

Background: The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) is key to Ca2+ homeostasis and is redox-regulated.
Results: SERCA 2 cysteine 674 mutation diminishes endothelial cell endoplasmic reticulum Ca2+ stores and impairs angiogenesis after hind limb ischemia and during hypoxia.
Conclusion: SERCA cysteine 674 is required for maintaining Ca2+ homeostasis and the angiogenic response to ischemia.
Significance: This reveals a novel redox control of ischemic angiogenesis involving Ca2+ stores.
Keywords: Angiogenesis, Calcium, Calcium ATPase, Endothelial Cell, Hypoxia
Abstract
The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) is key to Ca2+ homeostasis and is redox-regulated by reversible glutathione (GSH) adducts on the cysteine (C) 674 thiol that stimulate Ca2+ uptake activity and endothelial cell angiogenic responses in vitro. We found that mouse hind limb muscle ischemia induced S-glutathione adducts on SERCA in both whole muscle tissue and endothelial cells. To determine the role of S-glutathiolation, we used a SERCA 2 C674S heterozygote knock-in (SKI) mouse lacking half the key thiol. Following hind limb ischemia, SKI animals had decreased SERCA S-glutathione adducts and impaired blood flow recovery. We studied SKI microvascular endothelial cells in which total SERCA 2 expression was unchanged. Cultured SKI microvascular endothelial cells showed impaired migration and network formation compared with wild type (WT). Ca2+ studies showed decreased nitric oxide (·NO)-induced 45Ca2+ uptake into the endoplasmic reticulum (ER) of SKI cells, while Fura-2 studies revealed lower Ca2+ stores and decreased vascular endothelial growth factor (VEGF)- and ·NO-induced Ca2+ influx. Adenoviral overexpression of calreticulin, an ER Ca2+ binding protein, increased ionomycin-releasable stores, VEGF-induced Ca2+ influx and endothelial cell migration. Taken together, these data indicate that the redox-sensitive Cys-674 thiol on SERCA 2 is required for normal endothelial cell Ca2+ homeostasis and ischemia-induced angiogenic responses, revealing a novel redox control of angiogenesis via Ca2+ stores.
Introduction
Vascular endothelial growth factor (VEGF)4 is a critical mediator of angiogenesis, stimulating multiple pro-angiogenic behaviors in endothelial cells(1). VEGF production by endothelial cells and macrophages is stimulated by hypoxic conditions in ischemic tissue (1). In addition to initiating kinase cascades through VEGF receptor phosphorylation, VEGF also stimulates nitric oxide (·NO) production by increasing the activity of nitric oxide synthase (2). The central role for ·NO in the angiogenic response is illustrated by studies showing decreased blood flow recovery following hind limb ischemia and decreased angiogenic growth in the Matrigel plug assay in endothelial nitric oxide synthase (eNOS)-deficient mice (3–6). Although the mechanisms of action of ·NO in endothelial cells and pro-angiogenic pathways are poorly understood, it is known that in addition to signaling via cyclic-GMP, reactive oxygen and reactive nitrogen species such as ·NO can signal through changes in protein function induced by thiol modifications (7), although many of the important target proteins are not identified.
One target of ·NO induced post-translational modifications is the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), which plays multiple roles in the cardiovascular system (8). SERCA catalyzes the hydrolysis of ATP and couples it to the translocation of free cytosolic Ca2+ into SR/ER stores (9). Although expression patterns vary among cell types, SERCA 2b is expressed in all cells of the body, and we have previously reported that SERCA 2 is the key isoform responsible for ER Ca2+ uptake in human aortic endothelial cells (10).
Physiological levels of reactive oxygen and nitrogen species such as ·NO and hydrogen peroxide (H2O2), key signaling molecules in the cardiovascular system, increase reversible GSH adducts on cysteine 674 (Cys-674), the most reactive cysteine within the SERCA protein (7). In endothelial cells, reversible S-glutathiolation (GSH) at Cys-674 enhances maximal ATP-dependent Ca2+ uptake activity, decreases cytoplasmic Ca2+ levels, and promotes endothelial angiogenic responses in an overexpression system in vitro (10).
As a next crucial step in establishing the role of cysteine thiol cell signaling and oxidation in physiology and disease, we made a SERCA 2 C674S knock-in (SKI) mouse lacking the key thiol, which enables SERCA 2 to be activated by S-glutathiolation. Here we show that developmental and ischemic angiogenesis were impaired in SKI animals. In cardiac microvascular endothelial cells isolated from the SKI mouse, ER Ca2+ stores were depleted, and endothelial angiogenic behavior was impaired, but these defects could be rescued by directly increasing Ca2+ stores by overexpressing calreticulin, an ER Ca2+ store-binding protein. These studies indicate that redox-sensitive Ca2+ store maintenance via S-glutathione adducts on the key SERCA 2 Cys-674 thiol is required for normal angiogenic endothelial cell function and blood flow recovery after hind limb ischemia.
EXPERIMENTAL PROCEDURES
Materials
Endothelial basal medium-2 (EBM-2) was purchased from Lonza (Walkersville, MD). Endothelial mitogen (ECGS) was purchased from Biomedical Technologies, Inc. (Stoughton, MA). Fetal bovine serum (FBS), antibiotic-antimycotic, TRIzol, TaqMan primers, quantitative PCR (qPCR) reagents, Fura-2, pluronic F127, and ionomycin were purchased from Invitrogen (Grand Island, NY). Probenecid was from AAT Bioquest (Sunnyvale, CA). The SERCA 2 (IID8) and GAPDH antibodies as well as protein A/G PLUS-agarose beads were purchased from Santa Cruz Biotechnology (Dallas, TX), and the SERCA 2 C498 antibody was a custom polyclonal antibody from Bethyl Laboratories, Inc. (Montgomery, TX). Calreticulin antibody was purchased from Cell Signaling Technology (Danvers, MA). Anti-GSH antibody was from Virogen (Watertown, MA). Recombinant human VEGF-165 was purchased from R&D Systems (Minneapolis, MN). Diethylenetriamine NONOate (DETA NONOate, ·NO donor) was from Cayman Chemical (Ann Arbor, MI). Mouse calreticulin adenovirus was purchased from Vector Biolabs (Philadelphia, PA). Polybrene for adenoviral transduction, ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid tetrasodium salt (EGTA) for Ca2+ imaging experiments and DNase I for cell isolation were from Sigma-Aldrich. For protein harvest, cell lysis buffer and SDS sample buffer were from Boston Bioproducts (Ashland, MA), and phenylmethanesulfonyl fluoride (PMSF) and protease inhibitor mixture were from Sigma-Aldrich. SDS-PAGE gels were from Bio-Rad. Red blood cell lysis buffer and goat anti-rat IgG microbeads were from Miltenyi Biotec, Inc. (Auburn, CA). Collagenase type II was from Worthington Biochemical Corporation (Lakewood, NJ). Immobilon-FL PVDF protein transfer membranes were from Millipore (Billerica, MA). Gelatin and bovine serum albumin (BSA) were from Fisher Scientific (Waltham, MA). For Ca2+ uptake assays, 45Ca2+ and Ultima GoldTM XR scintillation fluid were from PerkinElmer. Rat anti-mouse CD31 antibody and MatrigelTM were from BD Biosciences (San Jose, CA).
