

Background: Cobra venom consists of diverse toxin homologues important for prey capture through variable levels of gene duplication.
Results: Comparative structural and cellular investigations revealed sensitivity of toxin homologues for extra- and intracellular targets via sulfated glycoconjugates.
Conclusion: Variation in positively charged and hydrophobic domains in CTX homologues altered their target specificities and endocytosis.
Significance: We provide a rationale for toxin homologues at cellular level to gain an evolutionary advantage.
Keywords: Cholesterol, Endocytosis, Heparan Sulfate, Snake Venom, X-ray Crystallography, Cardiotoxin, Sulfatide
Abstract
Cobra cardiotoxins (CTX) are a family of three-fingered basic polypeptides known to interact with diverse targets such as heparan sulfates, sulfatides, and integrins on cell surfaces. After CTX bind to the membrane surface, they are internalized to intracellular space and exert their cytotoxicity via an unknown mechanism. By the combined in vitro kinetic binding, three-dimensional x-ray structure determination, and cell biology studies on the naturally abundant CTX homologues from the Taiwanese cobra, we showed that slight variations on the spatial distribution of positively charged or hydrophobic domains among CTX A2, A3, and A4 could lead to significant changes in their endocytotic pathways and action mechanisms via distinct sulfated glycoconjugate-mediated processes. The intracellular locations of these structurally similar CTX after internalization are shown to vary between the mitochondria and lysosomes via either dynamin2-dependent or -independent processes with distinct membrane cholesterol sensitivity. Evidence is presented to suggest that the shifting between the sulfated glycoconjugates as distinct targets of CTX A2, A3, and A4 might play roles in the co-evolutionary arms race between venomous snake toxins to cope with different membrane repair mechanisms at the cellular levels. The sensitivity of endocytotic routes to the spatial distribution of positively charged or hydrophobic domains may provide an explanation for the diverse endocytosis pathways of other cell-penetrating basic polypeptides.
Introduction
Cardiotoxins (CTX)3 are cobra venom toxins known to cause severe tissue necrosis and systolic heart arrest in the envenomed victims in vivo (1). They are cationic polypeptides consisting of 60–62 amino acids in a three-fingered looped structure with a distinct spatial distribution of their positively charged and hydrophobic domains among various CTX homologues (or isomers). The specific targets of demonstrated cell toxicity have been shown to be cell surface carbohydrates of glycosaminoglycans (GAG), membrane lipids of sulfatides, and/or membrane proteins of integrin, depending on the biological assay used for the investigations (1–3). The exact biological reasons for the diverse targets of the various CTX homologues are not clear, although it has been demonstrated that the toxin levels of cobra venom proteins exhibit significant geographic variation for Taiwanese cobras caught in the wild (4). For instance, CTX A2 and A4 toxins are present in cobra venoms from the western coast but are absent in the snakes from the eastern coast. In contrast, a similar level of CTX A3, the major CTX in the Taiwanese cobra, can be identified in both western and eastern Taiwanese cobra venom.
On top of the diverse cell surface targets for CTX homologues, the intracellular locations of CTX after their internalization also appear to vary significantly. Depending on the type of CTX used for the study and experimental conditions, different CTX homologues have been shown to be located either in mitochondria or lysosomes. For instance, a sulfatide lipid-dependent cell-penetrating mechanism has been shown for CTX A3 to target mitochondria in cardiomyocytes and H9C2 cells (3, 5). In contrast, several other reports have suggested that other CTX homologues can target lysosomes after co-incubation of the CTX with the studied cell line (6, 7). Because cell-penetrating basic polypeptides have also been shown to be internalized with diverse endocytotic routes, we were interested to see whether the distinct endocytotic routes could depend on the type of cell surface targets for various CTX homologues. Endocytosis is important for the passage of extracellular cargo over plasma membrane barriers, thus exhibiting their biological activities. The modulated location and concentration of internalized macromolecules affects several cellular events such as cell proliferation, differentiation, tumorigenesis, and pathogen malignancy (8–10). Different endocytotic pathways are classified according to the specific machinery used, including clathrin-dependent, caveolae-dependent, phagocytotic, marcopinocytotic, and other mechanisms (CLIC/GEEC-dependent, flotillin-dependent, and Arf6-dependent mechanisms). Some of these processes are mediated by dynamin2, the GTPase involved in both clathrin-mediated endocytosis and caveolae-dependent pathway (11–13).
