

Background: Immune activation through surface expression of the human NKG2D ligand MICA is important in clearance of virus-infected or cancerous cells.
Results: Molecular characterization of N-glycosylation in regulation of MICA cell surface expression.
Conclusion: Surface expression of MICA alleles vary in dependence for N-glycosylation.
Significance: We identify N-glycosylation as an allele-specific regulatory mechanism of MICA and pinpoint the essential residues.
Keywords: Genetic Polymorphism, Glycosylation, Herpesvirus, Innate Immunity, Melanoma, Natural Killer Cells (NK Cells)
Abstract
NKG2D is an activating receptor expressed on several types of human lymphocytes. NKG2D ligands can be induced upon cell stress and are frequently targeted post-translationally in infected or transformed cells to avoid immune recognition. Virus infection and inflammation alter protein N-glycosylation, and we have previously shown that changes in cellular N-glycosylation are involved in regulation of NKG2D ligand surface expression. The specific mode of regulation through N-glycosylation is, however, unknown. Here we investigated whether direct N-glycosylation of the NKG2D ligand MICA itself is critical for cell surface expression and sought to identify the essential residues. We found that a single N-glycosylation site (Asn8) was important for MICA018 surface expression. The frequently expressed MICA allele 008, with an altered transmembrane and intracellular domain, was not affected by mutation of this N-glycosylation site. Mutational analysis revealed that a single amino acid (Thr24) in the extracellular domain of MICA018 was essential for the N-glycosylation dependence, whereas the intracellular domain was not involved. The HHV7 immunoevasin, U21, was found to inhibit MICA018 surface expression by affecting N-glycosylation, and the retention was rescued by T24A substitution. Our study reveals N-glycosylation as an allele-specific regulatory mechanism important for regulation of surface expression of MICA018, and we pinpoint the residues essential for this N-glycosylation dependence. In addition, we show that this regulatory mechanism of MICA surface expression is likely targeted during different pathological conditions.
Introduction
Generation of adaptive immunity is dependent on activation of antigen-presenting cells that can sense foreign pathogenic molecules through pathogen recognition receptors (1). Malfunctioning or stressed cells from the body itself rely on other pathways for immune activation, such as host cells that can alert the immune system through expression of stress molecules (2, 3). The natural killer group 2D (NKG2D)3 and NKG2D ligand sensing system is involved in regulation of both innate and adaptive immunity and is recognized as a prominent player in immune recognition of abnormal cells (4–6).
Ligands for NKG2D are induced on the surface of inflamed or stressed cells where they activate immune responses through interaction with NKG2D-expressing effector cells. The NKG2D receptor is expressed on NK, NKT, and CD8 T-cells and some CD4 T-cells (5, 7, 8). There are multiple ligands for NKG2D divided into two families: MHC class I chain-related (MIC) and UL16-binding protein (ULBP) (9, 10). Currently, MICA, MICB, and ULBP1–6 have been described in humans (11). MIC and ULBP proteins are homologs of the heavy chain of the classical MHC class I molecules; however, they do not associate with β2-microglobulin or present peptides (12–14). MICA and MICB are encoded from the MHC locus and are remarkably polymorphic (8, 9). At present, 76 different alleles of MICA have been identified, the majority being highly homologous. The 008 allele of MICA is unusual because it encodes a molecule lacking a cytoplasmic tail caused by a frameshift mutation that leads to a premature stop codon (15, 16). The MICA008 allele is frequent in most studied human populations, and it has been speculated that the high frequency is linked to protection from certain diseases (17).
Different forms of cell stress ranging from tumor transformation and viral infection to chronic inflammation and autoimmunity can induce NKG2D ligand surface expression (5, 6, 8). Cancer and viruses use various strategies to prevent engagement with NKG2D-positive effector cells, highlighting the importance of the NKG2D/NKG2D ligand system in the host response to viral infection and transformation. Multiple viruses encode proteins that affect NKG2D ligand transport to the cell surface and thereby evade immune activation. Inhibition of MICA, MICB, and ULBP1–2 by different viruses is well documented (10, 18–22). A recently discovered example is the human herpesvirus 7 (HHV7), which causes persistent infection in up to 90% of human populations (23). HHV7 encodes an immunoevasin, U21, which was found to down-regulate cell surface ULBP1, MICB, and MICA (24).
Some viruses can inhibit full-length MICA alleles, but not the truncated MICA008. An example is the human cytomegalovirus encoded glycoprotein UL142, which binds and retains only full-length MICA alleles in the cis-Golgi complex (22, 25). Also the human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus (HHV8/KSHV) K5 ubiquitin ligase hinders surface expression of MICB and full-length MICA through ubiquitination, but not MICA008 (26). The adenovirus E3/19K protein retains MICA and MICB in the endoplasmic reticulum (ER) (27) and is until now the only example of a virus targeting both full-length MICA alleles and the truncated MICA008.
Cancer cells can escape immune recognition by cleaving NKG2D ligands from their cell surfaces or by intracellular retention (28–31). Melanomas have been shown to restrict MICA cell surface expression despite possessing significant intracellular levels of MICA protein (32).
We have previously shown that intact N-glycosylation machinery is important for cell surface expression of MICA and MICB. Blocking general N-glycosylation did not affect the transcriptional activation of MICA, implying that N-glycosylation of MICA or other proteins involved in the intracellular transport is required for MICA cell surface expression (33). It remains unknown whether N-glycosylation of MICA itself directly regulates surface expression of MICA.
N-Glycosylation of proteins has multiple effects, ranging from intracellular sorting to regulation of half-life and bioactivity (34–36). However, many proteins that are normally glycosylated show no loss of function when glycosylation is prevented (37). N-Glycosylation is initiated by a cotranslational process in the lumen of ER. The N-glycans are added to consensus sequences Asn-Xaa-(Thr/Ser) and further trimmed and remodeled upon transit through the Golgi. Cancer and infection often result in altered glycosylation of cellular proteins (38–40). Although the purposes may be numerous, it is likely that the changes in N-glycosylation participate in escape of immune recognition.
Based on our previous studies, which showed that 2-deoxy d-Glucose (2DG) inhibited MIC surface expression by blocking cellular N-glycosylation, the aim of this project was to investigate the importance of the molecular N-glycan pattern of MICA itself for cell surface expression of MICA. Furthermore, we sought to identify residues in MICA important for this N-glycosylation-mediated regulation. We show that N-glycosylation is an allele-specific post-translational mechanism directly regulating MICA018 surface expression. We found that abrogation of N-glycosylation at position Asn8 inhibited cell surface expression of MICA018. This regulatory mechanism did not apply to MICA008 surface expression, which was unaffected by changes in N-glycosylation. Mutational analysis focusing on the differences between MICA018 and MICA008 alleles showed that a single amino acid (Thr24) in the extracellular domain of MICA018 was essential for N-glycosylation dependence, whereas the cytoplasmic tail was not involved. These regulatory properties of MICA018 sensitivity to N-glycosylation were found to be targeted by cells expressing HHV7-U21. Furthermore, MICA008 surface expression was also inhibited in HHV7-U21-expressing cells, possibly through a novel post-translational regulatory mechanism.
EXPERIMENTAL PROCEDURES
Cells
Human peripheral blood mononuclear cells (PBMCs), Jurkat Tag-9 (T-ALL), Granta-519 (Mantle cell lymphoma), FM78, FM86, FM55p, FM82, and SK-mel-28 (melanoma cell lines), U373 (glioblastoma-astrocytoma), and pHM-U21 (U373 cells stably transfected with U21) were kindly provided by Amy Hudson (Medical College of Wisconsin). PBMCs. Suspension cell lines and melanomas were cultured as previously described (33, 43). U373 and pHM-U21were cultured in DMEM with GlutaMAX (Invitrogen) supplemented with 10% FBS (Sigma-Aldrich) and 2 mmol/liter streptomycin/penicillin (Sigma-Aldrich). Stable expression of U21 in U373 cells was carried out using lentivirus-mediated gene transfer, using the pHAGE vector pHM, which carries the selectable marker for hygromycin resistance. Cells were subcloned from hygromycin-resistant colonies to ensure uniformity of expression (41). Jurkat Tag-9 cells were kindly provided by C. Geisler (University of Copenhagen, Copenhagen, Denmark). Melanoma cell lines were kindly provided by Dr. Per Thor Straten (University Hospital Herlev, Copenhagen, Denmark). PBMCs were purified from buffy coats from healthy human volunteer donors, obtained from the State Hospital (Copenhagen, Denmark).
Purification and Stimulation of Primary PBMCs
PBMCs were purified from healthy donors by centrifugation in Histopaque-1077 (Sigma) and stimulated using plate-bound anti-CD3 (16-0037; eBioscience) and soluble CD28 (16-0289; eBioscience), according to the protocol of the manufacture.
Constructs
The GFP-Myc-MICA018 and MICA008 plasmids, which contain the coding sequences of the full-length allele MICA018 or the truncated MICA008 allele downstream of a generic leader, a GFP cassette, and a Myc tag, were kindly provided by Dr. M. Wills (University of Cambridge, Cambridge, UK) (25).