Generation of the SKI Heterozygote Mouse
The SERCA 2 C674S heterozygote knock-in mouse was generated on a C57BL/6J background by inGenious Targeting Laboratory, Inc. (Ronkonkoma, NY). A mutated 14.4 kb SERCA exon 14 containing the Cys-674 to Ser-674 codon (TGT→TCC) was inserted into the endogenous SERCA2 gene locus. In this construct, a neomycin cassette was introduced into the intron between exon 14 and exon 15 for embryonic stem cells selection. The neomycin cassettes were removed in vivo by breeding in flp recombinase, and the recombinase was bred out by another round of breeding, all while maintaining the C57BL6 genetic background of the original embryonic stem cells used. This mouse was validated for absence of extra DNA inserts. Expression of the mutant allele is governed by the unaltered upstream native SERCA 2 promoter. The presence of the C674S mutation was confirmed by sequencing of both genomic DNA and cDNA from cardiac endothelial cells. Homozygous knock-in fetuses died in utero just prior to the initial period of vascular development (8–10.5 days) (11) so only heterozygous adult animals expressing 50% of SERCA 2 C674S allele were studied. All animal usage was approved by Boston University's Institutional Animal Care and Use Committee in accordance with the provisions of the Animal Welfare Act, Public Health Service Animal Welfare Policy, the principles of the NIH Guide for the Care and Use of Laboratory Animals, and the policies and procedures of Boston University Medical Campus. Animals were maintained in an AALAC approved Laboratory Animal Science Center staffed with licensed veterinarians.
Embryonic Development
Timed mating was carried out with male and female SKI mice. Pairs were housed together and females were examined each morning for the presence of a copulatory plug. The morning on which a copulatory plug appeared was considered day 0.5 post-fertilization. Females were sacrificed beginning at 7.5 days post-fertilization, and embryos were carefully dissected from the uterus. DNA was isolated from embryos for genotyping.
Hind Limb Ischemia Surgical Procedure and LASER Döppler Perfusion Imaging
8–12-week-old male littermate wild type (WT) and heterozygous SKI mice were anesthetized by intraperitoneal injection of xylazine (40 mg/kg) and ketamine (100 mg/kg). The left femoral artery, vein, and nerve proximal to the popliteal bifurcation site and distal to the inguinal ligament were ligated with 6–0 silk suture and excised. Perfusion in both the right and left lower limbs was recorded using LASER Döppler imaging pre-operatively, immediately post-operatively, and at the additional indicated post-operative time points. Blood flow was expressed as the ratio of ischemic to non-ischemic hind limb perfusion. Muscle tissue from ischemic and non-ischemic limbs was harvested at the indicated post-operative times and snap frozen in liquid nitrogen for RNA or protein analysis.
Isolation and Culture of Murine Cardiac Endothelial Cells
We have attempted to isolate hind limb muscle endothelial cells. However, yields were unacceptably low to be feasible. As an alternative, we have established methods for isolating primary cardiac microvascular endothelial cells from mice at greater than 90% purity using a Miltenyi AutoMACS Pro magnetic cell separator (Miltenyi Biotec, Auburn, CA). These cells have typical cobblestone appearance and express the endothelial cell antigens CD31 and eNOS, and at passage 4 are 87% CD31 positive. Briefly, mice were euthanized via inhaled anesthetic overdose (isoflurane) followed by cervical dislocation. Three hearts per genotype were pooled from mice aged 4–12 weeks. Hearts were rinsed in Hank's Balanced Salt Solution (HBSS) and minced with 3,000 units collagenase type II and 300 units DNase I. Samples were incubated at 37 °C with gentle agitation for 30 min, minced again then filtered through a 70 μm strainer. Samples were washed with Miltenyi AutoMACs running buffer and red blood cells were lysed. Cells were incubated with rat anti-mouse CD31 antibody followed by goat anti-rat IgG micro-beads, then labeled cells were separated according to the manufacturer's protocol. The resulting cells were resuspended in EBM-2 supplemented with 10% FBS, 50 μg/ml endothelial mitogen, and 1% antibiotic-antimycotic, then seeded on 0.5% gelatin-coated plates at a density of 20,000 cells per cm2. Cells were cultured and early passages used in in vitro experiments.
Immunoprecipitation
Following hind limb ischemia at the designated time points or 24 h in vitro hypoxia treatment of endothelial cells, whole muscle tissue or cell lysates were incubated with a custom polyclonal SERCA 2 antibody overnight then protein A/G beads for 1 h at 4 °C. Samples were then immunoblotted with a monoclonal GSH antibody. Following visualization of the bands using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE), membranes were stripped and re-probed with a monoclonal SERCA 2 IID8 antibody. Band density was evaluated using NIH ImageJ software, and the ratio of GSH to SERCA was used to determine relative amounts of SERCA-bound glutathione.