Recent genomic investigation of the King cobra has indicated that dynamic gene evolution and adaptation actually is in operation in the snake venom system (14). For the fast evolving biological weapon systems such as snake toxins, a comparative structural and functional study of naturally abundant CTX homologues should shed light on how venom proteins evolve at the molecular and cellular levels. Because some endocytotic cargos, such as Tat protein or arginine-rich polypeptide, have been used as efficient vehicles to deliver anionic nucleotides for therapeutic purposes (15–17), it was important to perform a similar study on basic polypeptides with more well defined structures, such as CTX homologues. Our results suggested that a relatively small spatial variation of either positively charged or hydrophobic domains within the well defined three-fingered structure could be significant enough to meet the fast evolutionary need of the biological “arms race.”
EXPERIMENTAL PROCEDURES
Materials
CTX were purified from Naja atra venom (purchased from a local snake farm in Tainan, Taiwan) by ion exchanger chromatography and reverse phase high performance liquid chromatography (18). FITC- or rhodamine-labeled CTX were prepared as described previously (3, 5). The antibodies against CTX were isolated from horse antiserum (Center for Disease Control, Taiwan) by a CTX-conjugated affinity column. If not specifically indicated, the other chemicals or heparinases were purchased from Sigma-Aldrich. De-N-sulfated heparin, de-2-O-sulfated heparin, and de-6-O-sulfated heparin were derived from high molecular weight heparin (∼18 kDa) as described earlier (19). De-N-acetylated heparins were prepared by hydrazinolysis of 35 mg of native heparin with 0.6 ml of 1% (w/v) hydrazine sulfate in anhydrous hydrazine at 100 °C for 10 h (20). Dropwise additions of 1.2 ml of HIO3 (0.25 m) at −18 °C were followed by extraction with diethyl ether to remove I2 product. The water fraction was harvested and lyophilized. All heparin derivatives were characterized by one-dimensional 13C NMR to determine the levels of chemical modification.
Cell Culture, Viability Assay, and Capillary Retention Assay
H9C2 cells and Chinese hamster ovary (CHO) cells were maintained in DMEM and modified McCoy's 5A medium (Sigma-Aldrich), respectively, supplemented with 10% (v/v) fetal bovine serum (Thermo Scientific HyClone), 100 units/ml penicillin, and 100 μg/ml streptomycin. Cell viability was reported by MTT assay for the reduction of MTT into purple formazan precipitates. After treatment with CTX for 30 min at 37 °C, the cells were treated with the MTT solution (2 mg/ml in culture serum) for 4 h. Absorbance at 570 nm was measured after removal of the MTT solution and PBS wash. The extent of CTX retention in the capillary tube plated with CHO cells was investigated as described previously (19).
CTX Endocytosis
For the endocytosis experiments, the cells were plated onto a 12-mm glass coverslip 1 day before the experiment. After washing with Hanks' balanced salt solution, fluorescently labeled CTX (1 μm), as well as organelle markers (Alexa Fluor 488 conjugate albumin, 50 μg/ml; FITC-dextran, 1 mg/ml; Alexa Fluor 488 conjugate transferrin, 25 μg/ml; and Hoechst 33342, 0.5 μg/ml), was preincubated at 4 °C for 30 min. After extensive, rapid washing of cells and incubation at 37 °C, the images were recorded using a Zeiss LSM 510 or an Olympus FV1000 confocal microscope using a pinhole size of full-width at half-maximum for quantification of internalized CTX (size 5 μm) or the percentage of colocalization (size 0.7 μm). The percentage of CTX A2/A4 colocalization was estimated based on the ratio of the area of the yellow spots versus red (or green) spots. The average intensity was quantified and averaged from 10 to 20 cells in three to five fields by integration of the fluorescence intensity using ImageJ software (21).
Pretreatments for Cell Studies
CTX endocytosis or CTX-induced cytotoxicity was blocked under the pretreatment of 80 μm dynasore for 15 min at 37 °C before CTX application. For cholesterol depletion or incorporation, cells were incubated with the indicated concentration of methyl-β-cyclodextran (MβCD) or a mixture of MβCD and cholesterol as indicated in Fig. 6C, respectively, for 15 min at 37 °C. The cells were then washed twice by 1% (w/v) BSA and once by Hanks' balanced salt solution. The heparinase pretreatments (8 mIU/ml) were performed at 37 °C for 1 h before CTX treatments.
FIGURE 6.