Prediction of N-Glycosylation Sites and Site-directed Mutagenesis
Prediction of N-glycosylation sites in MICA and MICB was done using the prediction server NetNGlyc,4 based on sequences obtained from the NCBI MHC sequence database. Site-directed mutagenesis was performed using a QuikChange Lightning multisite-directed mutagenesis kit (Agilent Technologies) following the manufacturer's instructions for templates larger than 5 kb. Primer design was performed according to the instructions in the mutagenesis protocol using the free web-based mutagenic primer design programs PrimerX (Lapid) and Primo (Chang Bioscience) in parallel.
Primers used for mutagenesis were as follows: N8Q, 5′-CCACAGTCTTCGTTATCAGCTCACGGTGCTG-3′; N56Q, 5′-GAAGATGTCCTGGGACAGAAGACATGGGACAG-3′;N102Q, 5′-GAGATCCATGAAGACCAGAGCACCAGGAGCTC-3′; N187Q, 5′-GCCCCCCATGGTGCAGGTCACCCGCAGCGAG-3′; N197Q, 5′-GCCTCAGAGGGCCAGATTACCGTGACATGCAGGGC-3′; N211Q, 5′-CTGGCTTCTATCCCTGGCAGATCACACTGAGCTGGCG-3′; N238Q, 5′-GTCCTGCCTGATGGGCAGGGAACCTACCAGACCTG-3′; N266Q, 5′-GAACACAGCGGGCAGCACAGCACTCACC-3′; K309STOP, 5′-TGTCCGTTGTTGTTAGAAGAAAACATCAG-3′; E319STOP, 5′-GCAGAGGGTCCATAGCTCGTGAGCC-3′; Q341STOP, 5′-AGGGATGCCACATAGCTCGGATTTCA-3′; MICA018to008, 5′-GCTGTTGCTGCTGGCTGCTGCTATTTTTGTTATTATTATTTTC-3′; KK309–310RR, 5′-CTATGTCCGTTGTTGTAGGAGGAAAACATCAGCTGCA-3;T24A, 5′-GCAGTCAGGGTTTCTTGCTGAGGTACATCTGG-3′; C36Y, 5′-GTCAGCCCTTCCTGCGCTATGACAGGCAGAAATGCAGG-3′; M129V, 5′-GAGACTGAGGAATGGACAGTGCCCCAGTCCTCCAGAG-3′; K172E, 5′-GGAACTACGGCGATATCTAGAATCCGGCGTAGTCCTGAGG-3′;G205S, 5′-CCGTGACATGCAGGGCTTCCAGCTTCTATCCC-3′; W210R-T213I-S215T, 5′-GGGCTTCTGGCTTCTATCCCCGGAATATCATACTGACCTGGCGTCAGGATGGGGTATC-3′; Q251R, 5′-CACCAGGATTTGCCGAGGAGAGGAGCAGAG-3′; and A24T, 5′-CTGTGCAGTCAGGGTTTCTCACTGAGGTACATCTGGATGG-3.
Selection was performed on Luria Broth agar plates containing 100 μg/ml ampicillin (Bistrol-Myers Squibb). Single colonies were picked for growth, and plasmids were purified using the NucleoSpin Plasmid Quikpure kit (Macheney-Nagel) and sequenced at Eurofins MWG Operon. Analysis of the sequences was performed using the free software CLC sequence viewer, version 6.4. Plasmids containing the required mutations were amplified through growth of bacteria in selective medium and purified using the NucleoBond Xtra Midi EF kit (Macheney-Nagel).
Transient Transfections
Transient transfections of all cell lines were done using the Amaxa Nucleofector Device (Lonza) according to the protocol of the manufacturer. Short descriptions for each cell line follow. For Jurkat cells, 3 × 106 cells were resuspended in 100 μl of cell line Nucleofector solution V or Ingenio electroporation solution (MIR 50117; Mirus Bio LLC), mixed with 1 μg or 3 μg of plasmid, and pulsed using the Nucleofector program G-010. For Granta-519 cells, 2 × 106 cells were resuspended in 100 μl of Ingenio electroporation solution, mixed with 5 μg of plasmid, and pulsed using the Nucleofector program T-001. The expression of the plasmids was measured 20–24 h after transfection. For melanoma cell lines, 1.5–2 × 106 cells were resuspended in 100 μl of Nucleofector solution T, mixed with 1.5–2 μg of plasmid, and pulsed using the Nucleofector program U-020. The expression of the plasmids was measured after 18–24 h. For U373 and pHM-U21 cells, 1 × 106 cells were resuspended in 100 μl of Nucleofector solution T, mixed with 0.25 μg of plasmid, and pulsed using the Nucleofector program T-020. The expression of the plasmids was measured after 18–20 h. For stimulated PBMCs, 3 × 106 cells were resuspended in 100 μl of Nucleofector solution T, mixed with 2 μg of plasmid, and pulsed using the Nucleofector program T-020. Cells were prepared for analysis by flow cytometry, fluorescence microscopy, and Western blot 18–24 h after transfection.
Reagents
The HDAC inhibitor FR901228 (Romidepsin, FK228, or Depsipeptide) was provided by the National Cancer Institute (Bethesda, MD). The inhibitors mannosidase I and II, kifunensine (K1140), and swainsonine (S6640), respectively, were obtained from Sigma-Aldrich.
Mannosidase Inhibitor Treatment
Cells were treated with 0–500 nm swainsonine or 0–10 μg/ml kifunensine (KF). Transfected cells were treated 5 h after transfection. 18 h after treatment, the cells were analyzed by flow cytometry for surface expression.
Flow Cytometry
Cell surface staining was done as previously described (43). The antibodies used for detection of surface expression of the GFP-Myc-MICA fusion protein were anti-Myc tag, clone 4A6 (05–724; Millipore), and the APC-coupled anti-mouse IgG1 (405308; Biolegend) as the secondary antibody and isotype control. For L-PHA staining, Alexa-fluor-647-coupled lectin PHA (L32457; Invitrogen) was used. For evaluation of MICA binding to NKG2D, recombinant human NKG2D-Fc chimera (1299; R&D Systems), and Zenon Alexa-fluor 647 human IgG labeling kit (Z-25408; Invitrogen), as well as isotype controls, were used. For surface staining of endogenous MICA/B and MHC class I, cells were stained with PE-conjugated anti-human MICA/B (558352; BD Biosciences), PE-conjugated anti-human MHC class I clone W6/32 (Sc-32235; Santa Cruz Biotechnology), or isotype controls. All isotype controls were purchased from BD Biosciences.
For transfected cells, gating was carried out on viable and GFP-positive cells (Gate 2), and the grid was set according to an empty control (pUC18). Transfection efficiency was measured as GFP intensity, and surface expression of the MICA fusion proteins was analyzed by Myc tag surface staining of GFP-positive cells. For analysis of endogenous surface molecules, gating was carried out on viable cells in forward-side scatter plots, and the grid was set according to isotype controls. For transfected U373 and pHM-U21 cells, gating was carried out on viable cells and with a GFP threshold, excluding heavily overexpressed MICA fusion protein. U373 cells are very efficiently transfected, and it is not straightforward to lower the transfection efficiency. The endogenous regulatory mechanisms are only working within a reasonably range of overexpression, which is the reason for the GFP expression threshold applied to U373 cells. Jurkat T-cells are not transfected with a similar high efficiency, and it is therefore not necessary to apply a threshold gate; applying the gate would, however, not affect the result.
Fluorescence Microscopy
Images of transfected cells were captured by use of a fluorescence microscope (Eclipse 80i; Nikon) with a 100× oil objective lens (NA 1.30; Nikon). Images were captured using NIS-Elements basic research imaging software (Digital sight DS-5M-L1; Nikon) and analyzed using Photoshop CS4 (Adobe). Antibodies used were anti-human ICAM-1 R-PE (C170; Leinco Technologies) and anti-mouse IgG1 eFlour 615 (42–4015; eBioscience).
Western Blotting
Whole cell lysates from U373 and pHM-U21 cells were prepared in 1% Triton X-100, 10 mm Tris-HCl (pH 7.4), 150 mm NaCl, and normalized to total protein concentration as determined by BCA assay (Pierce). Lysates were resolved by SDS-PAGE electrophoresis and transferred to BA-85 nitrocellulose membrane (Whatman, Florham Park, NJ). Western blotting of whole cell lysates from the other cell lines were done as previously described (43). The antibodies used in this study were GFP polyclonal antibody (3999-100; Biovision), polyclonal ERK1 antibody clone C-16 (Sc-93; Santa Cruz Biotechnology), polyclonal antibody MCW50 directed against the cytoplasmic tail of U21 (56), and a monoclonal anti-GAPDH (Imgenex, San Diego, CA). Appropriate HRP-conjugated secondary antibodies (Dako and Bio-Rad) were used, and bands were visualized using SuperSignal reagent (Pierce). IRDye 800CW goat anti-rabbit IgG secondary antibody (926-32211; LI-COR Biosciences) was used and visualized using (Odyssey Fc Imager; LI-COR Biosciences). To access changes in electrophoretic mobility, dilutions of samples for Western blot were performed to obtain the similar GFP intensity in each loaded sample. This was done to control for variations in transfection efficiencies. GFP intensity of transfected cells was measured by flow cytometry, and samples were diluted to obtain similar GFP intensity (mean fluorescent intensity) in each lane. Detection of minor changes in electrophoretic mobility (like for example a single N-glycan) is sensitive to variations in GFP intensity. On these Western blots, no endogenous loading control was included because our scope was to compare molecular size and not quantity.
