At this concentration, three derivatives (17, 30, and 39) were less efficient, while the other compounds did not significantly modulate IGABA (see Figure 2A,B and Table 1).
Figure 2.

(A, B) Modulation of chloride currents through GABAA receptors composed of α1, β2, and γ2S subunits by 100 μM piperine and the indicated derivatives (dotted line indicates cutoff for highly active compounds). (C, D) Concentration-dependent IGABA (EC3–7) enhancement through α1β2γ2S GABAA receptors, (C) for 22 (▲), 23 (●), 25 (◆), and 35 (■), ranked by efficiency, and (D) for 25 (○) and 43 (●), ranked by potency, compared to piperine (dotted line). (E, F) Representative IGABA modulated by (E) 23 and (F) 25. Data represent mean ± SEM from at least three oocytes and two oocyte batches. Asterisks indicate statistically significant differences from zero: *p < 0.05, **p < 0.01. Data for piperine were taken from ref (31).
Table 1. IGABA Modulation through α1β2γ2S GABAA Receptors by Indicated Compounds (100 μM)a.
| compd | modulation of IGABA (%) | n | compd | modulation of IGABA (%) | n |
|---|---|---|---|---|---|
| 1 | 0 ± 0 | 3 | 25 | 506 ± 74** | 3 |
| 2 | 10 ± 0 | 3 | 26 | 0 ± 0 | 3 |
| 3 | 5 ± 5 | 3 | 27 | 13 ± 13 | 3 |
| 4 | –15 ± 9 | 3 | 28 | 294 ± 66* | 3 |
| 5 | –2 ± 2 | 3 | 29 | 0 ± 0 | 3 |
| 6 | –7 ± 3 | 3 | 30 | 113 ± 17* | 3 |
| 7 | 8 ± 7 | 3 | 31 | –20 ± 20 | 3 |
| 8 | –8 ± 6 | 3 | 32 | –5 ± 5 | 3 |
| 9 | 0 ± 0 | 3 | 33 | 359 ± 50* | 3 |
| 10 | 1 ± 7 | 3 | 34 | 439 ± 31* | 3 |
| 11 | 51 ± 11 | 3 | 35 | 568 ± 54 | 3 |
| 12 | –6 ± 6 | 3 | 36 | 33 ± 9 | 3 |
| 13 | 33 ± 17 | 3 | 37 | 26 ± 14 | 3 |
| 14 | 0 ± 0 | 3 | 38 | 218 ± 43* | 3 |
| 15 | –1 ± 17 | 3 | 39 | 183 ± 20** | 3 |
| 16 | –6 ± 6 | 3 | 40 | 12 ± 8 | 3 |
| 17 | 79 ± 8* | 3 | 41 | 5 ± 5 | 3 |
| 18 | 66 ± 30 | 3 | 42 | 48 ± 12 | 3 |
| 20 | 61 ± 28 | 3 | 43 | 445 ± 74** | 3 |
| 21 | 258 ± 28 | 3 | 44 | 17 ± 17 | 3 |
| 22 | 986 ± 244* | 3 | 45 | –16 ± 14 | 3 |
| 23 | 1091 ± 257* | 3 |
All data are given as mean ± SEM. Asterisks indicate statistically significant differences from zero: *p < 0.05, **p < 0.01.
Five derivatives of this first set (22, 23, 25, 35, and 43) with amide modifications enhanced IGABA through α1β2γ2S GABAA receptors with higher efficiency (IGABA-max: 23 > 22 > 25 > 35) and/or higher potency (EC50: 25 < 43) than piperine (Figure 2C,D and Table 2).
Table 2. IGABA Modulation through α1β2γ2S GABAA Receptors by Indicated Compounds (100 μM)a.
| compd | modulation of IGABA (%) | n | compd | modulation of IGABA (%) | n |
|---|---|---|---|---|---|
| 46 | 42 ± 1** | 3 | 62 | 13 ± 2 | 3 |
| 47 | 364 ± 55** | 3 | 63 | 12 ± 4 | 3 |
| 48 | 49 ± 7 | 3 | 64 | 4 ± 4 | 3 |
| 49 | 30 ± 15 | 3 | 65 | 105 ± 18 | 3 |
| 50 | 178 ± 32* | 3 | 66 | 67 ± 23 | 3 |
| 51 | 280 ± 52** | 3 | 67 | 18 ± 9 | 3 |
| 52 | 63 ± 12* | 3 | 68 | –1 ± 12 | 3 |
| 53 | 298 ± 31** | 3 | 69 | 74 ± 1* | 3 |
| 54 | 34 ± 8 | 3 | 70 | 32 ± 12 | 3 |
| 55 | 79 ± 24 | 3 | 71 | 32 ± 10 | 3 |
| 56 | 114 ± 11 | 3 | 72 | 334 ± 23** | 3 |
| 57 | 15 ± 15 | 3 | 73 | 514 ± 76** | 3 |
| 58 | –5 ± 12 | 3 | 74 | 60 ± 17 | 2 |
| 59 | 134 ± 39 | 3 | 75 | 58 ± 29* | 3 |
| 60 | 51 ± 21 | 3 | 76 | 122 ± 26* | 3 |
| 61 | 11 ± 2 | 3 | 77 | 138 ± 29* | 3 |
All data are given as mean ± SEM. Asterisks indicate significant differences from zero: *p < 0.05, **p < 0.01.
N,N-Dipropyl-Substituted Compounds 22 And 23 Display the Highest Efficiency
Compounds 22 (N,N-dipropyl) and 23 (N,N-diisopropyl) modulated IGABA most efficiently (IGABA-max for 22, 1581% ± 74%; IGABA-max for 23, 1673% ± 146%; IGABA-max for piperine, 302% ± 27%). Compounds 35 (IGABA-max 733% ± 60%) and 25 (IGABA-max 760% ± 47%) were less efficient, underscoring the important role of a noncyclic disubstituted amide motif (Figure 2C).
N,N-Dibutyl-Substituted Compound 25 Displays the Highest Potency
Figure 2D illustrates IGABA modulation by the most potent N-substituted piperine derivative (EC50 for 25, 13.8 ± 1.8 μM < EC50 for 43, 23.1 ± 3.3 μM < EC50 for piperine, 52.4 ± 9.4 μM31). Based on the modifications at the amide group, it can be concluded that installation of noncyclic substituents bearing 3–4 carbons each at the tertiary amide improves both efficacy and potency of the analogue compounds.
Rigidification of the Linker Region Has No Significant Effect on IGABA Modulation
The influence of linker rigidity on IGABA modulation was studied by means of a library comprising 32 linker derivatives. According to Zaugg et al.31 and Pedersen et al.,32 a carbon chain containing at least four carbons, a conjugated double bond adjacent to the amide group, and a bulky amine moiety seem to facilitate efficient receptor binding and/or IGABA modulation.
Based on previous reports by Zaugg et al.,31 we hypothesized that rigidification of the linker part of the structure may beneficially affect biological activity.31 This assessment was based in particular on a decrease in modulatory capacity when partially saturated linkers were installed or when structural flexibility was increased by extending the linker length.
Three major structural modifications were envisaged (Scheme 1). (i) Replacement of the linker by an aryl ring (phenyl, heteroaryl, naphthyl): in this arrangement, both alkene groups of the diene system of the linker would be integrated into the rigid aromatic core. (ii) Integration of one linker double bond into a naphthyl ring: this compound class was expected to render more flexibility but still adopt a more rigidified system compared to the piperine diene structure; moreover, arrangement should allow for different angles of the aryl core relative to the amide anchoring group depending on the substitution site at the naphthyl system. (iii) “Ring closure” of the diene motif with the aryl part, consequently generating a carboxylate-substituted naphthyl lead structure: in this arrangement the double bond adopts a bent geometry, and again different angles of the aryl and amide parts can be obtained depending on the substitution site.
For the synthesis of aryl-bridged compounds, two different methods were utilized. For a number of products (46, 49, 53, and 58–64) (Scheme 3), the corresponding bromo-substituted aromatic carboxylic acids were reacted with 3,4-(methylenedioxy)phenylboronic acid under Suzuki–Miyaura cross-coupling conditions.36 The resulting bis(aryl)carboxylic acids were converted to the final amide products via the corresponding acid chloride intermediates. Alternatively, the corresponding bromobenzoic acid amides were prepared prior to the coupling step. Subsequent Suzuki–Miyaura coupling with 3,4-(methylenedioxy)phenylboronic acid afforded the final products 47, 48, 50–52, and 54–57 (Scheme 3).
Scheme 3. Synthesis of Piperine Analogues Containing an Aryl Spacer.

Conditions: (d) Boronic acid, Pd(PPh3)4 2 mol %, K2CO3, DME/EtOH/water, 140 °C, mw, 1 h. (e) Either (COCl)2, cat. DMF, and DCM or EDCI·HCl, HOBt, and dry DCM, followed by amine.
In order to access the 5-position of the naphtho[2,3-d]dioxole core, naphtho[2,3-d]dioxol-5-ol triflate was chosen as a precursor.37 Heck coupling38 employing methyl acrylate afforded 65a, which gave acrylic acid 65b after cleavage of the methyl ester (Scheme 4). Amide formation yielded the final products 65–67.
Scheme 4. Synthesis of Piperine Analogues with (Partial) Integration of the Spacer Motif into an Aryl Core.

Conditions: (e) Either (COCl)2, cat. DMF, and DCM or EDCI·HCl, HOBt, and dry DCM, followed by amine. (f) CO, Pd(OAc)2, dppp, Hünig’s base, DMF/water, 70 °C. (g) Methyl acrylate, Pd(OAc)2 5 mol %, phenanthroline monohydrate 5.5 mol %, NEt3, dry DMF. (h) LiOH, THF/water, rt. (i) B2pin2, [Ir(OMe)cod]2 1.5 mol %, 4,4′-di-tert-butyl-2,2′-bipyridine 3 mol %, cyclohexane, reflux. (j) CuBr2, MeOH/water. (k) Methyl acrylate, Pd(OAc)2 3 mol %, (o-tolyl)3P 6 mol %, NEt3, 80 °C. (l) Methyl acrylate, NaI, dry DMF, 90°C. (m) DDQ, benzene, 80 °C.
Iridium-catalyzed direct borylation39 of naphtho[2,3-d]dioxole allowed direct access to the 6-position of the naphtho[2,3-d]dioxole core. Boronic acid ester 68a obtained in this step was converted into the corresponding bromide4068b and coupled under standard Heck cross-coupling conditions to afford acrylate 68c (Scheme 4). The methyl ester was hydrolyzed, and acid 68d was converted into products 68–70 (Scheme 4).
Naphthodioxol-5-ol triflate was also used in a palladium-catalyzed hydroxycarbonylation reaction41 to provide access to carboxylic acid 71a, which was further converted to products 71–74 (Scheme 4). A different route was chosen to synthesize derivatives of naphtodioxole-6-carboxylic acid: By treating bis(bromomethyl)benzodioxole with iodide, a highly reactive diene was generated in situ,42 which was intercepted with methyl acrylate in a Diels–Alder reaction. The resulting decaline derivative 75a was oxidized with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to afford naphthaline 75b. Saponification of the methyl ester gave carboxylic acid 75c, which was further converted to final products 75–77 (Scheme 4).
At 100 μM, five compounds (47, 51, 53, 72, and 73) modulated IGABA more efficiently than piperine (see Figure 3A,B and Table 2). IGABA potentiation ranged from 280% ± 52% (51) to 514% ± 76% (72). IGABA enhancement by 46, 50, 52, 69, 75, 76, and 77 was less pronounced compared to piperine [IGABA potentiation range 42% ± 1% (46) to 178% ± 30% (50)]. None of the other derivatives induced significant IGABA enhancement (see Figure 3A,B and Table 2).