Effect of membrane cholesterol concentration on the internalization and cytotoxicity of CTX homologues. A and B, confocal images and quantifications of CTX endocytosis upon cholesterol (chol.) depletion (10 mm MβCD) or cholesterol enrichment (0.2 mm cholesterol/2 mm MβCD). Filipin, the cholesterol chelator, is used to reflect the decrease or increase in cholesterol concentration as a result of the depletion or enrichment process. Bar = 20 μm. Data were mean ± S.E. (n = 5); *, p < 0.05; **, p < 0.01. C, CTX A3-induced cytotoxicity and endocytosis as a result of cholesterol depletion and incorporation. D, inhibition of CTX A3-induced leakage of 6-CF-loaded vesicles by elevated cholesterol content in sulfated glycosphingolipid (SGC)-containing vesicles.
Extracellular Ca2+ Influx
Monitoring of extracellular Ca2+ influx was done using the following procedure. Subconfluent cells were loaded with 5 μm Fluo-3 AM (Invitrogen), 0.5% (w/v) pluronic F-127 (Invitrogen), and 0.5 mm probenecid for 45 min in a culture medium. Time-lapsed images were recorded on a confocal microscope for 5 min (with 1-s intervals) at 37 °C immediately after the CTX (3 μm) addition.
Membrane Repair Study
The level of plasma membrane repair was assayed by decreased propidium iodide influx as visualized by confocal microscopy (Olympus Fv1000). Before the CTX A3 addition, cells were repeatedly washed in a cold Ca2+-free Hanks' balanced salt solution buffer. CTX A3 was pretreated for 5 min at 4 °C in a Ca2+-free Hanks' balanced salt solution buffer, which was then replaced by warm buffers (at 37 °C) with or without 1.8 mm Ca2+. After a 5-min incubation at 37 °C, cells were fixed in 4% paraformaldehyde and stained with 50 μg/ml propidium iodide (Sigma-Aldrich).
Lysosomal Membrane Permeabilization Assay
The effect of CTX A2 and A4 on the lysosome integrity of H9C2 cells was monitored by red fluorescence of acridine orange (5 μg/ml) with reference monitoring using LysoTracker Green DND-26 (0.5 μm), as well as by the green fluorescence of LysoSensor Green DND-189 (1 μm). Briefly, after pretreatment with fluorescent dye at 37 °C for 30 min, the cells were extensively washed and incubated with 3 μm CTX A2 or A4 at 37 °C for an additional 30 min. The images were recorded using an Olympus FV1000 confocal microscope. For detection of cathepsin D, after incubation with CTX the cells were fixed with 4% formaldehyde and stained with anti-human cathepsin D antibody (Santa Cruz Biotechnology) followed by a Cy3-conjugated goat anti-rabbit IgG antibody (Santa Cruz Biotechnology) according to the manufacturer's instructions.
Surface Plasmon Resonance (SPR) Experiments
SPR experiments were performed using a Biacore X instrument (GE Healthcare) at 25 °C under continuous flow (40 μl/min) of a running buffer (10 mm HEPES-NaOH, pH 7.4, 150 mm NaCl, and 0.005% surfactant P20) as described previously (19). To investigate the heparin binding specificity toward CTX A2 or A4, we also performed SPR competition experiments. First, the retained CTX were achieved by injecting CTX (5 μm) over immobilized heparin (200 RU). Upon stable CTX retention (CTX A2, 90 RU; CTX A4, 250 RU) after a 4 min-dissociation phase, extrinsic heparin was injected through the retained CTX surface to enable dissociation of the retained CTX. The retained CTX was prepared by 2-min injections of 5 μm CTX A2 or A4 over immobilized heparin surfaces with 4-min dissociation phases to achieve stable CTX retentions. The retained CTX were competed out of the immobilized heparin surface by extrinsic heparin derivatives and exhibited accelerated dissociation behaviors. Second, competition SPR experiments were performed by using the strategy of CTX binding onto immobilized heparin in the presence of heparin mimetics as competitors in solution, as described previously (22) with a slight modification. Premixing the CTX and heparin at the desired concentrations was done prior to the injection, and the reduced amount in Req (response at equilibrium binding) as compared with the Req in the absence of competitor was normalized as the percentage of relative response as shown in Fig. 3A.
FIGURE 3.

SPR studies for CTX A2 and A4 binding to immobilized heparin with sulfate specificity. A, effect of distinct sulfation groups on the binding of heparin mimetics to CTX A2 (open symbols) and CTX A4 (close symbols) as revealed by the SPR binding sensograms in the presence of the related desulfated heparin mimetics. The arrows emphasized the difference in Ki values for de-6-O-sulfated heparin binding to CTX A2 and A4. B, the retained CTX were dissociated from immobilized heparin surfaces by injecting heparin through competition. Insert, indicates that the competition of injected heparin was concentration-dependent, reflecting the competitive ability of injected heparin. C, effects of heparin modifications on their competitive abilities as revealed by the dissociation of the retained CTX in the presence of indicated heparin mimetics. de2OS, de-2-O-sulfation; de6OS, de-6-O-sulfation; deNS, de-N-sulfation.