PNGase F Treatment
Treatment of cell lysates with PNGase F (P0704S; New England Biolabs) was performed as previously described (33).
Cytolytic Assay
The cytolytic assay was performed as previously described (44). As target cells, Granta-519 cells transfected with GFP-Myc-MICA018 or GFP-Myc-MICA008 with or without the mutation N8Q were used. In brief, Granta-519 cells were transfected and allowed to recover for 2–4 h. The cells were then treated with 0.5 μm dilinoleyloxycarbocyanine (Invitrogen) for 24 h. As effector cells, we used purified PBMCs cultured for 3 days at a concentration of 5 × 106 cells/ml in RPMI 1640 supplemented with 10% human serum (Lonza) and 20 ng/ml IL-15 (R&D Systems). Effector and target cells were mixed, and after 4 h of incubation, the cells were stained with propidium iodide and immediately analyzed by flow cytometry. To measure cytolytic activity, we gated on dilinoleyloxycarbocyanine-positive cells and recorded the percentage of propidium iodide-positive cells. The percentage killing was calculated by subtracting the number of dead target cells (measured by propidium iodide staining) in a sample without effector cells, from a sample with target cells.
RESULTS
N-Glycosylation of MICA018 Is Necessary for Cell Surface Expression
MICA is a glycoprotein containing eight possible N-glycosylation sites (12). We have previously shown that general inhibition of N-glycosylation hinders cell surface expression of MICA and MICB (33). We therefore hypothesized that N-glycosylation of MICA itself could potentially regulate cell surface expression.
To test this, we abrogated N-glycosylation of MICA by substituting asparagine residues in the consensus sequences for N-glycosylation to glutamine residues using site-directed mutagenesis of a plasmid encoding the full-length MICA018 allele N-terminally tagged with GFP and Myc. We transiently transfected Jurkat T-cells with the constructs and analyzed the effect of the mutations on cell surface expression of the GFP-Myc-MICA018 fusion protein by flow cytometry. Transfection intensity was measured by total GFP expression, and the cell surface expression of the MICA fusion protein was measured by Myc tag surface staining. Because inhibition of N-glycosylation affects surface expression of both MICA and MICB, we expect that N-glycosylation sites necessary for surface expression should be present in both MICA and MICB (33). We therefore concentrated our initial screening on four sites shared by MICA and MICB (Asn187, Asn197, Asn211, and Asn238). We included a fifth position (Asn8) because N-glycosylation of MICA at this position was found by structural study to be the site in MICA most likely N-glycosylated (12). As shown in Fig. 1A, mutation of the N-glycosylation sites at positions Asn8, Asn187, Asn197, Asn211, and Asn238 (MICA018 5× N-mut) resulted in near total abrogation of cell surface expression of MICA018, which suggests that N-glycosylation is required for cell surface expression of MICA018. As expected, no cells were positive for Myc and GFP in the mock transfected control sample (Fig. 1A). To investigate translation of the mutant MICA constructs, we performed a Western blot of the transfected cells detecting the MICA fusion proteins with a GFP-specific antibody. The MICA018 WT fusion protein was detected at ∼90 kDa, corresponding to the predicted mobility of the MICA018 WT fusion protein (Fig. 1A). The electrophoretic mobility of the MICA018 5× N-mut fusion protein was lowered to ∼75 kDa, as would be expected upon lack of N-glycosylation (Fig. 1A). Furthermore, when analyzing MICA018 5× N-mut on SDS-PAGE, we did not observe a band corresponding to free GFP (30 kDa), indicating that the fusion protein is not immediately degraded but rather retained intracellularly. However, we did observe less pronounced bands in the 5× N-mut sample, suggesting that the mutant fusion protein is less stable than the WT. These data show that MICA018 is dependent of N-glycosylation for surface expression and indicate that the MICA018 construct is expressed and translated upon mutations in five selected N-glycosylation sites.
FIGURE 1.

Abrogation of N-glycosylation in MICA018 results in impaired cell surface expression. A, JTag9 cells were transfected with an empty plasmid (pUC18) or plasmids encoding GFP-Myc-tagged MICA018 fusion proteins (MICA018 WT and MICA018 5× N-mut). Gating was carried out on viable and GFP-positive cells (gate 2), and the grid was set according to pUC18 transfected cells. Left panel shows representative results from five independent experiments presented as dot plots. The upper row shows GFP intensity, and the lower row shows Myc surface staining (percentages inserted upper right). The right panels show representative Western blots of transfected cells detecting GFP and ERK. B, JTag9 cells were transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 fusion proteins. Gating was carried out on viable and GFP-positive cells, and the grid was set according to pUC18 transfected cells. Representative results from three independent experiments are presented as dot plots showing Myc surface staining (percentages inserted upper right). FSC, forward-side scatter.
To test whether one of the N-glycosylation sites was more important for the cell surface expression than others, we made constructs containing single mutations of the eight individual N-glycosylation sites. Interestingly, mutation of position Asn8 alone (MICA018 N8Q) was almost as efficient in blocking cell surface expression as the abrogation of N-glycosylation at all five sites (Fig. 1B). Moreover, mutation of any of the other N-glycosylation sites did not influence the cell surface expression significantly, suggesting that lack of glycosylation at Asn8 is the primary source of inhibiting cell surface expression of MICA018 5× N-mut.
MICA008 Is Not Dependent on N-Glycosylation for Cell Surface Expression
The truncated MICA008 allele is known to be regulated different from full-length alleles. For example, it has been shown to be more resistant to down-regulation by different viruses (22, 25, 26).
Preliminary studies showed that treatment of transfected cells with 2DG inhibited MICA018 cell surface expression but only minimally affected MICA008 surface expression (data not shown). 2DG is known to inhibit glycolysis as well as N-glycosylation, and the inhibition of N-glycosylation can be specifically alleviated by addition of d-mannose (33, 45). Inhibition of MICA018 surface expression could be rescued by addition of d-mannose, suggesting that MICA008 is less dependent on N-glycosylation for cell surface expression compared with MICA018 (data not shown). Therefore, we investigated the effect of abrogating N-glycosylation of MICA008. As illustrated in Fig. 2A, the distribution of N-glycosylation sites in MICA008 is almost identical to MICA018, except for position Asn211. Two different constructs were made, one containing the N8Q mutation (MICA008 N8Q) and one containing four mutations at Asn8, Asn187, Asn197, and Asn238 (MICA008 4× N-mut). The MICA008 constructs were transiently expressed in Jurkat T-cells. Interestingly, mutation of all four N-glycosylation sites or Asn8 alone did not affect cell surface expression of MICA008 (Fig. 2B). These data suggest that in contrast to MICA018, MICA008 is not dependent on N-glycosylation for surface expression. As illustrated in Fig. 2C, the Western blot confirmed expected mobility and expression of the N-glycosylation mutants. MICA018 N8Q showed increased electrophoretic mobility, indicating a lack of N-glycan structure at position Asn8. To confirm this, we digested cell lysates from MICA018 WT and MICA018 N8Q transfected JTag9 cells with peptide N-glycosidase F (PNGase F), which cleaves N-glycans. Mobility of these samples was investigated in a SDS-PAGE of higher density, allowing a more clear separation of the proteins. As shown in Fig. 2D, there was an increased mobility of MICA018 N8Q compared with MICA018 WT; this difference was not observed after PNGase F treatment, suggesting the mobility change is caused by altered N-glycosylation.
FIGURE 2.

Dependence for N-glycosylation differs between MICA alleles. A, left panel shows protein sequence alignment of MICA018 and MICA008. Predicted N-glycosylation sites (green), sequence variations (pink), and predicted transmembrane region (bold) are highlighted as indicated. Illustrations on the right show overall structure and N-glycosylation sites (green) of MICA018 and MICA008. Throughout the paper, MICA illustrations carrying mutations are highlighted as: N-glycosylation sites (black) and/or position 24 (pink). B, JTag9 cells were transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 or MICA008 fusion proteins. Gating was carried out on viable and GFP-positive cells, and the grid was set according to pUC18 transfected cells. Representative results from three independent experiments are shown as dot plots showing Myc surface staining (percentages inserted upper right). C, representative Western blot of lysates from JTag9 cells transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 or MICA008 fusion proteins. Mobility of indicated MICA fusion proteins was investigated by detection of GFP in a 4–12% gel. Samples were diluted according to GFP intensity as described under “Experimental Procedures.” D, representative Western blot of lysates from JTag9 cells transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 WT or MICA018 N8Q fusion proteins. Mobility of MICA018 WT and MICA018 N8Q fusion proteins untreated or digested with PNGase F was investigated by detection of GFP in a 10% gel. Samples were diluted according to GFP intensity as described under “Experimental Procedures.” FSC, forward-side scatter.