Figure 3.

(A, B) Modulation of chloride currents through GABAA receptors composed of α1, β2, and γ2S subunits by 100 μM piperine and the indicated derivatives (dotted line indicates cutoff for highly active compounds). (C, D) Concentration-dependent IGABA (EC3–7) enhancement through α1β2γ2S GABAA receptors: (C) by 47 (■), 53 (▲), and 72 (●), ranked by efficiency, and (D) by 56 (▲) and 73 (●), ranked by potency, compared to piperine (dotted line). (E, F) Representative IGABA modulated by (E) 72 and (F) 73. Data represent mean ± SEM from at least three oocytes and two oocyte batches. Asterisks indicate statistically significant differences from zero: *p < 0.05, **p < 0.01. Data for piperine were taken from ref (31).
Concentration–response curves of IGABA modulation by linker-modified derivatives 47, 53, 56, 72, and 73 are illustrated in Figure 3C,D. The combination of N,N-dipropyl amide from the series 1–45 with the two most efficient modifications in the linker region (1,4-phenylene and naphthodioxol-5-yl) resulted in 47 (IGABA-max = 603% ± 87%, EC50 = 70.8 ± 21.1 μM), 72 (IGABA-max = 706% ± 58%, EC50 = 102.0 ± 11.2 μM), and 73 (IGABA-max = 480% ± 85%, EC50 = 31.8 ± 5.3 μM) inducing stronger IGABA enhancement than piperine (Table 3). These findings underscore the general validity of favorable N,N-functionalization also for this series of linker-modified compounds. However, none of the modifications led to compounds with a higher activity than the initial parent compound 23.
Table 3. Efficiency and Potency of Further Characterized Piperine Derivatives and Piperinea.
| compd | IGABA-max (%) | EC50 (μM) | nH | n |
|---|---|---|---|---|
| piperine | 302 ± 27 | 52.4 ± 9.3 | 1.5 ± 0.2 | 3 |
| 22 | 1581 ± 74** | 86.7 ± 13.9 | 2.3 ± 0.2 | 6 |
| 23 | 1673 ± 146** | 51.7 ± 9.5 | 3.1 ± 0.8 | 6 |
| 25 | 760 ± 47** | 13.8 ± 1.8** | 1.8 ± 0.1 | 6 |
| 35 | 733 ± 60** | 67.7 ± 11.0 | 1.9 ± 0.3 | 6 |
| 43 | 505 ± 24** | 23.1 ± 3.3* | 1.6 ± 0.2 | 6 |
| 47 | 603 ± 87* | 70.8 ± 21.1 | 1.2 ± 0.2 | 3 |
| 53 | 388 ± 64 | 55.3 ± 17.6 | 1.5 ± 0.2 | 3 |
| 56 | 165 ± 4** | 36.8 ± 2.0 | 1.2 ± 0.0 | 3 |
| 72 | 706 ± 58** | 102.0 ± 11.2 | 1.9 ± 0.2 | 5 |
| 73 | 480 ± 85 | 31.8 ± 5.3 | 2.7 ± 0.2 | 6 |
From ref (31), including number of experiments n. Asterisks indicate significant differences from piperine: *p < 0.05; **p < 0.01.
Selectivity Profile
Previously, we have shown that 24(34) [(2E,4E)-5-(1,3-benzodioxol-5-yl)-N,N-diisobutyl-2,4-pentadienamide] similarly modulates GABAA receptors containing either β2/3 or β1 subunits, in contrast to the preferential modulation of β2/3 receptors by piperine.34
In the present study, analysis of the most efficient piperine derivative (23) revealed that GABAA receptors composed of α1β2γ2S (IGABA-max = 1673% ± 146%) and α5β2γ2S (IGABA-max = 1624% ± 156%) subunits were more efficiently modulated than receptors containing α3β2γ2S subunits (IGABA-max = 1284.6% ± 142%; see Table 4). Significantly weaker potentiation was observed for receptors composed of α2β2γ2S (IGABA-max = 980% ± 129%) and α4β2γ2S subunits (IGABA-max = 1316% ± 55%). Replacing the β2 subunits by β3 subunits did not significantly alter the strength of IGABA potentiation, whereas modulation of GABAA receptors containing β1 subunits was significantly less pronounced (IGABA-max = 1157% ± 69%; p < 0.05). In comparison with α1β2γ2S receptors, 23 displayed an increased potency for α2β2γ2S receptors, followed by α1β3γ2S, α3β2γ2S, and α4β2γ2S receptors. EC50 values for the other receptor subtypes did not differ from those for α1β2γ2S (see Figure 4A,B and Tables 4 and 5).
Table 4. Efficiency and Potency of 23 and 25 on GABAA Receptors of Different Subunit Compositionsa.
| receptor subtype | IGABA,max (%) | EC50 (μM) | nH | n |
|---|---|---|---|---|
| Compound 23 | ||||
| α1β1γ2S | 1157 ± 69* | 57.5 ± 7.3 | 1.8 ± 0.1 | 5 |
| α1β2γ2S | 1673 ± 146 | 51.7 ± 9.5 | 3.1 ± 0.8 | 6 |
| α1β3γ2S | 1240 ± 128 | 34.7 ± 5.7 | 1.9 ± 0.2 | 5 |
| α2β2γ2S | 980 ± 129** | 26.4 ± 6.6 | 1.9 ± 0.4 | 6 |
| α3β2γ2S | 1285 ± 142 | 36.6 ± 7.2 | 1.9 ± 0.3 | 5 |
| α4β2γ2S | 1316 ± 55* | 34.7 ± 3.8 | 1.7 ± 0.1 | 7 |
| α5β2γ2S | 1624 ± 156 | 61.9 ± 10.4 | 1.4 ± 0.1 | 7 |
| Compound 25 | ||||
| α1β1γ2S | 152 ± 30** | 15.9 ± 4.9 | 1.3 ± 0.6 | 5 |
| α1β2γ2S | 760 ± 47 | 13.8 ± 1.8 | 1.8 ± 0.1 | 8 |
| α1β3γ2S | 587 ± 8** | 29.5 ± 2.9** | 1.5 ± 0.1 | 4 |
| α2β2γ2S | 512 ± 26** | 14.8 ± 1.9 | 2.2 ± 0.3 | 4 |
| α3β2γ2S | 617 ± 42* | 16.0 ± 2.7 | 1.8 ± 0.1 | 6 |
| α4β2γ2S | 419 ± 73** | 56.7 ± 21.0 | 1.3 ± 0.3 | 4 |
| α5β2γ2S | 387 ± 20** | 17.2 ± 1.4 | 1.7 ± 0.2 | 5 |
Asterisks indicate significant differences from α1β2γ2S receptor subtype as follows: *p < 0.05; **p < 0.01.
Figure 4.

Analysis of subunit preferential IGABA enhancement by (A, B) the most efficient (23) and (C, D) the most potent (25) piperine derivatives. (E, F) Representative IGABA through seven GABAA receptor subtypes by 23 at 100 μM. Data represent mean ± SEM from at least three oocytes and two oocyte batches.
Table 5. Comparison of Potency and Efficiency of 23 for GABAA Receptors of Different Subunit Compositionsa.
| α1β2γ2S |
α1β1γ2S |
α1β3γ2S |
α2β2γ2S |
α3β2γ2S |
α4β2γ2S |
α5β2γ2S |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P | E | P | E | P | E | P | E | P | E | P | E | P | E | |
| α1β2γ2S | * | ** | * | |||||||||||
| α1β1γ2S | ** | * | * | * | ||||||||||
| α1β3γ2S | * | |||||||||||||
| α2β2γ2S | ** | * | ** | |||||||||||
| α3β2γ2S | ||||||||||||||
| α4β2γ2S | * | |||||||||||||
| α5β2γ2S | ||||||||||||||
Potency (P), expressed as EC50, and efficiency (E), expressed as IGABA-max, are compared. Asterisks indicate statistical significance as follows: *p < 0.05, **p < 0.01.
Like 23, derivative 25 most efficiently enhanced IGABA through GABAA receptors composed of α1β2γ2S subunits (IGABA-max = 760% ± 47%; see Table 4 and Figure 4C,D). Replacing the α1 subunit by α2/3/4/5 subunits significantly reduced IGABA potentiation by 25 (see Table 4 and Figure 4C). Notably, 25 displayed a more pronounced β2/3 preference compared to piperine or 23 [inducing a 3.9-fold (α1β3γ2S) to 5-fold (α1β2γ2S) stronger IGABA enhancement compared to α1β1γ2S receptors]. Compound 25 showed comparable potency for most of the tested receptor subtypes ranging from 13.8 ± 1.8 μM to 56.7 ± 21.0 μM; significantly higher EC50 values were estimated for α1β3γ2S receptors (see Tables 4 and 6).
Table 6. Comparison of Potency and Efficiency of 25 for GABAA Receptors of Different Subunit Compositionsa.
| α1β2γ2S |
α1β1γ2S |
α1β3γ2S |
α2β2γ2S |
α3β2γ2S |
α4β2γ2S |
α5β2γ2S |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P | E | P | E | P | E | P | E | P | E | P | E | P | E | |
| α1β2γ2S | ** | ** | ** | ** | * | ** | ** | |||||||
| α1β1γ2S | * | ** | ** | ** | * | ** | ||||||||
| α1β3γ2S | ** | * | ** | ** | ** | |||||||||
| α2β2γ2S | ** | |||||||||||||
| α3β2γ2S | * | ** | ||||||||||||
| α4β2γ2S | ||||||||||||||
| α5β2γ2S | ||||||||||||||
Potency (P), expressed as EC50, and efficiency (E), expressed as IGABA-max, are compared. Asterisks indicate statistical significance as follows: *p < 0.05, **p < 0.01.
These data support the previous observation that when the cyclic piperidine residue is replaced by N,N-dialkyl moieties such as N,N-dipropyl (23), N,N-diisopropyl (24),34 or N,N-dibutyl (25), efficiency and potency can be significantly enhanced. However, while 24(34) lost its ability to distinguish between the β-subunit isoforms, preferential modulation of β2/3 receptors by 23 was comparable to piperine, and it was even more pronounced for 25 (see Figure 4 B,D and Tables 4–6). Thus, 23 and 25 display—compared to classical GABAA receptor modulators such as benzodiazepines—a distinct subunit selectivity profile. Unlike benzodiazepines, 23 and 25 also modulate GABAA receptors containing α4 subunits with high efficiency and are not dependent on the presence of a γ2S subunit (data not shown). Whether this subunit selectivity profile has any pharmacological relevance has to be clarified in further studies.
Structure–Activity Relationships: General Trends
When the whole data set was analyzed, several distinct SARs could be deduced. They are mostly related to the substitution pattern at the amide nitrogen atom, as this was the main point of variation in the data set. Thus, concerning N,N-dialkyl-substituted amides, there is evidence that IGABA enhancement is related in a nonlinear (parabolic) function to the number of carbon atoms (Figure 5), with the optimum being dipropyl (23). This type of parabolic relationship is quite common, especially when it refers to a parameter that is linked to lipophilicity of the compounds and activity data obtained in a cellular assay. It has, for example, also been observed for a series of capsaicin analogues with respect to their TRPV1 activation.43 Interestingly, whether the alkyl chains are linear or branched does not reverse the order: 20 (dimethyl) < 21 (diethyl) < 23 (dipropyl)/22 (diisopropyl) < 25 (dibutyl)/2434 (diisobutyl) < 26 (dihexyl)/27 (dicyclohexyl). With respect to compounds where the amide nitrogen atom is part of a ring, methylpiperines 33, 34, and 35 induced the strongest IGABA potentiation, followed by azepane amide 28 and piperine. Interestingly, the dimethylpiperine 38 was comparably active to the parent compound. Introduction of a second heteroatom into the ring led to almost complete loss of IGABA enhancement (N-alkylpiperazine amides 31, 32, 40, 41, and 42 and morpholine amide 29).