Migration Assay
Endothelial cells were grown to >90% confluence then serum starved in EBM-2 overnight. Scratch wounds were applied to the monolayer using a sterile pipette tip. Cells were washed once with PBS then treated with serum-free medium, VEGF (50 ng/ml) or DETA NONOate (30 μm) at the time of scratch application. Images were taken at fixed positions along the scratch at 0 and 6 h. Using NIH ImageJ software, images were analyzed by comparing the width of the scratch (averaged from three measurements per condition) at the two time points.
Capillary-like Network Formation Assay
In vitro endothelial cell network formation was assayed using BD MatrigelTM. The Matrigel assay (12, 13) takes advantage of the phenomenon that cultured endothelial cells will spontaneously form network structures when seeded on this complex solubilized extracellular matrix rich in laminin, collagen IV, and heparan sulfate proteoglycans. Briefly, 96-well plates were coated with BD MatrigelTM for 30–60 min according to the manufacturer's instructions prior to seeding endothelial cells at a density of 1 × 104 cells per well. Cells were exposed to serum-free medium or VEGF (50 ng/ml) in serum-free medium for 6 or 16 h. Network formation scores on a scale of 0 to 4 were assigned by blinded observers.
Hypoxia
An in vitro hypoxic environment (0.5% O2, 5% CO2, 70% humidity, 37 °C) was generated using a LiveCellTM Pathology Devices, Inc. (Westminster, MD) chamber system. Cells were seeded in complete medium at a of density 500,000 cells/well in 6-well plates, and incubated under hypoxic conditions for the time points indicated.
Western Blotting
Samples in SDS sample buffer were run on 10% electrophoresis gels or 4–20% SDS-PAGE gels. Proteins were transferred to supported PVDF membranes and blocked in 5% milk/0.1% Tween 20 phosphate-buffered saline (PBST). Primary antibodies were incubated overnight at 4 °C in 1% BSA. Horseradish peroxidase-conjugated and infrared dye-conjugated secondary antibodies were used, and blots were imaged using film or the Odyssey infrared imager, respectively. Band density was evaluated using NIH ImageJ software and normalized to the GAPDH loading control. Final densitometry data were normalized to WT controls for each experiment.
qPCR
Total RNA was isolated from cells using TRIzol according to the manufacturer's protocol. Complementary DNA was synthesized using the Invitrogen High Capacity RNA-to-cDNA kit. qPCR was performed using pre-validated gene-specific FAM-NFQ-conjugated TaqMan probes multiplexed with a VIC-NFQ conjugated 18 S probe. Expression was analyzed using the comparative CT method with StepOnePlusTM Real-Time PCR software (Invitrogen), with specific mRNA expression levels normalized to 18 S. Final expression data were normalized to WT control cells for each experiment.
45Ca2+ Uptake Assay
Ca2+ uptake into the ER was measured using an oxalate-dependent ER 45Ca2+ uptake assay in cells in which the cell membrane was permeabilized. Cells were treated with DETA NONOate (30 μmol/liter) for 2 h. Medium was replaced with Ca2+ uptake solution (in mmol/liter: 30 Tris-HCl, 100 KCl, 5 NaN3, 6 MgCl2, 0.15 EGTA, 0.12 CaCl2, 10 oxalate) and endothelial cells were permeablized with saponin (250 μg/ml) prior to treatment with thapsigargin (TG, 10 μmol/liter, 20 min) such that extravesicular Ca2+ concentration was controlled by buffer content. Following treatment, cells were trypsinized then incubated in solution with 45Ca2+ (1 μCi) and ATP (2 mmol/liter). After 30 min, cells were filtered through Whatman GF/C glass filters under vacuum and washed with physiological saline solution. Radioactivity was measured on a HIDEX 300 SL Automatic TDCR Liquid Scintillation Counter (LabLogic, Brandon, FL). 45Ca2+ uptake was evaluated by counting radioactivity on the filters and normalized to protein concentration (14).
Intracellular Ca2+ Imaging
Endothelial cells were seeded on several glass coverslips for each treatment group. On each coverslip, 15 to 25 cells were identified and individually recorded. The combined data from 3 coverslips for a treatment group in a single day was taken as an independent n = 1. In adenovirus experiments, separate transfections were performed for each n. For intracellular Ca2+ measurements, the cells on coverslips were loaded with 3.3 μm Fura-2 AM in the presence of 0.02% pluronic F127 in serum free EBM-2 at 37 °C for 30 min. Following a 15-min wash in serum-free EBM-2, cells were transferred to a nominally Ca2+-free physiological saline solution supplemented with 2.5 mmol/liter probenecid. Changes in intracellular Ca2+ were monitored as previously described (10, 15–17) For Ca2+ influx experiments, after cells equilibrated for 1 min, they were treated with VEGF (50 ng/ml) or ·NO gas in saline solution (10 μmol/liter) for 2 min (baseline reading), before Ca2+ was reconstituted by adding 2 mmol/liter CaCl2 to the solution. For background fluorescence subtraction, at the end of each experiment, maximal Ca2+ was assessed with ionomycin (2 μmol/liter) and MnCl2 (8 mmol/liter) was then added to quench the Fura-2 signal. For Ca2+ store release experiments, cells were placed in EGTA (1 mmol/liter) for 3 min and allowed to equilibrate. Ionomycin (1 μmol/liter) was then added to the solution. Multiple cells were studied simultaneously using a dual-excitation fluorescence imaging system (InCyte2 Intracellular Imaging, Cincinnati, OH). The imaging system was calibrated so that Rmax = 2.205 at 602 nmol/liter Ca2+ and Rmin = 0.66 at 0 nmol/liter Ca2+. Changes in cytosolic Ca2+ were recorded as changes in the ratio of fluorescence at 340 nm and 380 nm [Δ(F340/F380)]. Changes in intracellular Ca2+ were expressed as the difference in F340/F380 ratio between baseline prior to Ca2+ addition and peak ratio following addition.
Adenoviral Transduction of Endothelial Cells with Mouse Calreticulin
WT and SKI endothelial cells were transfected with LacZ or mouse calreticulin adenoviral vectors to achieve ∼4-fold overexpression. Cells were transduced in serum-free EBM-2 containing 10 μg/ml polybrene for 4 to 6 h before adding two volumes of complete medium for 24–48 h. Where indicated, cells were either incubated in complete medium or serum-free medium for an additional 24 h for a total of 48–72 h of transfection.