Because the constructs did not show same transfection efficiency, samples were diluted to obtain similar mean fluorescent intensity of GFP intensity in each lane. A loading control against an endogenous protein is thus not informative and was not included (see “Experimental Procedures” for further explanation).
The Intracellular Tail Is Not Involved in the Intracellular Retention of MICA018 upon Abrogation of N-Glycosylation
Lack of a cytoplasmic tail in MICA008 is a prominent difference between MICA008 and MICA018 (Fig. 2A). Earlier studies have identified sites in the cytoplasmic tail of MICA that are involved in the regulation of cell surface expression (26, 46, 47). In particular, a triple-lysine motif at positions 308–310 has been shown to be involved in the retention of full-length MICA alleles in cells infected with KSHV despite induced transcription of MICA upon infection (26). In addition, a dihydrophobic leucine-valine motif at positions 324–325 has been shown to sort full-length MICA alleles to the basolateral surface of human intestinal epithelium, whereas the lack of a cytoplasmic tail results in expression on the apical surface (46).
To test whether any of these known regulatory sites are involved in the intracellular retention of MICA018 upon abrogation of N-glycosylation, we produced GFP-Myc-MICA018 constructs with mutations in the cytoplasmic tail. Three different mutants were made introducing stop codons at different sites in the cytoplasmic tail (K309STOP, E319STOP, and Q341STOP). The stop codons were strategically placed to abrogate the known regulatory motifs. Furthermore, a mutant construct was made to test the role of the triple-lysine motif (K309R,K310R).
As seen in Fig. 3A, neither the introduction of stop codons nor the mutation of the triple-lysine motif increased cell surface expression of the MICA018 constructs with five mutated N-glycosylation sites, indicating that neither is involved in the retention of MICA018 with incomplete N-glycosylation. To verify the independence of the intracellular domain in N-glycosylation-mediated regulation of MICA018, we introduced the frameshift mutation present in MICA008 (018to008). In line with above results, this mutation did not change the sensitivity of MICA018 surface expression to changes in N-glycosylation (Fig. 3A). Western blot analysis of the mutants confirmed expression and changes in electrophoretic mobility of the fusion proteins. As expected, the MICA018 to 008 mutation and stop codon mutations caused mobility shifts of the protein, resulting in bands corresponding to MICA008 (Fig. 3A). These data strongly suggest that the cytoplasmic tail is not involved in the retention of MICA018 upon abrogation of N-glycosylation.
FIGURE 3.

A single position in the extracellular domain is essential for the N-glycosylation dependence of MICA018. A–F, JTag9 cells were transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 or MICA008 fusion proteins. Gating was carried out on viable and GFP-positive cells, and the grid was set according to pUC18 transfected cells. A–C, the upper panels show bar graphs presenting mean and standard deviation of the percentage of Myc-positive cells, each graph represent results from two independent experiments. The lower panels show representative Western blots of transfected cells detecting GFP, samples were diluted according to GFP intensity as described under “Experimental Procedures.” D, upper panel shows representative results from three independent experiments presented as dot plots showing Myc surface staining (percentages inserted upper right). The lower panel shows representative Western blot of transfected cells detecting GFP, samples were diluted according to GFP intensity as described under “Experimental Procedures.” E, representative images showing GFP expression and ICAM-1 surface staining. Bar, 10 μm. F, upper panel shows representative results from three independent experiments presented as dot plots showing Myc surface staining (percentages inserted upper right). The lower panel shows representative Western blot of transfected cells detecting GFP; samples were diluted according to GFP intensity as described under “Experimental Procedures.” FSC, forward-side scatter.
Threonine 24 in the α1-Domain of MICA018 Is Responsible for Inhibiting Cell Surface Expression of Incompletely N-Glycosylated MICA018
Having excluded involvement of the cytoplasmic tail, we turned our attention to the extracellular domains of MICA018 and MICA008. Alignment of the two alleles shows differences in nine extracellular amino acids (Fig. 2A). To investigate the role of the different residues, four groups of mutations were made in the MICA018 WT and MICA018 5× N-mut constructs. Specific residues in MICA018 were changed to corresponding residues present in MICA008.
As shown in Fig. 3B, cell surface expression of the construct encoding MICA018 5× N-mut with substitutions in T24A, C36Y, and M129V (018 5× N-mut T24A,C36Y,M129V) was comparable to surface expression of MICA018 WT and MICA018 WT with substitutions in T24A, C36Y, and M129V (018 T24A,C36Y,M129V). In contrast, changing the six other residues in MICA018 to the corresponding MICA008 amino acids (K172E, G205S, W210R, T213I, S215T, and Q251R) did not significantly affect cell surface expression of the MICA018 5× N-mut construct (Fig. 3B). This strongly indicates involvement of one or more of the sites Thr24, Cys36, and Met129 in the retention of MICA018 with incomplete N-glycosylation. As expected, the mutations did not alter the mobility of the MICA fusion proteins in SDS-PAGE (Fig. 3B, bottom panel).
Next we made MICA018 constructs with single mutations in the three residues of interest. As seen in Fig. 3C, substitution of the threonine at position 24 (T24A) results in a similar phenotype as mutation of all three sites (T24A, C36Y, and M129V). In line with this, single mutations of the two other sites (C36Y and M129V) had only negligible effects (Fig. 3C). This suggests that Thr24 is responsible for retaining incompletely N-glycosylated MICA018 and that changing this site into alanine, as found in MICA008, confers resistance to this retention. In addition, the ability of the single T24A substitution to rescue surface expression of MICA018 5× N-mut suggests that the inhibition of surface expression is not due to improper folding of incompletely N-glycosylated MICA. To further substantiate these findings, we made a MICA018 mutant containing both the T24A and N8Q mutations (MICA018 T24A+N8Q). As shown in Fig. 3D, the T24A substitution completely restored cell surface expression of MICA018 N8Q, confirming the regulatory role of this site for MICA018 cell surface expression. In addition, we verified surface expression or intracellular retention of different MICA018 constructs in Jurkat T-cells, by fluorescence microscopy. GFP expression in Jurkat T-cells transiently expressing MICA018 WT colocalized with ICAM-1 surface staining, whereas GFP expression in cells transfected with MICA018 N-mut constructs was observed intracellularly (Fig. 3E). Expression of T24A substituted MICA018 N-mut constructs was found primarily on the surface (Fig. 3E), in line with our previously findings. Importance of the Thr24 residue as a regulatory site for MICA surface expression was underlined by changing alanine in MICA008 to threonine, as found in MICA018 (MICA008 A24T). As previously observed, the Asn8 mutation in MICA008 did not affect the surface expression level (Figs. 2B and 3F). However, introduction of an A24T substitution in MICA008 and MICA008 N8Q resulted in complete abrogation of surface expression (Fig. 3F). An explanation for this interesting finding could be that introduction of Thr24 makes MICA008 dependent on N-glycosylation for surface expression. MICA008 is normally not dependent on N-glycosylation for surface expression, and there has therefore not been a sufficient selection pressure to maintain the N-glycosylation required for surface expression of Thr24 MICA008. In support of this, we did not observe a clear change in electrophoretic mobility of MICA008 with abrogated Asn8 glycosylation (Fig. 3F). Expression and expected mobility of the other MICA008 fusion proteins were confirmed by Western blot (Fig. 3F).
The Difference in Dependence for N-Glycosylation of MICA018 and MICA008 Has Functional Implications
It is well described that activated primary T-cells can express MICA (30, 48, 49). To verify the differences in regulation of MICA018 and MICA008 in primary cells, we expressed the MICA018 and 008 constructs with or without the N8Q mutation in CD3-CD28-stimulated PBMCs. As illustrated in Fig. 4A, the cell surface expression of MICA018 N8Q was blocked, whereas the cell surface expression of the MICA008 construct was unaffected by the N8Q mutation. This confirms that MICA018 and MICA008 are also differently regulated with respect to N-glycosylation in activated primary T-cells.
FIGURE 4.