Figure 5.

Relation between log(potentiation of IGABA) of dialkyl-substituted piperine derivatives at the amide nitrogen and number of carbon atoms at this region. Data for 24* were taken from ref (34).
Replacement of the tertiary nitrogen atom for a secondary one, irrespective of alkyl or aryl substitution, led to a complete loss of activity (aryl-substituted N, 1–3, 5–7, 9, and 10; alkyl-substituted N, 4, 8, and 11–16). Reducing the H-bond acceptor strength of the amide by synthesizing the respective thioamide (44) abolished the modulatory activity. Reduction of the amide to the analogous amine changed the profile of the compound from potentiation (piperine at 100 μM, 226% ± 26%)31 to inactive (45 at 100 μM, −16% ± 14%; Table 1).
With respect to the linker region, shortening the distance by removing one vinylene unit significantly reduced IGABA enhancement (piperine vs 17 and 22 vs 18). All the other modifications, such as rigidification by inserting benzene, thiophene, or naphthalene moieties, reduced IGABA potentiation by at least a factor of 5 compared to 23. Interestingly, the modulatory activity did not seem to be related to distance of pharmacophoric substructures, such as the benzodioxole and the amide moiety. For naphthalene analogues 72 and 65, an increase in distance led to a decrease of activity, whereas in the case of 22 and 18, a decrease of distance led to a decrease of activity. Comparing 23 and 70, which show identical distance of these two moieties, 70 completely lacks activity (32% ± 12%, Table 1). In conclusion, the best compounds achieved in terms of efficiency were the piperine analogues 22 and 23.
Computational Analysis
In order to rationalize the trends observed in the SAR with respect to physicochemical properties and chemical substructures, we explored the possibility to apply quantitative structure–activity relationship (QSAR) methods. As IGABA potentiation does not allow classical QSAR analysis, binary classification models were built from five methods and three descriptor sets. For these studies, all 76 piperine derivatives described above were employed. Sixteen compounds showing ≥200% IGABA potentiation were assigned to an active class, since they were at least as active as the lead compound piperine. The remaining 60 ligands were assigned to an inactive class. Classification methods comprised instance-based classifier (IBk), J48 decision tree (J48), naïve-Bayes classifier (NB), random forest (RF), and support vector machine (SMO) implemented in the software package WEKA.44 The software package Molecular Operating Environment (MOE) was used for calculation of 2D descriptors and fingerprints. The three descriptor sets used comprised six 2D descriptors obtained after applying a feature selection algorithm on the whole panel of 125 2D MOE descriptors (6D), 11 physical chemical properties (PHYSCHEM), and MACCS fingerprints (MACCS).
The statistical parameters obtained for the 15 best classification models are listed in Table 7. Most of the models possess reliable quality (except models 11 and 13); that is, values of the Matthews correlation coefficient (MCC) are higher than 0.4 and total accuracy varies from 0.7 to 0.9.
Table 7. Statistical Parameters of the 15 Best Models Obtained after 10-Fold Cross-Validation.
| model | classification method | TP, TN, FP, FNa | sensitivity | specificity | accuracy | MCC, ROC |
|---|---|---|---|---|---|---|
| Descriptor Set 6D | ||||||
| 1 | IBk | 12, 52, 8, 5 | 0.706 | 0.867 | 0.831 | 0.542, 0.825 |
| 2 | J48 | 15, 46, 14, 2 | 0.882 | 0.767 | 0.792 | 0.556, 0.818 |
| 3 | NB | 16, 49, 11, 1 | 0.941 | 0.817 | 0.844 | 0.659, 0.831 |
| 4 | RF | 13, 52, 8, 4 | 0.765 | 0.867 | 0.844 | 0.588, 0.838 |
| 5 | SMO | 16, 39, 21, 1 | 0.941 | 0.650 | 0.714 | 0.491, 0.796 |
| Descriptor Set PHYSCHEM | ||||||
| 6 | IBk | 10, 52, 8, 7 | 0.588 | 0.867 | 0.805 | 0.446, 0.749 |
| 7 | J48 | 15, 46, 14, 2 | 0.882 | 0.767 | 0.792 | 0.556, 0.828 |
| 8 | NB | 15, 40, 20, 2 | 0.882 | 0.667 | 0.714 | 0.457, 0.828 |
| 9 | RF | 15, 46, 14, 2 | 0.882 | 0.767 | 0.792 | 0.556, 0.811 |
| 10 | SMO | 15, 36, 24, 2 | 0.882 | 0.600 | 0.662 | 0.400, 0.741 |
| Descriptor Set MACCS | ||||||
| 11 | IBk | 9, 45, 15, 8 | 0.529 | 0.750 | 0.701 | 0.250, 0.619 |
| 12 | J48 | 12, 48, 12, 5 | 0.706 | 0.800 | 0.779 | 0.453, 0.797 |
| 13 | NB | 12, 42, 18, 5 | 0.706 | 0.700 | 0.701 | 0.345, 0.713 |
| 14 | RF | 13, 43, 17, 4 | 0.765 | 0.717 | 0.727 | 0.409, 0.730 |
| 15 | SMO | 10, 56, 4, 7 | 0.588 | 0.933 | 0.857 | 0.561, 0.761 |
TP = true positive, TN = true negative, FP = false positive, FN = false negative.
Models 3 and 4, although possessing the best statistical performance parameters, are not discussed further, as they are difficult to interpret. Instead, models 7 and 12 are discussed in more detail, because these models (i) show almost equal performance, (ii) were built using descriptors of physical chemical properties and MACCS fingerprints, (iii) provide clear separation between active and inactive instances, and (iv) allow us to trace back the decisive chemical and structural descriptors for the data set.
The decision tree obtained in model 7 with PHYSCHEM descriptors (Figure 6) uses as a first criterion for separation of active and inactive piperine derivatives: the topological polar surface area. By applying a threshold of 39, 25 inactive ligands exhibiting polar substituents at the amide nitrogen were filtered out. These include compounds 1–16 with monosubstituted amide function and compounds 29, 31, 32, 36, 37, 40–42, and 44 containing several heteroatoms (e.g., OH groups or an additional nitrogen as in piperazines or both). Thus, application of a single filter decreased the number of inactive ligands in the data set almost by half, from 60 to 35 compounds. In the next branch of the decision tree, 10 compounds with less than four rotatable bonds were excluded from the data set. These included highly rigid piperine derivatives with linker regions modified to either a single double bond (17) or to an aromatic system (46, 50, 54, 58, 62, 65, 68, 71, and 75). Furthermore, 11 compounds with high lipophilicity (log P > 5.2) were filtered out: 26 and 27 with n-hexyl and cyclohexyl sustituents at the amide nitrogen, as well as 48, 52, 56, 60, 64, 67, 70, 77, and 63, which have dibutyl and dipropyl substituents in the same region. The fact that the top-ranked compounds are either N,N-dipropyl-, N,N-dibutyl-, or N,N-diisobutyl-substituted is reflected in the next leaf, which assigns five compounds (23, 24,3425, 43, and 73) with more than seven rotatable bonds to the active class. The last two branches of the decision tree filter out compounds on the basis of their molecular weight and refractivity.
Figure 6.

Decision tree obtained for the data set of 76 piperine derivatives with PHYSCHEM descriptor set.
The decision tree obtained for model 12 with MACCS fingerprints (Figure 7) is fully in line with the one based on the PHYSCHEM descriptor set. The first filtering criterion was presence or absence of an NH group. It filtered 21 derivatives (1–16, 31, 32, 40, 42, and 45), most of which were those showing high polar surface area (TPSA). The next branching filter was presence of a sulfur atom, which removes six inactive ligands (30, 44, and 58–61) from the data set. The next leaf separates compounds that do not have a six-membered ring as in piperidinyl, cyclohexyl, and morpholinyl, which led to seven correctly classified active ligands (21–23, 24,3425, 28, and 43) and three missclassified inactives (18, 20, and 26). This criterion is in line with the filter “b_rotN > 7” for active compounds in the PHYSCHEM model.
Figure 7.

Decision tree obtained for the data set of 76 piperine derivatives with MACCS fingerprints.
To summarize, active piperine analogues are mainly characterized by a topological polar surface smaller than 39, have at least three rotatable bonds (better more than 7), and show a log P value smaller than 5.2.
Compounds 25 and 23 Induce Anxiolysis in Mice
Activation of TRPV1 by piperine and its derivatives may cause unwanted side effects, including changes in pain sensation and body temperature and induction of fear that would interfere with GABAA-mediated effects45,46 (for review see ref (47)). In order to rule out potential activation of TRPV1, selected compounds were studied in X. laevis oocytes for interaction with TRPV1 prior to in vivo characterization. The most potent (25) and most efficient (23) piperine analogues (Table 3, Figure 2C,D) did not activate TRPV1 expressed in Xenopus oocytes (upon application of 100 μM, data not shown). Both compounds were further characterized concerning their anxiolytic activity (see also ref (34)).
As illustrated in Figure 8A, male C57BL/6N mice treated with 23 at doses ≥0.3 mg/kg body weight spent significantly more time in the open arms (OA) of the elevated plus maze (EPM) test compared to a saline-treated control group (control, 28.7% ± 2.7% for n = 41; 23 at 0.3 mg/kg, 45.6% ± 3.2% for n = 17; p < 0.01). This effect was dose-dependent and reached its maximum at a dose of 3 mg/kg body weight, indicating strong anxiolytic effects of 23. Similarly, mice treated with 25 also spent significantly more time in the OA of the EPM test at doses ≥0.3 mg/kg body weight compared to saline-treated control littermates (control, 28.7% ± 2.7% for n = 41; 25 at 0.3 mg/kg, 39.8% ± 4.1% for n = 23; p < 0.05; Figure 8B). The anxiolytic effect of 25 reaching its maximum at a dose of 3 mg/kg body weight (25 at 3 mg/kg, 43.9% ± 4.3% for n = 12), however, was less pronounced compared to 23.
Figure 8.

Compounds 23 and 25 display anxiolytic effects in the EPM test and little sedation in the OF test. Bars indicate time spent in open arms (OA) as a percentage of the total time 30 min after ip application of the indicated dose (in milligrams per kilogram of body weight) of (A) 23 and (B) 25 and the total ambulation after application of (C) 23 and (D) 25. White bars illustrate the behavior of control mice. Bars represent means ± SEM from at least eight different mice. Asterisks indicate statistically significant differences to control *p < 0.05, **p < 0.01 [analysis of variance (ANOVA) with Bonferroni]. Shaded bars for the behavioral effects of piperine are taken from ref (34). Behavioral experiments comparing the sedative and anxiolytic potential of piperine, 23, and 25 have been conducted in parallel.