Statistical Analysis
All data are presented as means ± S.E. Statistical significance was determined using a Student's t test, or one- or two-way ANOVA in conjunction with a Bonferroni multiple comparisons post-hoc test where appropriate. Non-normally distributed data were evaluated using the Mann-Whitney U test. *, p < 0.05, **, p < 0.01, or ***, p < 0.001 was considered statistically significant. Statistical outliers were identified using the Grubb's outlier test and excluded from the analysis.
RESULTS
Redox-inactive SERCA C674S Mutant Impairs Angiogenesis in Vivo
In vivo vascular development was assessed in two ways, evaluating both fetal and adult angiogenic programs. Using standard breeding, no homozygous SKI pups were generated. In an assessment of embryonic development, genomic DNA sequencing of whole embryos (Fig. 1A) revealed that homozygous knock-in animals (serine 674: TCC) were present in utero up to day 8.5, just prior to the initial period of vascular development, but not afterward. Gross embryonic evaluation showed a lack of blood vessels in homozygous SKI animals, which were also smaller compared with heterozygotes (Fig. 1A). Ischemic angiogenesis was evaluated using the well-established hind limb ischemia model, comparing adult WT and heterozygous SKI animals. As shown in Fig. 1B, SKI mice had impaired blood flow recovery at 21 and 28 days post hind limb ischemia.
FIGURE 1.

SERCA Cys-674 is required for vascular development in the fetus and ischemic angiogenesis in the adult. A, genomic DNA at E8.5 of embryo in WT (C674/C674), heterozygote (C674/S674), and homozygote (S674/S674). B, LASER Döppler analysis of blood flow recovery in WT and SKI mice after femoral artery ligation at indicated times. Upper: representative LASER Döppler perfusion images of WT and SKI mice. Lower: summary of blood flow recovery after hind limb ischemia. *, p < 0.05, versus WT, n = 9–10.
Hind Limb Ischemia and Hypoxia in Vitro Induce SERCA S-Glutathione Adducts in WT, but Not SKI Muscle Tissue and Endothelial Cells
SERCA S-glutathiolation, assessed by immunoprecipitating SERCA protein and immunoblotting for GSH, increased rapidly in the ischemic limb muscle during the initial 24 h of ischemia (Fig. 2A) and was maintained for at least 3 days post femoral artery ligation (Fig. 2B). At 3 days post hind limb ischemia, WT mice had a significantly greater increase in SERCA glutathiolation than SKI mice in the ischemic hind limb muscle (Fig. 2B). Because the endothelium is a major contributor to angiogenesis after hind limb ischemia, we exposed endothelial cells to hypoxia to mimic acute ischemia and determined whether GSH adducts form specifically on the SERCA Cys-674 thiol. As shown in Fig. 2C, hypoxia significantly increased SERCA GSH adducts in WT, but not in SKI endothelial cells, indicating the major site of hypoxia-induced GSH adducts on SERCA 2 is Cys-674. In these isolated heterozygous SKI endothelial cells, sequencing of reverse transcribed mRNA confirmed expression of both WT (cysteine, TGT) and mutant (serine, TCC) at SERCA-674 (Fig. 3A). Total SERCA 2 mRNA (Fig. 3B) and protein levels (Fig. 3C) were similar in endothelial cells from WT and heterozygote SKI mice. However, despite expressing similar amounts of SERCA 2, SKI endothelial cells had no significant increase in Ca2+ uptake into the ER in response to the ·NO donor DETA NONOate (Fig. 3D), as demonstrated previously (10) in human aortic endothelial cells with adenoviral overexpression of WT and SERCA 2 C674S mutant protein.
FIGURE 2.

SERCA Cys-674 GSH adducts in ischemic hind limb and hypoxic mouse endothelial cells. A, SERCA S-glutathiolation (GSH) was assessed by immuno-precipitation of SERCA protein from whole muscle lysates from ischemic (I) or non-ischemic limbs (N) of WT mice following femoral artery ligation in 7 mice euthanized after increasing lengths of time during the first 24 h following artery ligation. B, comparison of SERCA GSH in the hind limb muscles between WT and SKI mice obtained 3 days after hind limb ischemia. For the densitometric analysis of SERCA S-glutathiolation, GSH was first corrected by SERCA. The fold induction of GSH after hind limb ischemia was determined by the ratio of GSH in the ischemic limb to non-ischemic limb and then normalized to WT. *, p < 0.05, versus WT, n = 5. C, mouse endothelial cells were subjected to 24 h of hypoxia, and whole cell lysates were immunoprecipitated and blotted for GSH and SERCA as above. *, p < 0.05, versus WT, n = 3.
FIGURE 3.

Redox-dependent ER Ca2+ uptake is impaired in SKI endothelial cells. A, sequencing of reverse transcribed mRNA from freshly isolated endothelial cells from WT and heterozygous SKI animals confirmed endothelial expression of the mutation. TCC = Serine Codon, TGT = Cysteine Codon. B, equal expression of SERCA 2 mRNA in freshly isolated WT and SKI endothelial cells. C, similar SERCA 2 protein levels in WT and SKI endothelial cells. D, redox-dependent ER Ca2+ uptake is impaired in cultured endothelial cells from SKI compared with WT. WT and SKI endothelial cells were stimulated with 30 μm DETA NONOate for 2 h. Thapsigargin-sensitive 45Ca2+ uptake is indicated as a ratio to unstimulated control. **, p < 0.01, versus WT, n = 7.