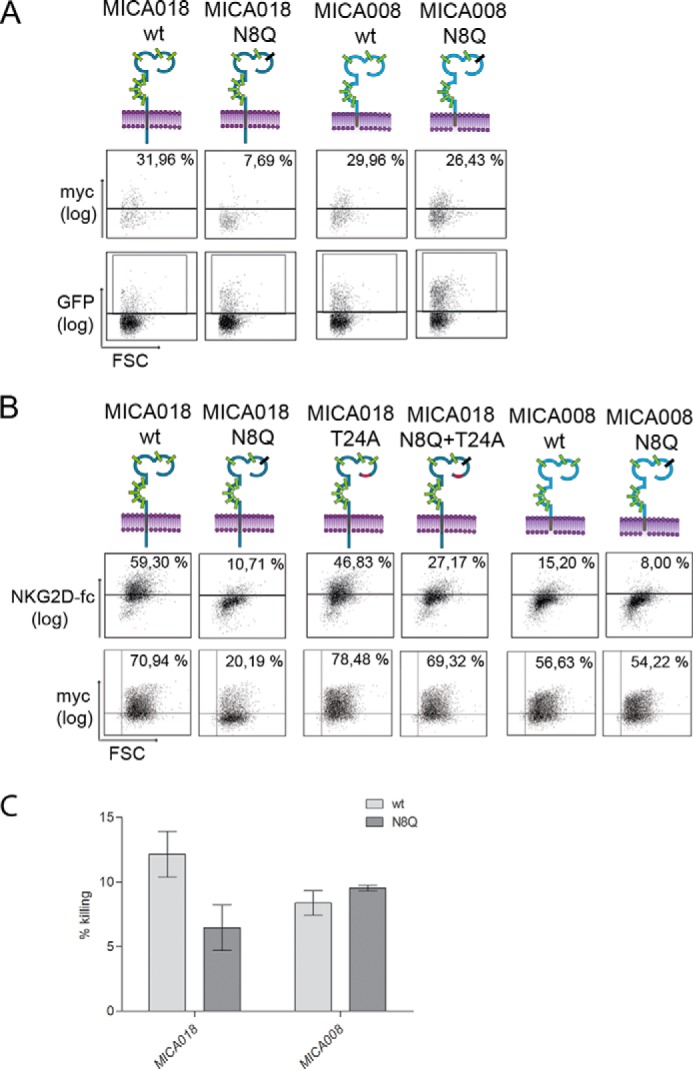
Difference in dependence for N-glycosylation between MICA018 and MICA008 has functional consequences. A, stimulated PBMCs were transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 or MICA008 fusion proteins. Gating was carried out on viable and GFP-positive cells, and the grid was set according to pUC18 transfected cells. Representative results from three independent experiments are shown as dot plots. The upper row shows Myc surface staining (percentages inserted upper right), and the lower row shows GFP intensity. B, JTag9 cells were transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 or MICA008 fusion proteins. Gating was carried out on viable and GFP-positive cells, and the grid was set according to pUC18 transfected cells. Representative results from three independent experiments are presented as dot plots showing NKG2D-fc staining (upper row) and Myc surface staining (lower row). The percentage of surface staining is inserted upper right. C, bar graph shows mean and standard deviation of the cytolytic activity of purified PBMCs (effector cells) targeting Granta-519 cells expressing indicated GFP-Myc-tagged MICA018 or MICA008 fusion proteins (target cells). Three repetitions of the experiment using PBMCs from the same donor was performed. An effector/target ratio of 10:1 was used in these experiments, based on prior titration analysis. Similar results were obtained using PBMCs from three different donors. FSC, forward-side scatter.
To investigate the functional effect of the mutation of Asn8 and Thr24, we performed cell surface staining of transfected cells with a soluble form of the native receptor NKG2D (NKG2D-fc). Fig. 4B shows that the cell surface expression of functional MICA018 is significantly reduced by the lack of N-glycosylation at position Asn8. The T24A substitution in MICA018 did not significantly alter the binding of NKG2D-fc compared with MICA018 WT (Fig. 4B). Abrogation of Asn8 glycosylation in MICA018 T24A reduced the NKG2D-fc binding compared with MICA018 T24A (Fig. 4B). In addition, the N8Q mutation in MICA008 results in a minor decrease in the NKG2D-fc binding when compared with MICA008 (Fig. 4B). These results suggest that N-glycosylation of MICA is not a necessity for the interaction with NKG2D but does influence the binding, which is in line with previous findings (13). Binding of NKG2D-fc to MICA008 WT was lower than observed for MICA018 WT (Fig. 4B). This is not surprising because MICA018 and 008 differ in position 129, which has previously been shown to influence the affinity to NKG2D (13). To further corroborate the functional significance of these findings, we performed a cytolytic assay with MICA transfected cells as target cells and IL-15-treated PBMCs as effector cells. Granta-519 B-cells were used because their endogenous expression level of NKG2D ligands is low (30). We transfected the Granta cells with constructs encoding MICA018 or 008 with or without the N8Q mutation. The cell surface expression of the fusion proteins expressed in Granta cells was found to be similar to the expression observed in Jurkat T-cells (data not shown). As illustrated in Fig. 4C, the N8Q mutation decreased the cytolytic killing of Granta cells transfected with the MICA018 construct. However, no difference was observed in the cytolytic killing of cells transfected with MICA008 N8Q when compared with MICA008 WT transfected cells (Fig. 4C). These experiments show less NKG2D interaction, as well as lower NKG2D-mediated killing of target cells caused by N-glycosylation-mediated inhibition of MICA018 cell surface expression.
Altered Branching of N-Glycans Is Not Directly Linked to Down-regulation of MICA Cell Surface Expression
It is well established that a change in the cellular N-glycan profile is associated with malignant transformation and increased metastasis (50, 51). A specific increase in the amount of highly complex N-glycans has been observed in melanomas, as well as breast and colon cancers (52).
We therefore examined whether the inhibition of MICA018 cell surface expression after mutation of the Asn8 glycosylation site could be due to defective trimming of mannose structures in Golgi. To address this question, we specifically inhibited the formation of complex N-glycans in MICA018 transfected Jurkat T-cells by treatment with either kifunensine or swainsonine, chemical inhibitors of Golgi α-mannosidase I and II, respectively (53, 54). The plant lectin L-PHA binds specifically to complex N-glycans (55) and was used to verify altered N-glycan branching. Treatment with kifunensine results in formation of high mannose N-glycans, whereas treatment with swainsonine results in formation of hybrid-type N-glycans, both leading to decreased L-PHA binding. As expected, decreased L-PHA staining was observed upon treatment with both swainsonine and kifunensine (Fig. 5A). However, neither treatment affected cell surface expression of MICA018, indicating that formation of complex N-glycans is not required for MICA018 cell surface expression.
FIGURE 5.

Qualitative alteration of N-glycans does not inhibit MICA/B cell surface expression. A, JTag9 cells were transfected with a plasmid encoding GFP-Myc-MICA018 and treated with 400 nm swainsonine (SW), 10 μg/ml kifunensine (KF), or culture medium as control (−). Gating was carried out on viable and GFP-positive cells, and the grid was set according to pUC18 transfected cells. Representative results from three independent experiments are shown as dot plots. The upper row shows Myc surface staining, and the lower row shows L-PHA staining (percentages inserted upper right). B, JTag9 cells were treated with 0–500 nm swainsonine or 0–10 μg/ml kifunensine. The figure shows staining of L-PHA and endogenous MICA/B represented as a function of the concentration of swainsonine and kifunensine in the culture medium. Representative results from three independent experiments are shown. FSC, forward-side scatter.
To corroborate these findings, untransfected Jurkat T-cells were treated with increasing concentrations of swainsonine and kifunensine, and the cell surface expression of endogenous MICA/B and complex N-glycans was evaluated by flow cytometry. As shown in Fig. 5B, treatment with both mannosidase inhibitors led to a dose-dependent decrease in the cell surface L-PHA binding, but the cell surface expression of MICA/B was not affected. These results show that formation of complex or hybrid-type N-glycans is not required for MICA surface expression and suggests that the dependence of N-glycosylation for translocation of MICA018 to the cell surface likely occurs in the ER before trimming of high mannose glycans.
Inhibition of MICA Cell Surface Expression by the Human Herpesvirus-7 U21 Is Mediated through N-Glycosylation and Threonine 24
Alterations in cellular N-glycosylation pathways have been associated with virus infection and cancer, possibly contributing to immune escape (38–40). The U21 protein expressed by HHV-7 is known to inhibit MHC class I surface expression (56, 57). In addition, U21 down-modulates cell surface expression of transfected MICA and MICB (24). MICB expression in U21 containing cells was associated with increased electrophoretic mobility (24). To test the possibility that altered N-glycosylation is involved in the U21-mediated retention of MICA, we used U373 cells stably expressing U21 (pHM-U21 cells). Functional U21 expression was confirmed by the ability to down-regulate MHC class I surface expression (Fig. 6A), and expression of U21 in U373 cells was verified by Western blot (Fig. 6A). To examine the regulation of endogenous MIC by U21, U373 and pHM-U21 cells were treated with the HDAC inhibitor FR901228, which is known to induce MICA/B surface expression (30). As shown in Fig. 6A, FR901228-induced MICA/B surface expression was abolished in pHM-U21 cells. Having verified U21-mediated down-regulation of endogenous MICA, we further investigated this regulatory mechanism through overexpression of different MICA constructs. As expected, surface expression of MICA018 5× N-mut and MICA018 N8Q was inhibited in U373 cells compared with MICA018 WT surface expression (Fig. 6B). In line with the regulatory ability of U21 on endogenous MICA, surface expression of the MICA018 WT fusion protein was significantly inhibited in pHM-U21 cells, and as expected, the expression pattern of the two N-mut constructs in pHM-U21 cells was low and comparable to levels observed in U373 cells (Fig. 6B). Western blotting showed an increased electrophoretic mobility of MICA018 WT expressed in pHM-U21 cells (Fig. 6B). This increase was not observed in MICA018 WT expressed in U373 cells (Fig. 6B), indicating a post-translational modification of MICA018 mediated by U21. There was no difference in the electrophoretic mobility of MICA018 5× N-mut expressed in U373 and pHM-U21 cells, strongly arguing that altered N-glycosylation is responsible for the mobility shift of MICA018 WT in U21-expressing cells. In addition, we found a similar reduction in molecular weight of MICA018 when transiently expressed in two melanoma cell lines, FM55p and FM82, indicating that MICA018 was modified post-translationally in these cells (data not shown). Interestingly, the decreased molecular weight of MICA018 was found in the melanomas with low intrinsic cell surface expression of endogenous MICA/B (data not shown). When digesting cell lysates with PNGase F, mobility of transiently expressed MICA018 was reduced to the same molecular weight (data not shown), showing that the increased electrophoretic mobility of MICA018 in the melanomas FM55p and FM82 was caused by an altered N-glycosylation pattern.