Application of doses ≥10 mg/kg of 23 or 25 did not further increase the anxiolytic effect in the EPM, which is presumably due to the concomitant occurring/developing of reduced locomotor activity (see Figure 8C,D for sedative effects in the open field test). Compared to piperine and the previously studied 24(34) (Figure 8A, shaded bars taken from ref (34)), anxiolysis induced by 23 was significantly (p < 0.05) more enhanced, which might reflect the stronger IGABA potentiation by 23 and/or the higher potency of 23 on receptors containing α2/3 and β3 subunits. Interestingly, the anxiolytic effect of the most potent and also more efficient derivative 25 did not differ from that of piperine and 24.34 It has, thus, to be clarified in further studies to what extent derivatization of the amide moiety affects the anxiolytic properties of piperine derivatives and whether receptors/channels other than GABAA receptors are targeted in vivo by these compounds.
Significant amounts of 23 and 25 were detected in mouse plasma after intraperitoneal (ip) application (see Table 8). The estimated plasma concentrations were below the micromolar concentrations required for significant IGABA potentiation of GABAA receptors expressed in Xenopus oocytes. However, drugs are commonly less potent on ion channels expressed in Xenopus oocytes as compared to channels expressed in either mammalian cells or even native tissues.48 The metabolite formation of 23 and 25 is currently unknown. At the current stage of our research, we cannot exclude that the observed anxiolytic and sedative effects are induced by more active metabolites. Furthermore, the currently unknown brain-barrier penetration of 23 and 25 and possible tissue accumulation warrants further research.
Table 8. Estimated Plasma Levels of Derivatives 23 and 25 after Intraperitoneal Applicationa.
| applied dose (mg/kg body weight) | mean plasma concn (ng/mL) | n |
|---|---|---|
| Compound 23 | ||
| 1 | 60.6 ± 14.5 | 3 |
| 3 | 194.0 ± 50.2 | 3 |
| 10 | 593.0 ± 92.4 | 3 |
| Compound 25 | ||
| 1 | 41.5 ± 8.7 | 3 |
| 3 | 172.0 ± 19.0 | 3 |
| 10 | 419.0 ± 37.2 | 3 |
Data are given as mean ± SEM; n indicates the number of animals used.
Conclusions
Piperine analogues modulating GABAA receptor with the highest efficiency show a tertiary amide nitrogen, substituted with flexible alkyl chains with a total of 6–8 carbon atoms. Polar substituents as well as rigid substituents give rise to a decrease of activity. Modifications of the linker region that lead to rigidification of the molecules also did not improve efficacy.
Compound 23 [(2E,4E)-5-(1,3-benzodioxol-5-yl))-N,N-dipropyl-2,4-pentadienamide] induced the strongest modulation of GABAA receptors (maximal GABA-induced chloride current enhancement IGABA-max = 1673.0% ± 146.3% and EC50 = 51.7 ± 9.5 μM, vs piperine, IGABA-max = 302% ± 27% and EC50 = 52.4 ± 9.4 μM), while 25 [(2E,4E)-5-(1,3-benzodioxol-5-yl))-N,N-dibutyl-2,4-pentadienamide] displayed the highest potency (EC50 = 13.8 ± 1.8 μM) but was less efficient than 23 (IGABA-max = 760% ± 47%). Both piperine analogues did not activate TRPV1 and induced pronounced anxiolytic action with little sedation, suggesting their potential use as scaffolds for drug development. The established determinants of efficacy may be used for future synthesis of improved GABAA modulators.
Experimental Section
Biological Activity
All experiments on animals were carried out in accordance with the Austrian Animal Experimental Law, which is in line with EU Directive 2010/63/EU. Every effort was made to minimize the number of animals used.
Expression of GABAA Receptors in Xenopus laevis Oocytes and Two-Microelectrode Voltage-Clamp Experiments
Preparation of stage V–VI oocytes from X. laevis and synthesis of capped runoff poly(A) cRNA transcripts from linearized cDNA templates (pCMV vector) was performed as previously described.49 Female X. laevis frogs (Nasco) were anesthetized by 15 min incubation in a 0.2% MS-222 (methanesulfonate salt of 3-aminobenzoic acid ethyl ester; Sigma–Aldrich, Vienna, Austria) solution before removal of parts of the ovaries. Follicle membranes from isolated oocytes were enzymatically digested with 2 mg/mL collagenase (type 1A, Sigma–Aldrich, Vienna, Austria).
Selected oocytes were injected with 10–50 nL of DEPC-treated water (diethyl pyrocarbonate, Sigma, Vienna, Austria) containing the different GABAA cRNAs at a concentration of approximately 300–3000 pg·nL–1·subunit–1.
To ensure expression of the γ2S subunit in the case of α1/2/3/5β2/3γ2S receptors, cRNAs were mixed in a ratio of 1:1:10. For expression of receptors composed of α4β2γ2S and α1β1γ2S, cRNAS were mixed in a ratio of 3:1:10. The amount of cRNAs was determined by means of a NanoDrop ND-1000 (Kisker-Biotech, Steinfurt, Germany).
Oocytes were stored at +18 °C in modified ND96 solution (90 mM NaCl, 1 mM CaCl2, 1 mM KCl, 1 mM MgCl2·6H2O, and 5 mM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], pH 7.4, all from Sigma–Aldrich, Vienna, Austria).
Chloride currents through GABAA receptors (IGABA) were measured at room temperature (+21 ± 1 °C) by means of a two-microelectrode voltage clamp technique making use of a Turbo TEC-05X amplifier (npi electronic, Tamm, Germany). IGABA were elicited at a holding potential of –70 mV. Data acquisition was carried out by means of an Axon Digidata 1322A interface using pCLAMP v.10 (Molecular Devices, Sunnyvale, CA). The modified ND96 solution was used as bath solution. Microelectrodes were filled with 2 M KCl and had resistances between 1 and 3 MΩ.
Fast Perfusion System
GABA and the studied derivatives were applied by means of the ScreeningTool (npi electronic, Tamm, Germany) fast perfusion system as described previously.50 To elicit IGABA, the chamber was perfused with 120 μL of GABA- or compound-containing solution at a volume rate of 300 μL/s.34 Care was taken to account for possible slow recovery from increasing levels of desensitization in the presence of high drug concentrations. The duration of washout periods was therefore extended from 1.5 min (<10 μM compounds) to 30 min (≥10 μM compounds). Oocytes with maximal current amplitudes >3 μA were discarded to exclude voltage clamp errors.
Data Analysis: GABAA Receptors
Stimulation of chloride currents by modulators of the GABAA receptor was measured at a GABA concentration eliciting between 3% and 7% of the maximal current amplitude (EC3–7). The GABA EC3–7 was determined for each oocyte individually. Enhancement of the chloride current was defined as (IGABA+compd/IGABA) – 1, where IGABA+compd is the current response in the presence of a given compound and IGABA is the control GABA current. IGABA-max reflects the maximal IGABA enhancement. Concentration–response curves were generated and the data were fitted by nonlinear regression analysis using Origin Software (OriginLab Corp.). Data were fitted to the equation 1/(1 + (EC50/[compound])nH), where nH is the Hill coefficient. Each data point represents the mean ± SEM from at least three oocytes and ≥2 oocyte batches. Statistical significance was calculated by paired Student t-test with a confidence interval of <0.05.
Molecular Modeling and Quantitative Structure–Activity Relationships
Data Set
The 2D structures of 76 piperine derivatives and piperine were drawn in the InstantJChem package for Excel (www.chemaxon.com/products/jchem-for-excel) and exported in sdf format. The LigPrep tool provided by Schrödinger in the Maestro package (Maestro, version 9.2; Schrödinger LLC, New York, 2011) was used to generate low-energy 3D structures and protonated states. All possible stereoisomers per ligand were computed and one low-energy conformation was generated per each stereoisomer in MMFF force field. The protonated states were determined at pH 7.4 (pH used in the experiments). For compounds 33, 34, 36, 38, and 39, several stereisomers were determined. Since these structures were not ionizable at this pH, the stereoisomers were considered equal in terms of 2D structure and duplicates were removed. Subsequently, the structures were imported into MOE, where partial atomic charges were calculated in the MMFF94 force field. Piperine (obtained from Sigma–Aldrich, Vienna, Austria) was used as a reference compound to determine the class labels of its derivatives. Potentiation of GABA current by piperine was 226% ± 26%;31 therefore, compounds with potentiation ≥200% were assigned to the active class, otherwise to the inactive. This led to an unbalanced data set with 17 “active” and 60 “inactive” compounds.
Descriptor Sets
One hundred forty-three 2D descriptors implemented in MOE were calculated. The full list is provided in Supporting Information (Table S1A). Descriptors showing no variance were removed from the data set, and the remaining 125 descriptors (Supporting Information, Table S1B) underwent feature selection by the BestFirst algorithm implemented in the software package WEKA version 3.7.9. Consequently, the six descriptors left (set 6D) were used for further classification studies (Table 9). Additionally, as a reference descriptor set, we used 11 descriptors of physicochemical properties (set PHYSCHEM) from the list of 125 descriptors described above (Table 10). These descriptors allow us to trace molecular features important for biological activity and have previously shown good performance in application to ligand-based studies.51 As an attempt to trace the structural features relevant to the activity of piperine derivatives, MACCS fingerprints (MACCS Keys; MDL Information Systems, Inc., San Leandro, CA) were computed in MOE. MACCS are a set of structural keys, where each key describes a small substructure consisting of up to 10 non-hydrogen atoms. A Python script (Supporting Information) was applied to divide the fingerprints into bit strings. The latter were further used in the classification studies as descriptor set “MACCS”.
Table 9. Set of Six 2D Descriptors Selected by BestFirst Algorithm for Classification Studies.
| name | definition |
|---|---|
| density | molecular mass density: weight divided by vdw_vol (amu/Å3) |
| lip_don | no. of OH and NH atoms |
| opr_brigid | no. of rigid bonds53 |
| PEOE_RPC+ numeric | relative positive partial charge: largest positive qi divided by the sum of positive qi |
| PEOE_VSA+3 | sum of vi where qi is in the range [0.15, 0.20) |
| SMR | molecular refractivity (including implicit hydrogens)a |
This property is an atomic contribution model54 that assumes the correct protonation state (washed structures). The model was trained on ∼7000 structures and results may vary from the mr descriptor.
Table 10. Eleven Descriptors of Physical Chemical Properties Used in the Study.
| name | definition |
|---|---|
| a_acc | no. of hydrogen-bond acceptor atoms |
| a_don | no. of hydrogen-bond donor atoms |
| b_rotN | no. of rotatable bondsa |
| log_P(o/w) | log of octanol/water partition coefficientb |
| mr | molecular refractivity (including implicit hydrogens)c |
| PEOE_VSA_HYD | total hydrophobic van der Waals surface area |
| TPSA | polar surface aread (Å2) |
| vsa_acc | approximate sum of VDW surface areas (Å2) of pure hydrogen-bond acceptors |
| vsa_don | approximate sum of VDW surface areas (Å2) of pure hydrogen-bond donors |
| vsa_hyd | approximate sum of VDW surface areas (Å2) of hydrophobic atoms |
| Weight | molecular weight (including implicit hydrogens) (amu) |
A bond is rotatable if it has order 1, is not a ring, and has at least two heavy neighbors.
Calculated from a linear atom-type model with r2 = 0.931.
Calculated from an 11-descriptor linear model with r2 = 0.997.
Calculated from group contributions to approximate the polar surface area from connection table.