Redox Inactive SERCA 2 Mutant Promotes Endothelial Dysfunction in Vitro through ·NO/VEGF Pathway Disruption
In an in vitro scratch wound assay, SKI endothelial cells had significantly impaired migratory responses to VEGF (Fig. 4A). Data from the Matrigel assay indicate that both basal and VEGF-induced capillary-like network formation were significantly impaired in SKI endothelial cells (Fig. 4B). Fura-2 Ca2+ signaling studies showed that neither ·NO nor VEGF detectably increased intracellular when added in Ca2+ solution, but that Ca2+ influx occurred following the reintroduction of extracellular Ca2+. The increase in Ca2+ was significantly less in SKI than in WT endothelial cells. (Fig. 5, A and B). As shown in the inset in Fig. 5A, the Ca2+ influx was agonist-dependent and not due to cellular leak as demonstrated by the absence of influx when the saline vehicle was added prior to readdition of Ca2+. In addition, in the absence of extracellular Ca2+ there was significantly decreased ionomycin-releasable Ca2+, indicating that SKI endothelial cells have diminished Ca2+ stores (Fig. 6A). Furthermore, pretreatment of WT endothelial cells with VEGF decreased ionomycin-releasable Ca2+ (Fig. 6B) indicating that the growth factor depletes Ca2+ stores. However, VEGF failed to further diminish the already depleted stores in SKI endothelial cells.
FIGURE 4.

SKI endothelial cells have impaired VEGF-mediated angiogenic behavior. A, cultured endothelial cell migration was evaluated using a monolayer scratch wound assay. Cells were exposed either to serum free medium or VEGF (50 ng/ml) for 6 h. *, p < 0.05, versus WT, n = 5. B, capillary-like network formation was evaluated in cultured endothelial cells seeded on BD MatrigelTM. Cells were exposed to serum-free medium or VEGF (50 ng/ml) for 6 h. *, p < 0.05, versus WT; **, p < 0.01, versus WT, n = 4.
FIGURE 5.

Ca2+ signaling is altered in SKI endothelial cells. Representative fura-2 traces and bar graphs show the effect of 10 μm ·NO (A) or 50 ng/ml VEGF (B) on Ca2+ influx following addition of 2 mmol/liter Ca2+ to Ca2+-free solution. *, p < 0.05, versus WT, n = 3–4. Representative traces are followed by quantification. The inset in A shows representative Fura-2 traces of WT and SKI endothelial cells showing addition of Ca2+ after addition of the saline vehicle for ·NO or VEGF.
FIGURE 6.

Ca2+ store maintenance is impaired in SKI endothelial cells. A, release of Ca2+ stores by ionomycin (1 μm) in the absence of extracellular Ca2+. ***, p < 0.001, versus WT, n = 4. B, ionomycin-induced Ca2+ store release after 45 min pre-treatment with VEGF (50 ng/ml). *, p < 0.05; **, p < 0.01 versus WT; *, p < 0.05, versus ionomycin alone, n = 4. Representative traces are followed by quantification.
Mitigating Ca2+ Store Deficiency Ameliorates SKI Endothelial Dysfunction
By assessing ionomycin-releasable Ca2+ stores, we determined that 4-fold overexpression of calreticulin (Fig. 7A), an ER resident Ca2+-binding protein that helps maintain ER Ca2+ levels (18, 19) restored ionomycin-releasable Ca2+ stores (Fig. 7B). The effect of calreticulin was independent of any significant effect on SERCA 2 mRNA (not shown) or protein expression (Fig. 7A). VEGF-induced Ca2+ influx, which is dependent on Ca2+ store operated pathways (10) was also restored by calreticulin overexpression (Fig. 8). Importantly, functional data demonstrate improved VEGF-induced endothelial cell migration following calreticulin overexpression (Fig. 9), directly implicating Ca2+ store depletion as the cause of impaired angiogenic responses of SKI endothelial cells.
FIGURE 7.

Calreticulin mitigates Ca2+ store deficiency. A, immunoblots and densitometric quantification of calreticulin and SERCA 2 protein following 48–72 h of calreticulin overexpression. *, p < 0.05, versus LacZ, n = 6. B, ionomycin releasable Ca2+ stores. *, p < 0.05, versus WT; ***, p < 0.001, versus LacZ, n = 5. A representative trace is followed by quantification.
FIGURE 8.

VEGF induced Ca2+ influx in SKI endothelial cells improves following Ca2+ store restitution. VEGF-induced Ca2+ influx. *, p < 0.05; ***, p < 0.001 versus LacZ, n = 5–6. Representative traces are followed by quantification.
FIGURE 9.
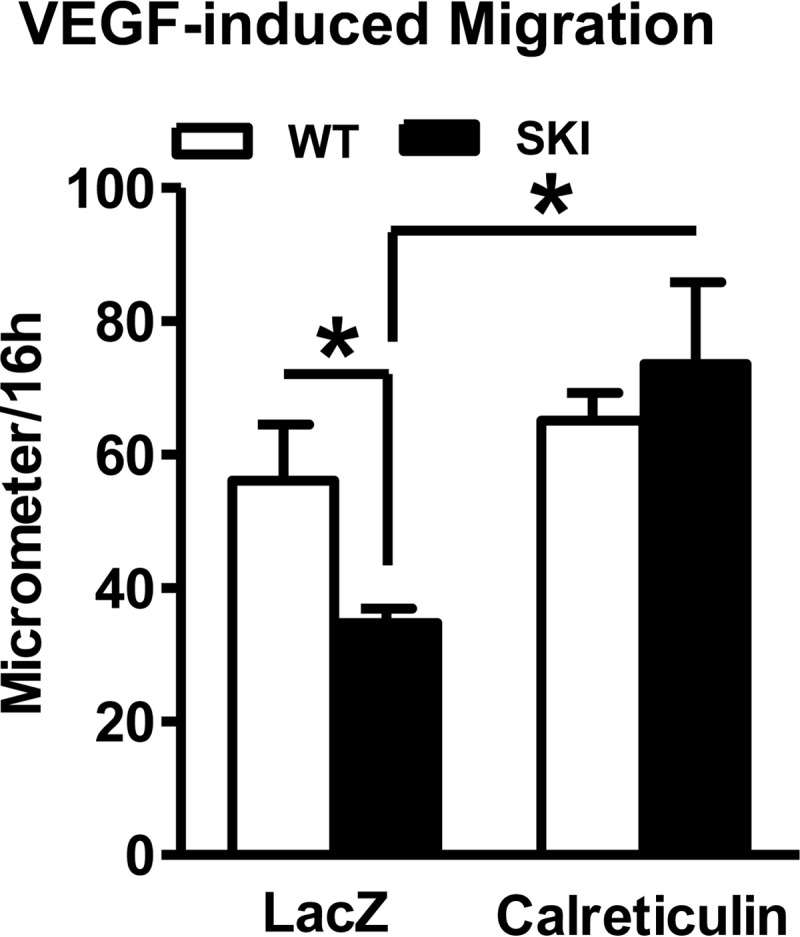
Migration of cultured endothelial cells was evaluated after LacZ or calreticulin overexpression in response to either serum-free medium or VEGF (50 ng/ml) for 16 h. *, p < 0.05, versus WT or LacZ, n = 7–8.