FIGURE 6.

N-Glycosylation-mediated inhibition of MICA018 by HHV7-U21 is rescued by a single substitution in position 24. A, U373 and pHM-U21 cells were stained for MHC class I surface expression (left panel). Western blot of whole cell lysates from U373 and pHM-U21 cells detecting U21 and GAPDH (middle panel). U373 and pHM-U21 cells treated with 20 μg/ml FR901228 or PBS as control (untreated) were stained for surface expression of endogenous MICA/B. The bar graphs show means and standard deviations of percentage of cells positive for surface staining from three independent experiments. B, U373 and pHM-U21 cells were transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 fusion proteins. The left panel shows representative results from three independent experiments presented as dot plots showing Myc surface staining (percentages inserted upper right). Gating was carried out on viable cells and at a GFP threshold, excluding heavily overexpressed MICA fusion protein (described under “Experimental Procedures”). The right panel shows representative Western blot of transfected cells detecting GFP; samples were diluted according to GFP intensity as described under “Experimental Procedures.” C, U373 and pHM-U21 cells were transfected with plasmids encoding indicated GFP-Myc-tagged MICA018 fusion proteins. The left panel shows representative results from three independent experiments presented as dot plots showing Myc surface staining (percentages inserted upper right). Gating was carried out on viable cells and at a GFP threshold, excluding heavily overexpressed MICA fusion protein (described under “Experimental Procedures”). The right panel shows representative Western blot of transfected cells detecting GFP; samples were diluted according to GFP intensity as described under “Experimental Procedures.” D, U373 and pHM-U21 cells were transfected with plasmids encoding indicated GFP-Myc-tagged MICA008 fusion proteins. The left panel shows representative results from three independent experiments presented as dot plots showing Myc surface staining (percentages inserted upper right). Gating was carried out on viable cells and at a GFP threshold, excluding heavily overexpressed MICA fusion protein (described under “Experimental Procedures”). The right panel shows representative Western blot of transfected cells detecting GFP; samples were diluted according to GFP intensity as described under “Experimental Procedures.” FSC, forward-side scatter.
Because U21-mediated inhibition of MICA018 surface expression was associated with altered N-glycosylation, we examined whether the T24A substitution could rescue the inhibition. When comparing MICA018 WT and MICA018 T24A surface expression in pHM-U21 cells, a robust increase in surface expression of MICA018 T24A was observed (Fig. 6C). This suggests that the U21-mediated inhibition of MICA018 was rescued by T24A substitution. In support of this, we observed similar rescue of MICA018 surface expression by the T24A substitution in the melanoma cell line FM55p (data not shown).
Western blotting of MICA018 T24A expressed in pHM-U21 cells showed a shift in electrophoretic mobility, similar to MICA018 WT expressed in pHM-U21 cells (Fig. 6C). This implies that the change in MICA018 N-glycosylation in pHM-U21 cells is not affected by the T24A substitution. Surface expression of MICA018 T24A in U373 cells compared with MICA018 WT was slightly increased (Fig. 6C), similar to the expression pattern observed in Jurkat T-cells (Fig. 3D). These data suggest that the retention of MICA018 by U21 is mediated by altered N-glycosylation through a Thr24-dependent mechanism.
Because MICA008 is not dependent on N-glycosylation for cell surface expression (Fig. 3F), we hypothesized that this allele would be resistant to U21-mediated down-modulation. As shown in Fig. 6D, similar surface expression of MICA008 and MICA008 N8Q was observed in U373 cells, which was expected according to our previous data. However, when expressed in pHM-U21 cells, MICA008 surface expression was inhibited, and strikingly this inhibition was partly alleviated by mutation at the Asn8 N-glycosylation site (Fig. 6D). MICA008 did not show an increase in electrophoretic mobility in pHM-U21 cells compared with U373 cells (Fig. 6D), suggesting that U21 does not change N-glycosylation of MICA008. When introducing the A24T substitution, we observed a similar surface expression profile of MICA008 in both U373 and pHM-U21 cells (Fig. 6D), as we have also previously shown in Jurkat T-cells (Fig. 3F). Together these results imply that U21 inhibits MICA008 cell surface expression through a novel post-translational regulatory mechanism, but not by directly changing MICA008 N-glycosylation.
DISCUSSION
Here we describe that Asn8 glycosylation is essential for cell surface expression of the full-length MICA018 allele. The MICA008 allele is, however, not affected by changes in N-glycosylation. MICA008 possesses a truncated cytoplasmic tail compared with full-length MICA alleles, and mutational analysis showed that the cytoplasmic tail is not involved in retention of MICA018 upon abrogation of N-glycosylation. Our study shows that regulation of MICA018 surface expression by N-glycosylation is critically dependent on expression of a threonine residue at position 24. MICA018 surface expression was thus not affected by altered N-glycosylation when position 24 was changed to alanine, the residue present in MICA008. Conversely it is possible that MICA008 surface expression became N-glycosylation-dependent upon introduction of a threonine residue at position 24.
We used the crystal structure of the extracellular domains of MICA001 as a model to examine the location of Asn8 and Thr24 within the MICA structure, because MICA001 and MICA018 only differ in position 126 in the amino acid sequence (14). Both position Asn8 and Thr24 are located in the α1 domain of MICA (Fig. 7A). Thr24 is buried in the protein structure positioned at the bottom of two narrow channels, whereas Asn8 is situated on the surface of the α1 domain (Fig. 7A). The two residues are placed in close proximity to each other, but on opposite sides of the molecule (Fig. 7, B and C). We suggest that the channels containing Thr24 in the bottom can bind an unknown protein that retains MICA until proper N-glycosylation has occurred at position Asn8. The T24A substitution abrogates this regulatory mechanism, likely because Thr24 is essential for binding to the unknown protein. This unretained phenotype is observed for MICA008 and MICA018 T24A. Several MICA alleles (*001, *012, *018, *060, and *061) possess a threonine residue at position 24, and it is likely that surface expression of these alleles will depend on N-glycosylation, although this requires experimental verification.
FIGURE 7.

Structure of the extracellular domains of MICA highlighting positions Asn8 and Thr24. A, structure of the ectodomain of the MICA molecule is shown from three different angles, highlighting positions Asn8 and Thr24 in the α1-domain. B, The position of Asn8 and Thr24 when looking into the protein mass from the top of the molecule. C, the position of Asn8 and Thr24 in space in a cartoon representation of the protein structure seen from the top of the molecule. The structure models are based on the crystal structure Protein Data Bank code 1B3J.
We examined the functional effect of MICA N-glycosylation and Thr24 for binding to the NKG2D receptor and found that none significantly influence the interaction. However, in line with previous findings, we observed a minor decrease in the interaction with NKG2D when N-glycosylation of MICA was absent (13).
MICA can be targeted by different pathogens to hamper their recognition; on the other hand unrestrained MICA expression can pave the road for autoimmunity (11, 42, 58). The different regulatory properties of individual MICA alleles are therefore likely an average of these opposing forces, with the high degree of polymorphism adding to the complexity of the entire NKG2D-mediated immune response. Regulation of MICA expression takes place at different levels, and here we show that N-glycosylation is a new post-translational mechanism important for direct regulation of MICA018 surface expression and possibly other Thr24-containing MICA alleles. When trying to map where in the N-glycosylation pathway, this regulation takes place, we found that inhibiting N-glycan trimming in Golgi with kifunensine or swainsonine did not affect MICA018 surface expression. Tunicamycin and 2DG inhibits N-glycosylation in the ER compartment and block MICA018 surface expression (33). Together these observations indicate that the inhibition is likely an early event occurring in the ER. In support of this, Fuertes et al. (32) have shown that melanomas preferentially retain MICA in a high mannose form associated with ER localization.
Our earlier data also suggest that ULBP2 can also be sequestered intracellularly after defective N-glycosylation (33), implying that N-glycosylation could be a general mechanism for post-translational regulation of NKG2D ligand surface expression. We speculate that it may be advantageous for pathogens to target the more uniform MHC class I-like protein structure through post-translational mechanisms and find it likely that other post-translational regulatory mechanisms remain to be elucidated.
It is well described that certain viruses and cancers facilitate changes in N-glycosylation often through unknown mechanisms (38, 39, 51). Some of these changes could block immune activation, for example through inhibition of surface expression of NKG2D ligands. Our current results indicate that this is the case for the U21 immunoevasin expressed by HHV7. We found that U21 inhibits surface expression of MICA018, and this inhibition was rescued by substitution of threonine 24. It could be argued that U21 inhibition of MICA is not a direct result of N-glycosylation modification and that it is an indirect result of intracellular sequestering of MICA in a partially N-glycosylated form. This is likely not the case because the MICA T24A substituted mutant is expressed on the cell surface with altered N-glycosylation in pHM-U21 cells. If the altered MICA N-glycosylation was a result of sequestering of an immature N-glycosylated form, the rescued cell surface expressed MICA should have been normally N-glycosylated, which is not the case as shown in Fig. 6C.