Computational Methods
As classification methods, instance-based classifier (IBk), J48 decision tree (J48), naïve-Bayes classifier (NB), random forest (RF), and Support vector machine (SMO) were used as implemented in Weka. All methods were used with the default parameter settings. Nevertheless, different costs were associated with misclassified compounds since the data set was unbalanced. The costs were evaluated by use of an in-house script (Supporting Information), which consequently built models with different costs of the false positive (FP) and false negative (FN) compounds (from 1 to 200 with step of 1 for FN and from 0 to 20 with step of 0.1 for FP). Moreover, inside the script the 10-fold cross-validation was applied and statistical parameters were computed. Subsequently, one model per method and descriptor set was selected on the basis of highest values of MCC, accuracy, sensitivity, and specificity and was taken for visual inspection and possible interpretation. The cost-sensitive parameters obtained for the best 15 models are listed in Table 11.
Table 11. Cost-Sensitive Parameters.
| method | cost FP | cost FN |
|---|---|---|
| Descriptor Set 6D | ||
| IBk | 1 | 1 |
| J48 | 6 | 1 |
| NB | 5 | 3 |
| RF | 9 | 5 |
| SMO | 52 | 19.1 |
| Descriptor Set PHYSCHEM | ||
| IBk | 1 | 1 |
| J48 | 18 | 11 |
| NB | 1 | 1 |
| RF | 21 | 2 |
| SMO | 49 | 18.1 |
| Descriptor Set MACCS | ||
| IBk | 3 | 2 |
| J48 | 29 | 12 |
| NB | 1 | 8 |
| RF | 4 | 1 |
| SMO | 3 | 2.2 |
Statistical Parameters
The statistical parameters of every model were calculated on the basis of values from confusion matrix (for details see ref (52)), where TP and TN stand for correctly classified active and inactive compounds and FP and FN for misclassified inactive and active ligands. The true-positive rates of active (sensitivity) and inactive (specificity) classes were calculated by the following formulas:
The accuracy of the model was defined as the ratio of correctly predicted compounds to the total amount of compounds.
Additionally, the Matthews correlation coefficient (MCC) was used to assess the quality of the obtained models. It was calculated from the formula
MCC is independent of the class sizes and therefore gives a rational evaluation of prediction in our case. It can return values from −1 to +1, where +1 determines perfect prediction, 0 means random classification, and −1 represents a total misclassification. The value of 0.4 was taken as a threshold to filter the best-performing models.
Behavioral Studies
Male mice (C57BL/6N) were obtained from Charles River Laboratories (Sulzfeld, Germany). For maintenance, mice were group-housed (maximum five mice per type IIL cage) with free access to food and water. At least 24 h before the commencement of experiments, mice were transferred to the testing facility, where they were given free access to food and water. The temperature in the maintenance and testing facilities was 23 ± 1 °C; the humidity was 40–60%; a 12 h light–dark cycle was in operation (lights on from 07:00 to 19:00). Only male mice aged 3–6 months were tested.
Compounds were applied by intraperitoneal (ip) injection 30 min before each test. Testing solutions were prepared in a solvent composed of 0.9% NaCl solution with 10% dimethyl sulfoxide (DMSO) and 3% Tween 80. Application of the solvent alone did not influence animal behavior. All doses are indicated as milligrams per kilogram of body weight of the animal.
Elevated Plus Maze Test
The animals’ behavior was tested over 5 min on an elevated plus maze 1 m above ground consisting of two closed and two open arms, each 50 × 5 cm in size. The test instrument was built from gray PVC; the height of closed arm walls was 20 cm. Illumination was set to 180 Lux. Animals were placed in the center, facing an open arm. Analysis of open and closed arm entries and time on open arm was automatically done with Video-Mot 2 equipment and software (TSE Systems, Bad Homburg, Germany).34
Open Field Test
Ambulation was tested over 10 min in a 50 × 50 cm flexfield box equipped with infrared rearing detection. Illumination was set to 150 Lux. The animals’ explorative behavior was analyzed by use of the ActiMot 2 equipment and software (TSE-systems, Bad Homburg, Germany). Arenas were subdivided into border (up to 8 cm from wall), center (20 × 20 cm, 16% of total area), and intermediate area according to the recommendations of EMPRESS (European Mouse Phenotyping Resource of Standardised Screens; http://empress.har.mrc.ac.uk).
Estimation of Plasma Levels
Trunk blood from male C57BL/6N (6 months) was taken 15, 30, and 60 min after ip application of 23 and 25 (doses 1, 3, and 10 mg/kg body weight; injection solutions were prepared as described for behavioral analysis). At each time point, mice were euthanized and blood samples (500–800 μL) were collected and compiled into ethylenediaminetetraacetic acid (EDTA)-coated microtubes (1.6 mg of EDTA/sample) and centrifuged at 12 000 rpm for 5 min at 4 °C. Plasma samples were transferred into 1.5 mL tubes and stored at −80 °C until analysis.
Materials
All solvents used were of UPLC grade. Acetonitrile and dimethyl sulfoxide (DMSO) were supplied by Scharlau (Barcelona, Spain). Methanol was from Lab-Scan (Gliwice, Poland). Ammonium formate, formic acid and trifluoroacetic acid (TFA) were purchased from BioSolve (Valkenswaard, Netherlands), and HPLC-grade water was obtained from an EASYpure II (Barnstead, Dubuque, IA) water purification system. Blank K3EDTA C57BL/6N mouse plasma was collected for generating plasma calibrators and quality controls (QC).
Preparations of Calibrators and Quality Control Samples
Two separate sets of 23 and 25 stock solutions were prepared in DMSO for making calibrators and quality control (QC) samples. Plasma calibrators were prepared by spiking corresponding stock solutions into a blank plasma sample. The following 23 and 25 concentrations were added: 20, 50, 100, 250, 500, 1000, and 2000 ng/mL. The same blank plasma and both stock solutions (for QC) were used to generate three level plasma QC samples at 60, 1000, and 1600 ng/mL for both 23 and 25.
Two internal standard (IS) stock solutions of 22 and 24 were prepared in DMSO in order to generate working solutions (WS) at 200 ng/mL in methanol.
Sample Preparation for UHPLC-MS/MS Analysis
Plasma proteins were precipitated by the addition of 50 μL of WS at 200 ng/mL of the corresponding IS: 22 (for 23) or 24 (for 25) and 500 μL of ice-cold acetonitrile to 20 μL of K3EDTA mouse plasma. Samples were vortexed at 1400 rpm for 10 min and then centrifuged at 13200g for 20 min at 10 °C. The supernatant was transferred into a 96-deep-well plate for drying under nitrogen gas flow (Evaporex EVX-96, Apricot Designs, Monrovia, CA) and redissolved in 200 μL of injection solvent (65% 10 mM ammonium formate + 0.05% formic acid, 35% acetonitrile + 0.05% formic acid) before MS/MS analysis.
LC-MS/MS Analyses
Quantification was performed on a 1290 Infinity LC system coupled with a 6460 triple quadrupole mass spectrometer with Jet Stream Technology, and data was processed with a MassHunter Workstation Software version B.06.00 (Agilent; Waldbronn, Germany). The 1290 Infinity LC system was equipped with a binary capillary pump, degasser, autosampler, autosampler thermostat, thermostated column compartment, and FlexCube. Separation was performed at 55 °C on a Kinetex XB-C18 column, 100 × 2.1 mm, 1.7 μm particle size (Phenomenex; Torrance, CA); mobile phase of (A) 0.05% formic acid in 10 mM ammonium formate and (B) 0.05% formic acid in ACN, gradient 40% B for 1 min, linear gradient to reach 88% B after 5.3 min, shifted to 100% B for 1 min, and back to equilibrium condition of 40% B for 0.7 min; flow rate of 0.5 mL/min; total run time of 7 min. Sample injected volume was 1 μL and autosampler was set at 10 °C. Needle wash solution was MeOH/ACN/IPA/H2O (1:1:1:1 v/v/v/v). Flexible cube was set at a flow rate of 1 mL/min for 20 s.
MS parameters were manually optimized as follow: drying N2 gas of 320 °C at a flow rate of 10 L/min, nebulizer pressure of 20 psi, sheath N2 gas of 400 °C at a flow rate of 11 L/min, nozzle voltage of 0 V, capillary voltage of 2.5 kV, and delta EMV 0 V. Quantification was determined in multiple reaction monitoring (MRM) mode with an ESI-MS/MS system in positive ionization mode. The MRM transitions of both 23 and 25 and corresponding internal standard were as shown in Table S2 (Supporting Information).
Syntheses
Details of synthesis and characterization of selected products 25, 51, and 62 and key intermediates 65a, 68a–c, 71a, and 75a,b are described below. Synthetic procedures and characterization data for all other compounds are included in Supporting Information. Purity was determined either by elemental analysis or by HPLC and was >95%. Unless otherwise noted, chemicals were purchased from commercial suppliers and used without further purification. Microwave reactions were performed on a Biotage Initiator Sixty microwave unit (Biotage AB, Uppsala, Sweden). Flash column chromatography was performed on silica gel 60 from Merck (40–63 mm), whereas most separations were performed by using a Büchi Sepacore medium-pressure liquid chromatography (MPLC) system with a 9g column (Buchi Labortechnik AG, Flawil, Switzerland). For thin-layer chromatography (TLC), aluminum-backed silica gel was used. Melting points were determined by using a Kofler-type Leica Galen III micro hot stage microscope (Aigner-Unilab Laborfachhandel GmbH, Vienna, Austria) and are uncorrected. For compounds unknown in the literature, either high-resolution mass spectrometry (HR-MS) or combustion analysis was performed. HR-MS was performed by E. Rosenberg at the Institute for Chemical Technologies and Analytics, Vienna University of Technology; all samples were analyzed by liquid chromatography/ion trap time-of-flight mass spectrometry (LC/IT-TOF-MS) in positive or negative ion detection mode with the recording of MS and MS/MS spectra. Combustion analysis was carried out in the Microanalytical Laboratory, Institute of Physical Chemistry, University of Vienna. NMR spectra were recorded on a Bruker AC 200 (200 MHz), a Bruker Avance DP160 (200 MHz), or a Bruker Avance 400 (400 MHz) spectrometer (Bruker GmbH, Vienna, Austria) and chemical shifts are reported in parts per million (ppm). For assignment of 13C multiplicities, standard 13C distortionless enhancement by polarization transfer (DEPT) or attached proton test (APT) spectra were recorded. HPLC analyses were performed on a Agilent 1200 HP-LC system with a Kinetex XB-C18, 2.6 μm, 50 × 2.1 mm column (Agilent Technologies GmbH, Vienna, Austria). The mobile phase was composed of ACN/water (gradient 50:50 up to 95:5 v/v) with 0.1% AcOH added. GC–MS runs were performed on a Thermo Finnigan Focus GC/DSQ II with a standard capillary column BGB 5 (30 m × i.d. 0.32 mm; Fisher Scientific GmbH, Vienna, Austria).
(2E,4E)-5-(1,3-Benzodioxol-5-yl)-N,N-dibutyl-2,4-pentadienamide (25)
Piperic acid chloride (218 mg, 1 mmol) was dissolved in 2.5 mL of dry THF. Dibutylamine (595 μL, 3.5 mmol) was added and the reaction mixture was stirred overnight at rt. After evaporation of the solvent, the residue was taken up in ethyl acetate (EtOAc; 40 mL) and washed two times each with 5% NaHCO3 and 2 N HCl. The organic layer was separated, dried with sodium sulfate, filtered, and evaporated. The pure product was obtained after recrystallization from ethanol.