DISCUSSION
Ischemia-induced oxidant production initiates signaling cascades essential for vascular repair (20), and our work here shows that redox regulation of SERCA 2 Cys-674 via reversible S-glutathiolation modification plays a significant role in ischemia-induced angiogenic responses in vivo. Furthermore, we provide new evidence that SERCA Cys-674 is post-translationally modified by GSH during ischemia and hypoxia, and that redox regulation by SERCA 2 of Ca2+ stores is essential for normal endothelial responses to VEGF and hypoxia.
In the present work, using the heterozygote redox inactive SERCA mutant SKI mouse lacking a full complement of the key Cys-674 thiol, we show that redox active SERCA Cys-674 is required for successful fetal angiogenic development. In addition, a clinically relevant and important parameter that reflects angiogenesis in the adult is the restoration of blood flow following ischemia. Although ischemic angiogenesis in the adult can occur with only 50% redox active SERCA, the response is significantly impaired. While the precise mechanisms to explain the difference in blood flow in vivo require further study, given that development to the adult was not severely affected by heterozygote expression of the mutant SERCA 2 C674S allele, it is possible that either developmental angiogenesis avoids redox-regulated mechanisms, or those changes in endothelial cells that occur during ischemia or during growth factor responses in vitro require them to a greater degree. We speculate that in vivo studies of SKI mice under various conditions of increased oxidant generation might further reveal the importance of redox-dependent regulation by SERCA of Ca2+ stores shown here.
Notably, early blood flow recovery did not differ between WT and SKI animals, and the impairment manifested itself 2 weeks after the ischemic insult. This lack of difference in blood flow early on despite differences in glutathiolation of SERCA is important. One possible explanation is that many cell types may contribute to the in vivo result, including macrophages and other immune cells, muscle cells, and fibroblasts, but the in vitro experiments focus only on endothelial cells. Additionally, the contribution of early endothelial signaling to later angiogenic potential is not fully understood, but we hypothesize that endothelial SERCA glutathiolation may play a significant role, and we have shown that increased SERCA glutathiolation is sustained for 3 days in vivo.
Furthermore, during the early stages of blood flow recovery, although endothelial SERCA glutathiolation may be an important angiogenic signal, we may not be directly evaluating angiogenic function in the first 2 weeks. Although the hind limb ischemia model is a well-established method of evaluating angiogenesis, like other methods, it is subject to several flaws. First, it has been demonstrated in the literature that inter-animal variability in the vasculature at baseline can potentially have a more profound impact on measured blood flow recovery than other experimental manipulations (22). Additionally, there exists controversy as to what the different phases of blood flow recovery actually represent. It has been suggested that early blood flow recovery, particularly in the first 7 days after ischemia, is the result of recruitment of existing collateral vessels, whereas later recovery is due to the development of new vessels through angiogenesis and arteriogenesis (23). The lack of difference in blood flow recovery in WT and SKI animals in the first 2 weeks may be the result of equal ability to recruit existing vessels. In contrast, because the SKI animals demonstrate a defect in angiogenic potential, they are unable to sufficiently restore blood flow at later stages when new vessel development is required under stress. Thus, early responses to severe ischemia may be maximal and as suggested above may be governed by structural factors whereas angiogenesis and arteriogenesis become important later.
In addition to the in vivo effects, redox inactivation of SERCA 2 at Cys-674 impaired Ca2+ store regulation and largely prevented ·NO-dependent 45Ca2+ uptake and VEGF-mediated angiogenic behavior in vitro. This work, which did not require adenoviral overexpression of SERCA used in our earlier studies, also verifies the importance of endogenous levels of the SERCA 2 redox-sensitive Cys-674 thiol for angiogenic behavior. Failure of homozygous mice to develop beyond the stage of prenatal vascular development is also consistent with a need for the SERCA 2 reactive thiol for normal developmental angiogenesis in vivo, but this interesting finding will be pursued in future work.
Expression of the SERCA 2 C674S Mutant Promotes Endothelial Dysfunction in Vitro through ·NO/VEGF Pathway Disruption
Angiogenesis is a complex process requiring endothelial cell activation and vascular basement membrane disruption followed by proliferation and migration of endothelial cells in response to chemotactic gradients of growth factors and VEGF signaling (21). The in vitro monolayer scratch wound assay showed that SKI endothelial cells had impaired migratory responses to VEGF, suggesting a critical defect in an early stage of the angiogenic process.
Although scratch wound assays are routinely used to evaluate the migratory behavior of many cell types, they are subject to several limitations and confounding factors. First, in an angiogenic context, cells migrate directionally in response to chemotactic gradients. In the scratch wound assay, cells can essentially only migrate in one direction, into the denuded area. Additionally, the formation and disruption of focal adhesions, integrin signaling, and cell spreading are all factors that can affect the measured outcome of cell movement, but may not be directly related to defects in growth factor signaling. These factors led us to supplement our findings with the network formation assay.
Following migration, another important step in the formation of the structure of a nascent vessel is organization of endothelial cells into a network. Network formation is a complex process that requires coordinated interaction not only between cells to achieve appropriate directional movement and juxtaposition, but also with the extracellular matrix for the disruption and reformation of adhesions to the basement membrane. The Matrigel assay data suggest that basal and VEGF-induced network formation are impaired in SKI endothelial cells, indicating a required role for SERCA redox regulation in the ability of endothelial cells to execute complex cell-cell and cell-matrix interactions in response to stimuli in vitro. VEGF-induced oxidants as well as direct application of ·NO stimulate S-glutathiolation and activation of SERCA Cys-674 in human aortic endothelial cells, thus promoting angiogenic responses in endothelial cells in vitro (10). In the present work we determined that hypoxia alone increased SERCA Cys-674 glutathiolation that is required for in vitro angiogenic behavior and that altered SERCA redox regulation in SKI endothelial cells leads to aberrant ·NO/VEGF pathway Ca2+ signaling, thereby diminishing angiogenic responses.