It has previously been shown that U21 inhibits MICB surface expression (24); the molecular mechanism behind the inhibition has, however, not been resolved. Our current data show that MICA Asn8 is critical for regulation by N-glycosylation. MICB Asn8 lacks the consensus site for N-glycosylation, and it is therefore not likely that MICB is regulated by the N-glycosylation mechanism pinpointed by our current studies on MICA.
Interestingly, the HHV7-U21 protein also retained MICA008; however, this was likely not through a direct change in MICA008 N-glycosylation. We are currently examining the structural requirements for U21 inhibition of MICA008, which likely constitutes a novel motif for post-translational regulation of MICA surface expression. MICA008 is a particularly frequent allele found in the Western part of the world (11, 17), and the high frequency of MICA008 is likely linked to protection against certain pathogens. Indeed, the gene product encoded by the MICA008 allele cannot be retained by certain viruses (22, 26). However, the difference in regulation between MICA008 and full-length MICA alleles is not straightforward. We found the polymorphism in the extracellular domains of MICA008 and MICA018 is responsible for the difference in regulation of the two alleles. McSharry et al. (27) showed that adenovirus E3/19K retain both MICA008 and several full-length MICA alleles in the ER. We now show that HHV7-U21 retains both the MICA018 and MICA008 alleles. Our study illustrates the complexity of MICA regulation, which possibly explains the need for numerous activating ligands for NKG2D, as previously speculated (17).
We provide evidence for N-glycosylation as an allele-specific post-translational regulatory pathway of MICA. We show that HHV7 through U21 likely use this mechanism in the course of avoiding immune surveillance. The N-glycosylation-mediated regulation possibly applies to all Thr24-containing MICA alleles, and we hypothesize that this particular site interacts with an undiscovered protein until proper N-glycosylation has occurred. We show that mutation of this regulatory site rescues inhibition of MICA018 surface expression in HHV7-U21-expressing cells. Between 8 and 19% of the human population carries a Thr24-expressing MICA allele. Our current results suggest that these individuals could have a diminished reaction toward, for example HHV7 infection. Individuals expressing MICA Thr24 may on the other hand have a better control of MICA surface expression, possibly counteracting development of autoimmunity.
Acknowledgments
We thank Dr. Per Thor Straten (University Hospital Herlev) for melanoma cell lines, Amy Hudson (Medical College of Wisconsin) for U373 and pHM-U21 cell lines, Caroline Strømme for initiating production of MICA018 N-mut constructs, and the Center for Advanced Bioimaging (University of Copenhagen) for fluorescence microscopy images.
This work was supported by Project DFF-1331-00169 from the Danish Council for Independent Research/Medical Sciences.
R. Gupta, E. Jung, and S. Brunak, manuscript in preparation.
- NKG2D
- natural killer group 2D
- MICA
- MHC class I-related protein A
- MICB
- MHC clas I-related protein B
- ULBP
- UL16-binding protein
- HHV
- human herpesvirus
- ER
- endoplasmic reticulum
- 2DG
- 2-deoxy-d-glucose
- PNGase F
- peptide N-glycosidase F
- L-PHA
- leukoagglutinin
- PBMC
- peripheral blood mononuclear cell.
REFERENCES
- 1. Reis e Sousa C. (2004) Toll-like receptors and dendritic cells: for whom the bug tolls. Semin. Immunol. 16, 27–34 [DOI] [PubMed] [Google Scholar]
- 2. Tesniere A., Panaretakis T., Kepp O., Apetoh L., Ghiringhelli F., Zitvogel L., Kroemer G. (2008) Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 15, 3–12 [DOI] [PubMed] [Google Scholar]
- 3. Kroemer G., Zitvogel L. (2007) Death, danger, and immunity: an infernal trio. Immunol. Rev. 220, 5–7 [DOI] [PubMed] [Google Scholar]
- 4. Guerra N., Tan Y. X., Joncker N. T., Choy A., Gallardo F., Xiong N., Knoblaugh S., Cado D., Greenberg N. M., Greenberg N. R., Raulet D. H. (2008) NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. González S., Groh V., Spies T. (2006) Immunobiology of human NKG2D and its ligands. Curr. Top. Microbiol. Immunol. 298, 121–138 [DOI] [PubMed] [Google Scholar]
- 6. López-Larrea C., Suárez-Alvarez B., López-Soto A., López-Vázquez A., Gonzalez S. (2008) The NKG2D receptor: sensing stressed cells. Trends Mol. Med. 14, 179–189 [DOI] [PubMed] [Google Scholar]
- 7. Groh V., Smythe K., Dai Z., Spies T. (2006) Fas-ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat. Immunol. 7, 755–762 [DOI] [PubMed] [Google Scholar]
- 8. Raulet D. H., Gasser S., Gowen B. G., Deng W., Jung H. (2013) Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 31, 413–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bahram S., Bresnahan M., Geraghty D. E., Spies T. (1994) A second lineage of mammalian major histocompatibility complex class I genes. Proc. Natl. Acad. Sci. U.S.A. 91, 6259–6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cosman D., Müllberg J., Sutherland C. L., Chin W., Armitage R., Fanslow W., Kubin M., Chalupny N. J. (2001) ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14, 123–133 [DOI] [PubMed] [Google Scholar]
- 11. Champsaur M., Lanier L. L. (2010) Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 235, 267–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li P., Willie S. T., Bauer S., Morris D. L., Spies T., Strong R. K. (1999) Crystal structure of the MHC class I homolog MIC-A, a gamma-delta T cell ligand. Immunity 10, 577–584 [DOI] [PubMed] [Google Scholar]
- 13. Steinle A., Li P., Morris D. L., Groh V., Lanier L. L., Strong R. K., Spies T. (2001) Interactions of the human NKG2D with its ligands MICA, MICB and homologs of the mouse RAE-1 protein family. Immunogenetics 53, 279–287 [DOI] [PubMed] [Google Scholar]
- 14. Li P., Morris D. L., Willcox B. E., Steinle A., Spies T., Strong R. K. (2001) Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat. Immunol. 2, 443–451 [DOI] [PubMed] [Google Scholar]
- 15. Robinson J., Pérez-Rodríguez M., Waller M. J., Cuillerier B., Bahram S., Yao Z., Albert E. D., Madrigal J. A., Marsh S. G. (2001) MICA sequences 2000. Immunogenetics 53, 150–169 [DOI] [PubMed] [Google Scholar]
- 16. Mizuki N., Ota M., Kimura M., Ohno S., Ando H., Katsuyama Y., Yamazaki M., Watanabe K., Goto K., Nakamura S., Bahram S., Inoko H. (1997) Triplet repeat polymorphism in the transmembrane region of the MICA gene: a strong association of six GCT repetitions with Behçet disease. Proc. Natl. Acad. Sci. U.S.A. 94, 1298–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stephens H. (2001) MICA and MICB genes: can the enigma of their polymorphism be resolved? Trends Immunol. 22, 378–385 [DOI] [PubMed] [Google Scholar]
- 18. Welte S. A., Sinzger C., Lutz S. Z., Singh-Jasuja H., Sampaio K. L., Eknigk U., Rammensee H. G., Steinle A. (2003) Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur. J. Immunol. 33, 194–203 [DOI] [PubMed] [Google Scholar]
- 19. Eagle R. A., Traherne J. A., Hair J. R., Jafferji I., Trowsdale J. (2009) ULBP6/RAET1L is an additional human NKG2D ligand. Eur. J. Immunol. 39, 3207–3216 [DOI] [PubMed] [Google Scholar]
- 20. Wu J., Chalupny N. J., Manley T. J., Riddell S. R., Cosman D., Spies T. (2003) Intracellular retention of the MHC class I-related chain B ligand of NKG2D by the human cytomegalovirus UL16 glycoprotein. J. Immunol. 170, 4196–4200 [DOI] [PubMed] [Google Scholar]
- 21. Dunn C., Chalupny N. J., Sutherland C. L., Dosch S., Sivakumar P. V., Johnson D. C., Cosman D. (2003) Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J. Exp. Med. 197, 1427–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chalupny N. J., Rein-Weston A., Dosch S., Cosman D. (2006) Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem. Biophys. Res. Commun. 346, 175–181 [DOI] [PubMed] [Google Scholar]
- 23. Blauvelt A. (2001) Skin diseases associated with human herpesvirus 6, 7, and 8 infection. J. Invest. Dermatol. Symp. Proc. 6, 197–202 [DOI] [PubMed] [Google Scholar]