Yield 76% (746 mg, 2.26 mmol), light brown crystals, mp 88–90 °C. 1H NMR (CDCl3, 200 MHz) δ 0.85–1.05 (m, 6H, CH3), 1.22–1.45 (m, 4H, CH2), 1.46–1.71 (m, 4H, CH2), 3.25–3.47 (m, 2H, CH2), 5.98 (s, 2H, O–CH2–O), 6.35 (d, J = 14.6 Hz, 1H, H2), 6.70–6.85 (m, 3H), 6.86–6.95 (m, 1H), 7.00 (d, J = 7.9 Hz, 1H), 7.36–7.54 (m, 1H). 13C NMR (CDCl3, 50 MHz) δ 14.1 (q, CH3), 14.1 (q, CH3), 20.3 (t, CH2), 20.5 (t, CH2), 30.3 (t, CH2), 32.2 (t, CH2), 46.8 (t, N–CH2), 48.1 (t, N–CH2), 101.5 (t, O–CH2–O), 105.9 (d), 108.7 (d), 120.5 (d), 122.7 (d), 125.6 (d), 131.2 (s), 138.6 (d), 142.6 (d), 148.3 (s, C–O), 148.4 (s, C–O), 166.3 (s, CO–N). Anal. Found, C 71.96, H 7.91, N 3.95; Calcd (·0.23H2O), C 72.01, H 8.30, N 4.20.
3-(Benzo[d][1,3]dioxol-5-yl)-N,N-dipropylbenzamide (51)
Benzodioxol-5-boronic acid (138 mg, 0.83 mmol, 1 equiv), 51a (237 mg, 0.83 mmol, 1 equiv), Pd(PPh3)4 (19 mg, 2 mol %), and sodium carbonate (615 mg, 5.81 mmol, 7 equiv) were charged into a microwave vial. Then a mixture of dimethyl ether (DME)/EtOH 5:1 (6.4 mL) and water (1.8 mL) was added, and the resulting suspension was degassed by passing through argon for 5 min. The vial was sealed and heated to 140 °C for 1 h in the microwave. After cooling to rt, the reaction mixture was extracted with dichloromethane (DCM), the solvent was evaporated, and the crude product was directly subjected to column chromatography with light petroleum (LP)/EtOAc mixture as eluent.
Yield 60% (163 mg, 0.50 mmol), colorless oil. TLC 0.24 (LP/EtOAc 4:1). 1H NMR (CDCl3, 200 MHz) δ 0.76–0.98 (br m, 6H, CH3), 1.57–1.67 (br m, 4H, CH2), 3.20–3.47 (br m, 4H, N–CH2−), 6.00 (s, 2H, O–CH2–O), 6.86–6.90 (m, 1H, ArH), 7.03–7.08 (m, 2H, ArH), 7.25–7.30 (m, 1H, ArH), 7.42 (t, J = 7.4 Hz, 1H, ArH), 7.48–7.54 (m, 2H, ArH). 13C NMR (CDCl3, 50 MHz) δ 11.1 (q, CH3), 11.4 (q, CH3), 20.7 (t, CH2), 22.0 (t, CH2), 46.3 (t, CH2), 50.7 (t, CH2), 101.2 (t, O–CH2–O), 107.6 (d), 108.6 (d), 120.7 (d), 124.8 (d), 124.9 (d), 127.4 (d), 128.8 (d), 134.8 (s), 137.9 (s), 141.1 (s), 147.3 (s), 148.2 (s), 171.6 (s, −CO–N). HR-MS [M + H]+m/z (pred) = 326.1751, m/z (meas) = 326.1749, difference = −0.61 ppm.
[5-(Benzo[d][1,3]dioxol-5-yl)naphthalen-1-yl](piperidin-1-yl)methanone (62)
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI·HCl; 65 mg, 0.34 mmol, 2 equiv) was added to a suspension of 62a (50 mg, 0.17 mmol, 1 equiv) and hydroxybenzotriazole (HOBt; 52 mg, 0.34 mmol, 2 equiv) in dry dichloromethane (2 mL) under argon at rt. After 2 h, the suspension was transformed into an opaque solution and TLC indicated full consumption of the starting material. Piperidine (0.5 mL) was added at rt and stirring was continued overnight. After full conversion was detected by TLC, the reaction mixture was diluted with EtOAc (30 mL); washed with 0.5 N HCl, saturated NaHCO3, and brine (20 mL each); dried with sodium sulfate; and evaporated. The crude product was purified by column chromatography with LP/EtOAc mixture as eluent.
Yield 62% (0.11 mmol, 38 mg), colorless solid, mp 150–153 °C. TLC 0.09 (LP/EtOAc 4:1). 1H NMR (CDCl3, 200 MHz) δ 1.40–1.50 (m, 2H, CH2), 1.66–1.80 (m, 4H, CH2), 3.15–3.21 (m, 2H, N–CH2), 3.87–3.93 (m, 2H, N–CH2), 6.04 (s, 2H, O–CH2–O), 6.93–6.95 (m, 3H, ArH), 7.39–7.56 (m, 4H, ArH), 7.84 (d, J = 8.3 Hz, 1H, ArH), 7.94 (dd, J1 = 7.2 Hz, J2 = 2.6 Hz, 1H, ArH). 13C NMR (CDCl3, 50 MHz) δ 24.6 (t, CH2), 25.9 (t, CH2), 26.7 (t, CH2), 42.7 (t, N–CH2), 48.3 (t, N–CH2), 101.2 (t, O–CH2–O), 108.2 (d), 110.6 (d), 123.3 (d), 123.4 (d), 124.4 (d), 125.3 (d), 126.2 (d), 126.9 (d), 127.4 (d), 120.9 (s), 131.9 (s), 134.3 (s), 135.2 (s), 140.3 (s), 147.0 (s), 147.5 (s), 169.4 (s, CO–N). HR-MS [M + H]+m/z (pred) = 360.1594, m/z (meas) = 360.1597, difference = 0.83 ppm.
(E)-Methyl 3-(Naphtho[2,3-d][1,3]dioxol-5-yl)acrylate (65a)
For synthesis of 65a, a modification of a previously published method38 was employed. A 8-mL vial with magnetic stirrer, screw cap, and septum was charged with naphtho[2,3-d][1,3]dioxol-5-yl trifluoromethanesulfonate (synthesized according to ref (37)) (480 mg, 1.5 mmol, 1 equiv), 1,10-phenanthroline monohydrate (16 mg, 0.083 mmol, 5.5 mol %), palladium(II) acetate (17 mg, 0.075 mmol, 5 mol %), and anhydrous N,N-dimethylformamide (DMF, 5 mL). Then triethylamine (250 μL, 1.8 mmol, 1.2 equiv) and methyl acrylate (680 μL, 7.5 mmol, 5 equiv) were added successively. The vial was flushed with argon and heated to 80 °C for 16 h. Reaction control by TLC showed full conversion. The solvent was evaporated, and the residue was taken up in DCM and adsorbed on silica. Column chromatography (45 g of SiO2, eluent LP/EtOAc, 5% isocratic) yielded the pure product.
Yield 95% (364 mg, 1.425 mmol), colorless solid, mp 125–126 °C. TLC 0.44 (LP/EtOAc 4:1). 1H NMR (CDCl3, 200 MHz) δ 3.82 (s, 3H, CH3), 5.99 (s, 2H, O–CH2–O), 6.43 (d, J = 15.7 Hz, 1H, H3), 7.04 (s, 1H, ArH), 7.25 (t, J = 7.7 Hz, 1H, H7′), 7.38 (s, 1H, ArH), 7.52 (d, J = 7.1 Hz, 1H, ArH), 7.61 (d, J = 8.1 Hz, 1H, ArH), 8.29 (d, J = 15.7 Hz, 1H, H2). 13C NMR (CDCl3, 50 MHz) 51.7 (q, CH3), 99.9 (d), 101.4 (t, O–CH2–O), 104.4 (d), 119.9 (d), 123.5 (d), 124.0 (d), 128.7 (s), 129.5 (d), 130.7 (s), 130.9 (s), 142.1 (d), 147.6 (s), 148.6 (s), 167.3 (s, COOR). HR-MS [M – MeOH]+m/z (pred) = 225.0546, m/z (meas) = 225.0553, difference = 3.11 ppm.
4,4,5,5-Tetramethyl-2-(naphtho[2,3-d][1,3]dioxol-6-yl)-1,3,2-dioxaborolane (68a)
For synthesis of 68a, a modification of a procedure published by Ishyama et al.39 was used. A three-necked flask with magnetic stirrer, septum, reflux condenser, and balloon was charged with naphtho[2,3-d][1,3]dioxole (1.72 g, 10 mmol, 1 equiv), bis(pinacolato)diboron (1.27 g, 5 mmol, 0.5 equiv), [Ir(OMe)cod]2 (100 mg, 0.15 mmol, 1.5 mol %), and 4,4′-di-tert-butyl-2,2′-bipyridine (81 mg, 0.3 mmol, 3 mol %) and flushed with argon. Then cyclohexane (60 mL) was added and the reaction was heated to reflux and monitored with GC/MS. After 24 h the reaction did not proceed any further. After evaporation of the solvent, the residue was redissolved in DCM, adsorbed on silica, and directly subjected to column chromatography (45 g of SiO2, eluent LP/EE 30:1), which yielded the pure product (683 mg of starting material could be reisolated in this step).
Yield 29% (48% based on recovered starting material, 874 mg, 2.9 mmol), colorless solid, mp 97–99 °C. TLC 0.18 (LP/EE 30:1). 1H NMR (CDCl3, 200 MHz) δ 1.38 (s, 12H, CH3), 6.03 (s, 2H, O–CH2–O), 7.10 (s, 1H), 7.64 (d, J = 8.2 Hz, 1H), 7.70 (d, J = 8.2 Hz, 1H), 8.16 (s, 1H, H5). 13C NMR (CDCl3, 50 MHz) 24.9 (q, 4C, CH3), 83.8 (s, B–O–CR3), 101.0 (t, O–CH2–O), 103.8 (d), 104.4 (d), 126.2 (d), 129.3 (d), 129.8 (s), 132.5 (s), 134.9 (d), 147.4 (s), 148.4 (s); C6 signal could not be detected due to low signal intensity.
6-Bromonaphtho[2,3-d][1,3]dioxole (68b)
For synthesis of 68b, a modification of a published procedure40 was used. In a three-necked flask with magnetic stirrer and reflux condenser, 68a (700 mg, 2.35 mmol, 1 equiv) was dissolved in methanol. Copper(II) bromide (1.57 g, 7 mmol, 3 equiv) was dissolved in water (20 mL) and added. The reaction was heated to reflux for 18 h and checked with TLC. The reaction mixture was cooled, diluted with water (200 mL), and extracted with 3 × 50 mL of DCM. The combined organic extracts were washed with 50 mL each water and brine, dried with anhydrous sodium sulfate, and evaporated.
Yield 94% (555 mg, 2.21 mmol), colorless solid, mp 135–138 °C. TLC 0.40 (LP/EE 30:1). 1H NMR (CDCl3, 200 MHz) δ 6.04 (s, 2H, O–CH2–O), 7.01 (s, 1H, ArH), 7.06 (s, 1H, ArH), 7.38 (dd, J1 = 8.7 Hz, J2 = 1.9 Hz, 1H, H7), 7.51 (d, J = 8.7 Hz, 1H, H8), 7.79 (d, J = 1.9 Hz, 1H, H5). 13C NMR (CDCl3, 50 MHz) 101.3 (t, O–CH2–O), 103.0 (d), 103.8 (d), 118.1 (s), 127.5 (d), 128.5 (d), 128.9 (d), 131.8 (s), 148.0 (s), 148.3 (s). One signal could not be detected due to low signal intensity.