Expression of the SERCA 2 C674S Mutant Diminishes Ca2+ Stores
Fura-2 Ca2+ signaling studies demonstrated impaired VEGF- and ·NO-induced Ca2+ influx, similar to previous studies in human aortic endothelial cells with SERCA 2 C674 WT and C674S mutant adenoviral overexpression (10). In these human cells, we found that regulation of the store-operated Ca2+ entry (SOCE) pathway to be critical for VEGF- and ·NO-induced Ca2+ influx. Specifically, knocking down the SOCE channel Orai1 significantly blunted the VEGF- and ·NO-induced Ca2+ influx (10). Also, high concentrations of ryanodine, which should prevent store emptying, blocked VEGF- and ·NO-induced Ca2+ influx (10). Together with the current studies in mouse endothelial cells, these data corroborate our conclusions that SERCA-dependent intracellular Ca2+ stores are important for ·NO/VEGF responses. In addition, here we show significantly decreased ionomycin-releasable Ca2+, indicating that SKI endothelial cells have diminished Ca2+ stores. Furthermore, pretreatment of WT endothelial cells with VEGF decreased ionomycin-releasable Ca2+, indicating that Ca2+ store release plays a role in the VEGF response. However, in SKI endothelial cells, VEGF failed to further diminish the already depleted stores. These data are consistent with a requirement for Ca2+ store depletion-mediated Ca2+ influx for VEGF-induced angiogenic behaviors, and suggest that SKI endothelial cells may have impaired angiogenic responses because of inadequate Ca2+ stores or inadequately responsive stores.
Notably, Fura-2 in the cytosol failed to detect any substantial Ca2+ rise upon application of VEGF in the absence of extracellular Ca2+. This result suggests that VEGF-induced Ca2+ release in endothelial cells is small and/or quickly compartmentalized, which may explain the inability of Fura2 to detect global cytosolic Ca2+ rise. However, the results showing that VEGF does partially deplete Ca2+ stores clearly indicate that no matter how small VEGF release is on the whole-cell scale, and that it is enough to evoke substantial Ca2+ entry, further supporting a role for a store-operated Ca2+ entry mechanism.
Also, although this study confirmed that cultured endothelial cells from SKI mice exhibit abnormal function, because the SKI mouse expresses the mutant SERCA in all cells, it is possible that other cells may contribute to the abnormal responses observed in the SKI mouse in vivo.
Ca2+ Store Restoration Averts Endothelial Dysfunction
Although the importance of Ca2+ signaling to basic endothelial functions such as proliferation, migration, and differentiation is well-established, the specific mechanisms of Ca2+ regulation are complex, multifaceted, and often poorly understood. Given that disturbances of Ca2+ signaling are deleterious to endothelial function and vascular health, we hypothesized that overexpressing calreticulin, an important ER resident Ca2+-binding protein that helps maintain ER Ca2+ levels (18, 19) might rescue the decreased Ca2+ stores and therefore cell dysfunction observed in the SKI endothelial cells. Although the exact mechanisms of the relationship between Ca2+ stores and signaling and endothelial function are as yet unclear, calreticulin overexpression enhanced SKI endothelial cell VEGF-induced Ca2+ influx and migration, further indicating that the lack of redox-dependent Ca2+ store filling is responsible for the abnormalities in Ca2+ stores and function. Thus, our results indicate that there is Ca2+ store-dependent modulation of VEGF responses, and impaired redox regulation of SERCA 2 results in Ca2+ disruption, critically retarding angiogenic behavior.
Taken together, these data indicate that redox regulation of SERCA 2 Cys-674 is critical for promoting endothelial angiogenic behaviors in response to VEGF, and for the first time elucidates a Ca2+-dependent mechanism whereby maintenance of Ca2+ stores by a redox-dependent post-translational modification on one SERCA cysteine thiol has a direct consequence for angiogenic signaling. Because Ca2+ store deficiency has signaling and functional consequences for SKI endothelial cells, and endothelial dysfunction is mitigated by rebuilding stores with calreticulin, Ca2+ store homeostasis is evidently a link between GSH adduct modification of SERCA and pro-angiogenic signaling. As redox-dependent activation of SERCA 2 depends upon ·NO, and ·NO is required for angiogenesis, it is apparent that there is normally a positive reinforcement of angiogenic signaling that requires SERCA Cys-674 redox-dependent Ca2+ store maintenance that has not been previously recognized. The present work contributes to an improved understanding of the causes and consequences of oxidants arising during ischemia, and may thus inform novel therapeutic development for ischemic cardiovascular diseases.
This work was supported, in whole or in part, by National Institute of Health Grants HL031607-30 (to R. A. C., X. T.), R37 HL104017 (to R. A. C.), and R01-HL071793 (to V. B.), NHLBI T32 Training Grant HL07969 (to M. D. T.) American Diabetes Association Award 7-09-JF-69 (to X. T.), and a Martin Luther King, Jr. Fellowship from Boston University (to M. D. T.), and the NHLBI-sponsored Boston University Cardiovascular Proteomics Center (Contract No. N01-HV-28178, to R. A. C.). These studies were also supported by the Calcium Affinity Research Collaborative of the Evans Center, Department of Medicine, Boston University Medical Center.
- VEGF
- vascular endothelial growth factor
- EBM-2
- endothelial basal medium-2
- eNOS
- endothelial nitric-oxide synthase
- ER
- endoplasmic reticulum
- GSH
- glutathione
- ·NO
- nitric oxide
- qPCR
- quantitative PCR
- redox
- reduction/oxidation
- SERCA 2
- sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2
- SKI
- SERCA 2 C674S knock-in
- TG
- thapsigargin.