- 24. Schneider C. L., Hudson A. W. (2011) The human herpesvirus-7 (HHV-7) U21 immunoevasin subverts NK-mediated cytoxicity through modulation of MICA and MICB. PLoS Pathogens 7, e1002362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ashiru O., Bennett N. J., Boyle L. H., Thomas M., Trowsdale J., Wills M. R. (2009) NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J. Virol. 83, 12345–12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thomas M., Boname J. M., Field S., Nejentsev S., Salio M., Cerundolo V., Wills M., Lehner P. J. (2008) Down-regulation of NKG2D and NKp80 ligands by Kaposi's sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 1656–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McSharry B. P., Burgert H. G., Owen D. P., Stanton R. J., Prod'homme V., Sester M., Koebernick K., Groh V., Spies T., Cox S., Little A. M., Wang E. C., Tomasec P., Wilkinson G. W. (2008) Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 82, 4585–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waldhauer I., Goehlsdorf D., Gieseke F., Weinschenk T., Wittenbrink M., Ludwig A., Stevanovic S., Rammensee H. G., Steinle A. (2008) Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 68, 6368–6376 [DOI] [PubMed] [Google Scholar]
- 29. Kaiser B. K., Yim D., Chow I. T., Gonzalez S., Dai Z., Mann H. H., Strong R. K., Groh V., Spies T. (2007) Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 447, 482–486 [DOI] [PubMed] [Google Scholar]
- 30. Skov S., Pedersen M. T., Andresen L., Straten P. T., Woetmann A., Odum N. (2005) Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 65, 11136–11145 [DOI] [PubMed] [Google Scholar]
- 31. Zwirner N. W., Fuertes M. B., Girart M. V., Domaica C. I., Rossi L. E. (2007) Cytokine-driven regulation of NK cell functions in tumor immunity: role of the MICA-NKG2D system. Cytokine Growth Factor Rev. 18, 159–170 [DOI] [PubMed] [Google Scholar]
- 32. Fuertes M. B., Girart M. V., Molinero L. L., Domaica C. I., Rossi L. E., Barrio M. M., Mordoh J., Rabinovich G. A., Zwirner N. W. (2008) Intracellular retention of the NKG2D ligand MHC class I chain-related gene A in human melanomas confers immune privilege and prevents NK cell-mediated cytotoxicity. J. Immunol. 180, 4606–4614 [DOI] [PubMed] [Google Scholar]
- 33. Andresen L., Skovbakke S. L., Persson G., Hagemann-Jensen M., Hansen K. A., Jensen H., Skov S. (2012) 2-Deoxy d-glucose prevents cell surface expression of NKG2D ligands through inhibition of N-linked glycosylation. J. Immunol. 188, 1847–1855 [DOI] [PubMed] [Google Scholar]
- 34. Fares F. (2006) The role of O-linked and N-linked oligosaccharides on the structure-function of glycoprotein hormones: development of agonists and antagonists. Biochim. Biophys. Acta 1760, 560–567 [DOI] [PubMed] [Google Scholar]
- 35. Elbein A. D. (1991) The role of N-linked oligosaccharides in glycoprotein function. TIBTECH 9, 346–352 [DOI] [PubMed] [Google Scholar]
- 36. Dennis J. W., Granovsky M., Warren C. E. (1999) Glycoprotein glycosylation and cancer progression. Biochim. Biophys. Acta 1473, 21–34 [DOI] [PubMed] [Google Scholar]
- 37. Benjamin L., Cassimeris L., Lingappa V., Plopper G. (2007) Cells, p. 120, Jones and Bartlett Publishers, Sudbury, MA [Google Scholar]
- 38. Marcos N. T., Magalhães A., Ferreira B., Oliveira M. J., Carvalho A. S., Mendes N., Gilmartin T., Head S. R., Figueiredo C., David L., Santos-Silva F., Reis C. A. (2008) Helicobacter pylori induces β3GnT5 in human gastric cell lines, modulating expression of the SabA ligand sialyl-Lewis x. J. Clin. Invest. 118, 2325–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lantéri M., Giordanengo V., Hiraoka N., Fuzibet J. G., Auberger P., Fukuda M., Baum L. G., Lefebvre J. C. (2003) Altered T cell surface glycosylation in HIV-1 infection results in increased susceptibility to galectin-1-induced cell death. Glycobiology 13, 909–918 [DOI] [PubMed] [Google Scholar]
- 40. Kobata A., Amano J. (2005) Altered glycosylation of proteins produced by malignant cells, and application for the diagnosis and immunotherapy of tumours. Immunol. Cell Biol. 83, 429–439 [DOI] [PubMed] [Google Scholar]
- 41. May N. A., Glosson N. L., Hudson A. W. (2010) Human herpesvirus 7 u21 downregulates classical and nonclassical class I major histocompatibility complex molecules from the cell surface. J. Virol. 84, 3738–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Groh V., Bruhl A., El-Gabalawy H., Nelson J. L., Spies T. (2003) Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 100, 9452–9457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andresen L., Jensen H., Pedersen M. T., Hansen K. A., Skov S. (2007) Molecular regulation of MHC class I chain-related protein A expression after HDAC-inhibitor treatment of Jurkat T cells, J. Immunol. 179, 8235–8242 [DOI] [PubMed] [Google Scholar]
- 44. Andresen L., Hansen K. A., Jensen H., Pedersen S. F., Stougaard P., Hansen H. R., Jurlander J., Skov S. (2009) Propionic acid secreted from propionibacteria induces NKG2D ligand expression on human-activated T lymphocytes and cancer cells. J. Immunol. 183, 897–906 [DOI] [PubMed] [Google Scholar]
- 45. Kurtoglu M., Maher J. C., Lampidis T. J. (2007) Differential toxic mechanisms of 2-deoxy-d-glucose versus 2-fluorodeoxy-d-glucose in hypoxic and normoxic tumor cells. Antioxid. Redox Signal. 9, 1383–1390 [DOI] [PubMed] [Google Scholar]
- 46. Suemizu H., Radosavljevic M., Kimura M., Sadahiro S., Yoshimura S., Bahram S., Inoko H. (2002) A basolateral sorting motif in the MICA cytoplasmic tail. Proc. Natl. Acad. Sci. U.S.A. 99, 2971–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nice T. J., Deng W., Coscoy L., Raulet D. H. (2010) Stress-regulated targeting of the NKG2D ligand Mult1 by a membrane-associated RING-CH family E3 ligase. J. Immunol. 185, 5369–5376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Molinero L. L. (2003) Up-regulated expression of MICA on activated T lymphocytes involves Lck and Fyn kinases and signaling through MEK1/ERK, p38 MAP kinase, and calcineurin. J. Leukocyte Biol. 73, 815–822 [DOI] [PubMed] [Google Scholar]
- 49. Zwirner N. W., Fernández-Viña M. A., Stastny P. (1998) MICA, a new polymorphic HLA-related antigen, expressed mainly by keratinocytes, endothelial cells, and monocytes. Immunogenetics 47, 139–148 [DOI] [PubMed] [Google Scholar]
- 50. Satomaa T., Heiskanen A., Leonardsson I., Angström J., Olonen A., Blomqvist M., Salovuori N., Haglund C., Teneberg S., Natunen J., Carpén O., Saarinen J. (2009) Analysis of the human cancer glycome identifies a novel group of tumor-associated N-acetylglucosamine glycan antigens. Cancer Res. 69, 5811–5819 [DOI] [PubMed] [Google Scholar]
- 51. Lau K. S., Dennis J. W. (2008) N-Glycans in cancer progression. Glycobiology 18, 750–760 [DOI] [PubMed] [Google Scholar]
- 52. Fernandes B., Sagman U., Auger M., Demetrio M., Dennis J. W. (1991) βl-6 branched oligosaccharides as a marker of tumor progression in human breast and colon neoplasia. Cancer Res. 51, 718–723 [PubMed] [Google Scholar]
- 53. Elbein A. D., Tropea J. E., Mitchell M., Kaushal G. P. (1990) Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J. Biol. Chem. 265, 15599–15605 [PubMed] [Google Scholar]
- 54. Tulsiani D. R., Harris T. M., Touster O. (1982) Swainsonine inhibits the biosynthesis of complex glycoproteins by inhibition of Golgi mannosidase II. J. Biol. Chem. 257, 7936–7939 [PubMed] [Google Scholar]
- 55. Cummings R. D., Kornfeld S. (1982) Characterization of the structural determinants required for the high affinity interaction of asparagine-linked oligosaccharides with immobilized Phaseolus vulgaris leukoagglutinating and erythroagglutinating lectin, J. Biol. Chem. 257, 11230–11234 [PubMed] [Google Scholar]
- 56. Glosson N. L., Gonyo P., May N. A., Schneider C. L., Ristow L. C., Wang Q., Hudson A. W. (2010) Insight into the mechanism of human herpesvirus 7 U21-mediated diversion of class I MHC molecules to lysosomes. J. Biol. Chem. 285, 37016–37029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hudson A. W., Howley P. M., Ploegh H. L. (2001) A human herpesvirus 7 glycoprotein, U21, diverts major histocompatibility complex class I molecules to lysosomes. J. Virol. 75, 12347–12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ogasawara K., Hamerman J. A., Ehrlich L. R., Bour-Jordan H., Santamaria P., Bluestone J. A., Lanier L. L. (2004) NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity 20, 757–767 [DOI] [PubMed] [Google Scholar]