(E)-Methyl 3-(Naphtho[2,3-d][1,3]dioxol-6-yl)acrylate (68c)
An 8-ml vial with magnetic stirrer, screw cap, and septum was charged with 68b (300 mg, 1.2 mmol, 1 equiv), methyl acrylate (163 μL, 1.8 mmol, 1.5 equiv), palladium(II) acetate (8 mg, 0.036 mmol, 3 mol %), and tri-o-tolylphosphine (22 mg, 0.072 mmol, 6 mol %) and flushed with argon. Triethylamine (0.85 mL) was added via syringe and the reaction was heated to 80 °C. TLC monitoring (eluent LP/EE 30:1) showed full conversion after 8 h. The reaction mixture was diluted with diethyl ether (30 mL). Due to low solubility of the product in diethyl ether, it was necessary to add ethyl acetate (20 mL) and DCM (10 mL) to obtain a clear solution. The organic phase was washed with 3 × 10 mL of 0.5 N HCl and 30 mL of brine and dried with sodium sulfate. Evaporation of the solvent gave the pure product in quantitative yield.
Yield 100% (310 mg, 1.2 mmol), colorless solid, mp 151–152 °C. TLC 0.16 (LP/EE 30:1). 1H NMR (CDCl3, 200 MHz) δ 3.81 (s, 3H, CH3), 6.06 (s, 2H, O–CH2–O), 6.49 (d, J = 16.0 Hz, 1H, H3), 7.10 (s, 1H, ArH), 7.12 (s, 1H, ArH), 7.50 (dd, J1 = 8.6 Hz, J2 = 1.6 Hz, 1H, H7′), 7.64 (d, J = 8.6 Hz, 1H, H8′), 7.74–7.83 (m, 2H, H2, H5′). 13C NMR (CDCl3, 50 MHz) δ 51.7 (q, CH3), 101.3 (t, O–CH2–O), 103.9 (d), 104.4 (d), 116.9 (d), 127.6 (d), 128.7 (d), 130.3 (s), 130.4 (s), 131.7 (s), 145.1 (d), 148.2 (s), 148.7 (s), 167.6 (d, COOR). HR-MS [M + H]+m/z (pred) = 257.0808, m/z (meas) = 257.0807, difference = −0.39 ppm.
Naphtho[2,3-d][1,3]dioxole-5-carboxylic acid (71a)
For synthesis of 71a, a modification of a published procedure41 was used. In a two-necked flask equipped with magnetic stirrer, septum, and balloon, naphtho[2,3-d][1,3]dioxol-5-yl trifluoromethanesulfonate42 (96 mg, 0.3 mmol, 1 equiv), 1,3-bis(diphenylphosphino)propane (dppp; 7 mg, 0.018 mmol, 6 mol %), and palladium(II) acetate (2 mg, 0.009 mmol, 3 mol %) were suspended in DMF/water 3:1 (1 mL). A steel cannula reaching to the bottom of the flask was used to bubble carbon monoxide through the solution for 10 min; after that, the balloon was filled with CO gas in order to maintain its supply throughout the reaction time. Hünig’s base (102 μL, 0.6 mmol, 2 equiv) was added via syringe and the reaction mixture was heated to 70 °C. After 3 h, reaction control with TLC indicated complete consumption of the starting material. The reaction mixture was diluted with ethyl acetate (10 mL) and extracted with 3 × 5 mL of saturated NaHCO3. The combined aqueous extracts were acidified to pH = 2 with 2 N HCl and extracted with 3 × 10 mL of ethyl acetate. The combined organic extracts were washed with 10 mL each water and brine and dried with sodium sulfate. Evaporation of the solvent gave the pure product.
Yield 67% (116 mg, 0.54 mmol), colorless solid, mp 259–263 °C. TLC 0.60 (CHCl3/MeOH 10%). 1H NMR (acetone-d6, 400 MHz) δ 6.17 (s, 2H, O–CH2–O), 7.33 (s, 1H, ArH), 7.41–7.45 (m, 1H, H7), 8.00 (d, J = 8.0 Hz, 1H, ArH), 8.18 (dd, J1 = 7.4 Hz, J2 = 1.1 Hz, 1H, ArH), 8.49 (s, 1H, ArH). 13C NMR (acetone-d6, 100 MHz) δ 101.7 (t, O–CH2–O), 102.2 (d), 104.1 (d), 123.1 (d), 125.7 (s), 128.9 (d), 129.2 (s), 131.6 (s), 132.4 (d), 147.6 (s), 149.5 (s), 168.2 (s, COOH).
Methyl 5,6,7,8-Tetrahydronaphtho[2,3-d][1,3]dioxole-6-carboxylate (75a)
For synthesis of 75a, a modification of a published method42 was used. A three-necked flask with magnetic stirrer, septum, reflux condenser, and balloon was charged with 5,6-bis(bromomethyl)benzo[d][1,3]dioxole (2.0 g, 6.5 mmol, 1 equiv), methyl acrylate (2.94 mL, 32.5 mmol, 5 equiv), and anhydrous DMF (50 mL) and was flushed with argon. Sodium iodide (3.9 g, 26 mmol, 4 equiv) was added and the reaction was heated to 90 °C overnight (in previous experiments on a smaller scale, full conversion had been reached after 2 h). Above 70 °C the reaction mixture began to turn red. The reaction was quenched with 200 mL of water, and then, sodium thiosulfate 5% was added until the mixture became colorless. The aqueous mixture was extracted with 5 × 50 mL of methyl tert-butyl ether (MTBE). The organic phase was washed with 50 mL each water and brine, dried with anhydrous sodium sulfate, and evaporated.
Yield 89% (1.35 g, 5.79 mmol), colorless solid, mp 71–72 °C. TLC 0.15 (LP/EE 30:1). 1H NMR (CDCl3, 200 MHz) δ 1.75–1.91 (m, 1H), 2.10–2.22 (m, 1H), 2.61–2.78 (m, 3H), 2.88–2.91 (m, 2H), 3.71 (s, 3H, CH3), 5.87 (s, 2H, O–CH2–O), 6.53 (s, 1H, ArH), 6.55 (s, 1H, ArH). 13C NMR (CDCl3, 50 MHz) δ 25.9 (t, CH2), 28.6 (t, CH2), 31.6 (t, CH2), 39.9 (d, C6), 51.8 (q, CH3), 100.6 (t, O–CH2–O), 108.5 (d), 108.6 (d), 127.6 (s), 128.5 (s), 145.7 (s), 145.9 (s), 175.8 (d, COOR)
Methyl Naphtho[2,3-d][1,3]dioxole-6-carboxylate (75b)
Compound 75a (100 mg, 0.43 mmol, 1 equiv) was dissolved in benzene (3 mL, p.a.) under argon. DDQ (242 mg, 1.07 mmol, 2.5 equiv) was added and the reaction mixture was heated to 80 °C for 2 h. TLC analysis was inconclusive due to very similar Rf values of starting material and product. Staining with cerium molybdophosphoric acid dip reagent indicated full conversion (The starting material is readily stained; the product only weakly). The reaction was quenched with 20 mL of 2 N NaOH and changed color to brown. The reaction was extracted with 3 × 10 mL of EtOAc. The organic phase was washed with water until the washings were colorless (5 × 10 mL) and subsequently washed with brine, dried over sodium sulfate, and evaporated.
Yield 73% (72 g, 0.31 mmol), colorless solid, mp 130–132 °C, sublimation above 105 °C. TLC 0.15 (LP/EE 30:1). 1H NMR (CDCl3, 200 MHz) δ 3.95 (s, 3H, CH3), 6.06 (s, 2H, O–CH2–O), 7.11 (s, 1H, ArH), 7.17 (s, 1H, ArH), 7.66 (d, J = 8.6 Hz, 1H, H8), 7.90 (dd, J1 = 8.6 Hz, J2 = 1.6 Hz, 1H, H7), 8.38 (d, J = 1.6 Hz, 1H, H5). 13C NMR (CDCl3, 50 MHz) δ 52.1 (q, CH3), 101.4 (t, O–CH2–O), 103.8 (d), 104.9 (d), 124.1 (d), 125.9 (s), 127.0 (d), 129.6 (s), 129.7 (d), 133.3 (s), 148.2 (s), 149.5 (s), 167.4 (d, COOR). HR-MS [M + H]+m/z (pred) = 231.0652, m/z (meas) = 231.658, difference = 2.60 ppm.
Acknowledgments
This work was supported by the Austrian Science Fund (FWF doctoral program “Molecular drug targets” W1232 to S.H., G.F.E., and M.D.M.). We thank Mihaela Coner for technical assistance.
Glossary
Abbreviations Used
- DEPC
diethyl pyrocarbonate
- EtOAc
ethyl acetate
- GABAA
γ-aminobutyric acid type A receptor
- IGABA
γ-aminobutyric acid-induced chloride current
- IGABA-max
maximum aminobutyric acid-induced chloride current potentiation
- LP
light petroleum
- rt
room temperature
- TRPV1
transient receptor potential vanilloid type 1 receptor
- VR1
vanilloid receptor 1
Supporting Information Available
Synthetic procedures and characterization data for piperine analogues; shell script for evaluation of costs; Python script to divide MACCS fingerprints into bite strings; parameters obtained for model 3; full composition of 10 trees in model 4; and a table with the full list of descriptors calculated. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
▽ (T.S.) Institute of Medical Genetics Medical University of Vienna, Waehringerstrasse 10, 1090 Vienna, Austria.