REFERENCES
- 1. Neufeld G., Tessler S., Gitay-Goren H., Cohen T., Levi B. Z. (1994) Vascular endothelial growth factor and its receptors. Prog. Growth Factor Res. 5, 89–97 [DOI] [PubMed] [Google Scholar]
- 2. Haeussler D. J., Pimentel D. R., Hou X., Burgoyne J. R., Cohen R. A., Bachschmid M. M. (2013) Endomembrane H-Ras controls vascular endothelial growth factor-induced nitric-oxide synthase-mediated endothelial cell migration. J. Biol. Chem. 288, 15380–15389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee P. C., Salyapongse A. N., Bragdon G. A., Shears L. L., 2nd, Watkins S. C., Edington H. D., Billiar T. R. (1999) Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 277, H1600–H1608 [DOI] [PubMed] [Google Scholar]
- 4. Tamarat R., Silvestre J.-S., Kubis N., Benessiano J., Duriez M., deGasparo M., Henrion D., Levy B. I. (2002) Endothelial nitric oxide synthase lies downstream from angiotensin II-induced angiogenesis in ischemic hindlimb. Hypertension 39, 830–835 [DOI] [PubMed] [Google Scholar]
- 5. Urano T., Ito Y., Akao M., Sawa T., Miyata K., Tabata M., Morisada T., Hato T., Yano M., Kadomatsu T., Yasunaga K., Shibata R., Murohara T., Akaike T., Tanihara H., Suda T., Oike Y. (2008) Angiopoietin-related growth factor enhances blood flow via activation of the ERK1/2-eNOS-NO pathway in a mouse hind-limb ischemia model. Arterioscler. Thromb. Vasc. Biol. 28, 827–834 [DOI] [PubMed] [Google Scholar]
- 6. Yu J., deMuinck E. D., Zhuang Z., Drinane M., Kauser K., Rubanyi G. M., Qian H. S., Murata T., Escalante B., Sessa W. C. (2005) Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc. Natl. Acad. Sci. U. S. A. 102, 10999–11004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adachi T., Weisbrod R. M., Pimentel D. R., Ying J., Sharov V. S., Schöneich C., Cohen R. A. (2004) S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nature Med. 10, 1200–1207 [DOI] [PubMed] [Google Scholar]
- 8. Adachi T. (2010) Modulation of vascular sarco/endoplasmic reticulum calcium ATPase in cardiovascular pathophysiology. Adv. Pharmacol. 59, 165–195 [DOI] [PubMed] [Google Scholar]
- 9. Vangheluwe P., Raeymaekers L., Dode L., Wuytack F. (2005) Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: cell biological implications. Cell Calcium 38, 291–302 [DOI] [PubMed] [Google Scholar]
- 10. Evangelista A. M., Thompson M. D., Weisbrod R. M., Pimental D. R., Tong X., Bolotina V. M., Cohen R. A. (2012) Redox Regulation of SERCA2 Is Required for Vascular Endothelial Growth Factor-Induced Signaling and Endothelial Cell Migration. Antioxid. Redox Signal. 17, 1099–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carmeliet P., Moons L., Collen D. (1998) Mouse models of angiogenesis, arterial stenosis, atherosclerosis and hemostasis. Cardiovasc. Res. 39, 8–33 [DOI] [PubMed] [Google Scholar]
- 12. Khoo C. P., Micklem K., Watt S. M. (2011) A comparison of methods for quantifying angiogenesis in the matrigel assay in vitro. Tissue Eng. Part C, Methods 17, 895–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagata D., Mogi M., Walsh K. (2003) AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J. Biol. Chem. 278, 31000–31006 [DOI] [PubMed] [Google Scholar]
- 14. Adachi T., Matsui R., Weisbrod R. M., Najibi S., Cohen R. A. (2001) Reduced sarco/endoplasmic reticulum Ca(2+) uptake activity can account for the reduced response to NO, but not sodium nitroprusside, in hypercholesterolemic rabbit aorta. Circulation 104, 1040–1045 [DOI] [PubMed] [Google Scholar]
- 15. Abdullaev I. F., Bisaillon J. M., Potier M., Gonzalez J. C., Motiani R. K., Trebak M. (2008) Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circulation Res. 103, 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Corda S., Spurgeon H. A., Lakatta E. G., Capogrossi M. C., Ziegelstein R. C. (1995) Endoplasmic reticulum Ca2+ depletion unmasks a caffeine-induced Ca2+ influx in human aortic endothelial cells. Circulation Res. 77, 927–935 [DOI] [PubMed] [Google Scholar]
- 17. Ziegelstein R. C., Xiong Y., He C., Hu Q. (2006) Expression of a functional extracellular calcium-sensing receptor in human aortic endothelial cells. Biochem. Biophys. Res. Commun. 342, 153–163 [DOI] [PubMed] [Google Scholar]
- 18. Coe H., Michalak M. (2009) Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 28, F96–F103 [PubMed] [Google Scholar]
- 19. Michalak M., Groenendyk J., Szabo E., Gold L. I., Opas M. (2009) Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417, 651–666 [DOI] [PubMed] [Google Scholar]
- 20. Maulik N. (2002) Redox signaling of angiogenesis. Antioxid. Redox Signal. 4, 805–815 [DOI] [PubMed] [Google Scholar]
- 21. Folkman J. (2003) Fundamental concepts of the angiogenic process. Curr. Mol. Med. 3, 643–651 [DOI] [PubMed] [Google Scholar]
- 22. Zbinden S., Clavijo L. C., Kantor B., Morsli H., Cortes G. A., Andrews J. A., Jang G. J., Burnett M. S., Epstein S. E. (2007) Interanimal variability in preexisting collaterals is a major factor determining outcome in experimental angiogenesis trials. Am. J. Physiol. Heart Circ. Physiol. 292, H1891–H1897 [DOI] [PubMed] [Google Scholar]
- 23. Lotfi S., Patel A. S., Mattock K., Egginton S., Smith A., Modarai B. (2013) Towards a more relevant hind limb model of muscle ischaemia. Atherosclerosis 227, 1–8 [DOI] [PubMed] [Google Scholar]