Author Contributions
⊥ A.S., L.W., and D.G. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Macdonald R. L.; Olsen R. W. GABAA Receptor Channels. Annu. Rev. Neurosci. 1994, 17, 569–602. [DOI] [PubMed] [Google Scholar]
- Sieghart W. Structure and Pharmacology of Gamma-Aminobutyric Acid A Receptor Subtypes. Pharmacol. Rev. 1995, 47, 181–234. [PubMed] [Google Scholar]
- Sigel E.; Steinmann M. E. Structure, Function, and Modulation of GABA(A) Receptors. J. Biol. Chem. 2012, 287, 40224–40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly C. N.; Wafford K. A. The Cys-Loop Superfamily of Ligand-Gated Ion Channels: The Impact of Receptor Structure on Function. Biochem. Soc. Trans. 2004, 32, 529–534. [DOI] [PubMed] [Google Scholar]
- Sigel E.; Kaur K. H.; Luscher B. P.; Baur R. Use of Concatamers to Study GABAA Receptor Architecture and Function: Application to Delta-Subunit-Containing Receptors and Possible Pitfalls. Biochem. Soc. Trans. 2009, 37, 1338–1342. [DOI] [PubMed] [Google Scholar]
- Smart T. G.; Paoletti P. Synaptic Neurotransmitter-Gated Receptors. Cold Spring Harbor Perspect. Biol. 2012, 4, a009662 10.1101/cshperspect.a009662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard E. A.; Skolnick P.; Olsen R. W.; Möhler H.; Sieghart W.; Biggio G.; Braestrup C.; Bateson A. N.; Langer S. Z. International Union of Pharmacology. XV. Subtypes of Gamma-Aminobutyric Acid A Receptors: Classification on the Basis of Subunit Structure and Receptor Function. Pharmacol. Rev. 1998, 50, 291–313. [PubMed] [Google Scholar]
- Simon J.; Wakimoto H.; Fujita N.; Lalande M.; Barnard E. A. Analysis of the Set of GABA(A) Receptor Genes in the Human Genome. J. Biol. Chem. 2004, 279, 41422–41435. [DOI] [PubMed] [Google Scholar]
- Olsen R. W.; Sieghart W. International Union of Pharmacology. LXX. Subtypes of Gamma-Aminobutyric Acid(A) Receptors: Classification on the Basis of Subunit Composition, Pharmacology, and Function. Update. Pharmacol. Rev. 2008, 60, 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.; Wang R.; Barot S.; Weiss D. S. Stoichiometry of a Recombinant GABA Receptor. J. Neurosci. 1996, 16, 5415–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V.; Ehya N.; Fuchs K.; Sieghart W. Stoichiometry and Assembly of a Recombinant GABAA Receptor Subtype. J. Neurosci. 1997, 17, 2728–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann S. W.; Baur R.; Sigel E. Individual Properties of the Two Functional Agonist Sites in GABA(A) Receptors. J. Neurosci. 2003, 23, 11158–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhler H. GABAA Receptors in Central Nervous System Disease: Anxiety, Epilepsy, and Insomnia. J. Recept. Signal Transduction Res. 2006, 26, 731–740. [DOI] [PubMed] [Google Scholar]
- Macdonald R. L.; Kang J. Q.; Gallagher M. J. Mutations in GABA Receptor Subunits Associated with Genetic Epilepsies. J. Physiol. 2010, 588, 1861–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U.; Knoflach F. Beyond Classical Benzodiazepines: Novel Therapeutic Potential of GABAA Receptor Subtypes. Nat. Rev. Drug Discovery 2011, 10, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin E.; Liu J.; Rudolph U. Alpha2-Containing GABA(A) Receptors: A Target for the Development of Novel Treatment Strategies for CNS Disorders. Pharmacol. Ther. 2012, 136, 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trincavelli M. L.; Da Pozzo E.; Daniele S.; Martini C. The GABA-BZR Complex as Target for the Development of Anxiolytic Drugs. Curr. Trends Med. Chem. 2012, 12, 254–269. [DOI] [PubMed] [Google Scholar]
- Whiting P. J. GABA-A Receptors: A Viable Target for Novel Anxiolytics?. Curr. Opin. Pharmacol. 2006, 6, 24–29. [DOI] [PubMed] [Google Scholar]
- Atack J. R. The Benzodiazepine Binding Site of GABA(A) Receptors as a Target for the Development of Novel Anxiolytics. Expert Opin. Invest. Drugs 2005, 14, 601–618. [DOI] [PubMed] [Google Scholar]
- Atack J. R. GABAA Receptor Subtype-Selective Modulators. I. Alpha2/Alpha3-Selective Agonists as Non-Sedating Anxiolytics. Curr. Top. Med. Chem. 2011, 11, 1176–1202. [DOI] [PubMed] [Google Scholar]
- Hosie A. M.; Wilkins M. E.; da Silva H. M.; Smart T. G. Endogenous Neurosteroids Regulate GABAA Receptors through Two Discrete Transmembrane Sites. Nature 2006, 444, 486–489. [DOI] [PubMed] [Google Scholar]
- Li G. D.; Chiara D. C.; Sawyer G. W.; Husain S. S.; Olsen R. W.; Cohen J. B. Identification of a GABAA Receptor Anesthetic Binding Site at Subunit Interfaces by Photolabeling with an Etomidate Analog. J. Neurosci. 2006, 26, 11599–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Hulst C.; Atack J. R.; Kooy R. F. The Complexity of the GABAA Receptor Shapes Unique Pharmacological Profiles. Drug Discovery Today 2009, 14, 866–875. [DOI] [PubMed] [Google Scholar]
- Forman S. A.; Miller K. W. Anesthetic Sites and Allosteric Mechanisms of Action on Cys-Loop Ligand-Gated Ion Channels. Can. J. Anaesth. 2011, 58, 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn B. G.; Brown A. R.; Lambert J. J.; Belelli D. Neurosteroids and GABA(A) Receptor Interactions: A Focus on Stress. Front. Neurosci. 2011, 5, 131. 10.3389/fnins.2011.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara D. C.; Dostalova Z.; Jayakar S. S.; Zhou X.; Miller K. W.; Cohen J. B. Mapping General Anesthetic Binding Site(S) in Human Alpha1beta3 Gamma-Aminobutyric Acid Type A Receptors with [(3)H]Tdbzl-Etomidate, a Photoreactive Etomidate Analogue. Biochemistry 2012, 51, 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter L.; de Graaf C.; Sieghart W.; Varagic Z.; Morzinger M.; de Esch I. J.; Ecker G. F.; Ernst M. Diazepam-Bound GABAA Receptor Models Identify New Benzodiazepine Binding-Site Ligands. Nat. Chem. Biol. 2012, 8, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston G. A.; Hanrahan J. R.; Chebib M.; Duke R. K.; Mewett K. N. Modulation of Ionotropic GABA Receptors by Natural Products of Plant Origin. Adv. Pharmacol. (San Diego, CA, U. S.) 2006, 54, 285–316. [DOI] [PubMed] [Google Scholar]
- Hanrahan J. R.; Chebib M.; Johnston G. A. Flavonoid Modulation of GABA(A) Receptors. Br. J. Pharmacol. 2011, 163, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J.; Sterner O. Modulation of GABA(A) Receptors by Natural Products and the Development of Novel Synthetic Ligands for the Benzodiazepine Binding Site. Curr. Drug Targets 2011, 12, 1674–1688. [DOI] [PubMed] [Google Scholar]
- Zaugg J.; Baburin I.; Strommer B.; Kim H. J.; Hering S.; Hamburger M. HPLC-Based Activity Profiling: Discovery of Piperine as a Positive GABA(A) Receptor Modulator Targeting a Benzodiazepine-Independent Binding Site. J. Nat. Prod. 2010, 73, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen M. E.; Metzler B.; Stafford G. I.; van Staden J.; Jager A. K.; Rasmussen H. B. Amides from Piper Capense with CNS Activity: A Preliminary SAR Analysis. Molecules 2009, 14, 3833–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara F. N.; Randall A.; Gunthorpe M. J. Effects of Piperine, the Pungent Component of Black Pepper, at the Human Vanilloid Receptor (TRPV1). Br. J. Pharmacol. 2005, 144, 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khom S.; Strommer B.; Schöffmann A.; Hintersteiner J.; Baburin I.; Erker T.; Schwarz T.; Schwarzer C.; Zaugg J.; Hamburger M.; Hering S. GABAA Receptor Modulation by Piperine and a Non-TRPV1 Activating Derivative. Biochem. Pharmacol. 2013, 85, 1827–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen I.; Clausen K.; Scheibye S.; Lawesson S. O. Thiation with 2,4-Bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-Disulfide: N-Methylthiopyrrolidone (2-pyrrolidinethione, 1-methyl-). Org. Synth. 1984, 62, 158–164. [Google Scholar]
- Hayashi K.; Yamazoe A.; Ishibashi Y.; Kusaka N.; Oono Y.; Nozaki H. Active Core Structure of Terfestatin A, a New Specific Inhibitor of Auxin Signaling. Bioorg. Med. Chem. 2008, 16, 5331–5344. [DOI] [PubMed] [Google Scholar]
- Blanchot M.; Candito D. A.; Larnaud F.; Lautens M. Formal Synthesis of Nitidine and Nk109 via Palladium-Catalyzed Domino Direct Arylation/N-Arylation of Aryl Triflates. Org. Lett. 2011, 13, 1486–1489. [DOI] [PubMed] [Google Scholar]
- Cabri W.; Candiani I.; Bedeschi A.; Santi R. 1,10-Phenanthroline Derivatives: A New Ligand Class in the Heck Reaction. Mechanistic Aspects. J. Org. Chem. 1993, 58, 7421–7426. [Google Scholar]
- Ishiyama T.; Takagi J.; Hartwig J. F.; Miyaura N. A Stoichiometric Aromatic C-H Borylation Catalyzed by Iridium(I)/2,2′-Bipyridine Complexes at Room Temperature. Angew. Chem., Int. Ed. 2002, 41, 3056–3058. [DOI] [PubMed] [Google Scholar]
- Thompson A. L. S.; Kabalka G. W.; Akula M. R.; Huffman J. W. The Conversion of Phenols to the Corresponding Aryl Halides under Mild Conditions. Synthesis 2005, 2005, 547–550. [Google Scholar]
- Grimm J. B.; Wilson K. J.; Witter D. J. Suppression of Racemization in the Carbonylation of Amino Acid-Derived Aryl Triflates. Tetrahedron Lett. 2007, 48, 4509–4513. [Google Scholar]
- Plunkett K. N.; Godula K.; Nuckolls C.; Tremblay N.; Whalley A. C.; Xiao S. Expeditious Synthesis of Contorted Hexabenzocoronenes. Org. Lett. 2009, 11, 2225–2228. [DOI] [PubMed] [Google Scholar]
- Klopman G.; Li J. Y. Quantitative Structure-Agonist Activity Relationship of Capsaicin Analogues. J. Comput.-Aided Mol. Des. 1995, 9, 283–294. [DOI] [PubMed] [Google Scholar]
- Hall M.; Frank E.; Holmes G.; Pfahringer B.; Reutemann P.; Witten I. H. The Weka Data Mining Software: An Update. SIGKDD Explorations 2009, 11110–18. [Google Scholar]
- Gunthorpe M. J.; Chizh B. A. Clinical Development of Trpv1 Antagonists: Targeting a Pivotal Point in the Pain Pathway. Drug Discovery Today 2009, 14, 56–67. [DOI] [PubMed] [Google Scholar]
- Marsch R.; Foeller E.; Rammes G.; Bunck M.; Kossl M.; Holsboer F.; Zieglgansberger W.; Landgraf R.; Lutz B.; Wotjak C. T. Reduced Anxiety, Conditioned Fear, and Hippocampal Long-Term Potentiation in Transient Receptor Potential Vanilloid Type 1 Receptor-Deficient Mice. J. Neurosci. 2007, 27, 832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y.; Szallasi A. Transient Receptor Potential (Trp) Channels: A Clinical Perspective. Br. J. Pharmacol. 2013, 10.1111/bph.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin A. L. In Expression and Analysis of Recombinant Ion Channels: From Structural Studies to Pharmacological Screening; Clare J. J., Trezise D. J., Eds.; Wiley–VCH Verlag: Weinheim, Germany, 2006; pp 1–25. [Google Scholar]
- Khom S.; Baburin I.; Timin E. N.; Hohaus A.; Sieghart W.; Hering S. Pharmacological Properties of GABAA Receptors Containing Gamma1 Subunits. Mol. Pharmacol. 2006, 69, 640–649. [DOI] [PubMed] [Google Scholar]
- Baburin I.; Beyl S.; Hering S. Automated Fast Perfusion of Xenopus Oocytes for Drug Screening. Pfluegers Arch. 2006, 453, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demel M. A.; Kraemer O.; Ettmayer P.; Haaksma E.; Ecker G. F. Ensemble Rule-Based Classification of Substrates of the Human ABC-Transporter ABCB1 Using Simple Physicochemical Descriptors. Mol. Inf. 2010, 29, 233–242. [DOI] [PubMed] [Google Scholar]
- Kohavi R.; Provost F. Glossary of Terms. Mach. Learn. 1998, 30, 271–274. [Google Scholar]
- Oprea T. I. Property Distribution of Drug-Related Chemical Databases. J. Comput.-Aided Mol. Des. 2000, 14, 251–264. [DOI] [PubMed] [Google Scholar]
- Wildman S. A.; Crippen G. M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Model. 1999, 39, 868–873. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.