In Vitro Vesicle Leakage Assay
The vesicle was prepared using POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; Avanti Polar Lipids) with different added POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine, Avanti Polar Lipids) or sulfatide (Sigma-Aldrich). To prepare the vesicles containing the fluorescent dye 6-CF (Sigma-Aldrich), lipids were dried under vacuum overnight and rehydrated with 10 mm Tris, pH 7.4, and 50 mm 6-CF. The reconstituted solution was then extruded through a polycarbonate filter with pore size 0.1 μm to form large homogeneous unilamellar vesicles. The vesicles were isolated from residual fluorescence dyes by a Sepharose CL-4B column. The percentage of 6-CF leakage was calculated using the formula % leakage = (F(t) − F0)/(F(t) − F∝), where Fo is the initial fluorescence intensity of vesicles before CTX was added, F(t) is the fluorescent intensity of the vesicle after CTX was added, and F∞ is the fluorescence intensity of the vesicle treated with 0.02% (v/v) Triton X-100.
Crystallization, Data Collection, and Processing
CTX A2 and A4 protein crystallizations were performed by the hanging-drop vapor diffusion method at 291 K. The drops of CTX (1 μl in 10 mg/ml) were mixed with the same volume of reservoir solution containing 5.5 m ammonium nitrate and 0.1 m bis-Tris, pH 7.2, for CTX A2, or 1.3 m ammonium sulfate, 100 mm Tris-HCl, pH 8.5, and 200 mm lithium sulfate for CTX A4, respectively. The CTX A2 and A4 crystals grew and appeared after 3 to 4 days. The crystals containing CTX A2 or A4 were then transferred to the respective cryoprotectant solutions (reservoir solution with 20% glycerol) and flash-cooled in liquid nitrogen for x-ray diffraction. All x-ray diffraction data were collected at 200 K at beamline BL13B1 at the National Synchrotron Radiation Research Center in Hsinchu, Taiwan or at BL26B2 at SPring-8, Japan. All data sets were indexed, integrated, and scaled using the program HKL2000 (23). The structure of CTX A2 was determined by molecular replacement with Phaser (24) using the structure of the CTX A3 complex with sulfogalactoceramide (PDB code 2BHI) as the search model. After several rounds of residue substitution and model building in COOT (25), the refinement procedures were accomplished with Refmac5 (26). The model validation was performed with PROCHECK (27). We used the CTX A2 structure as the search model for CTX A4 structure determination by molecular replacement with Phaser followed by refinement including TLS refinement with Refmac5. Data collection and refinement statistics are given in Table 1.
TABLE 1.
Data collection and refinement statistics of CTX A2 and A4 structures
Values in parentheses refer to statistics in the highest resolution shell.
| CTX A2 (PDB 4OM4) | CTX A4 (PDB 4OM5) | |
|---|---|---|
| Data collection | ||
| Space group | P43212 | P6522 |
| Cell dimensions | ||
| a, b, c (Å) | 107.412, 107.412, 100.256 | 72.933, 72.933, 249.422 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 120 |
| Resolution (Å) | 30-2.74 (2.84-2.74) | 50-2.55 (2.64-2.55) |
| Rmerge | 0.103 (0.494) | 0.15 (0.906) |
| I/σI | 25.78 (7.44) | 30.58 (6.09) |
| Completeness (%) | 99.9 (100) | 99.9 (100) |
| Redundancy | 15.5 (15.0) | 41.1 (41.8) |
| Refinement | ||
| Resolution (Å) | 25.62-2.74 | 31.03-2.552 |
| Rwork/Rfree | 0.1966/0.2596 | 0.2149/0.2623 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.015 | 0.011 |
| Bond angles (°) | 1.914 | 1.268 |
| Ramachandran analysis (%) | ||
| Favored/Allowed/Generous/Disallowed | 87.6/11.2/0.8/0.4 | 88.9/10.6/0.5/0.0 |
Molecular Docking
A model of heparin dp10 was built from PDB entry 1HPN (28). Various topologies of heparin dp10 were generated by the PRODRG server (29–30). The molecular docking of heparin dp10 binding to the CTX A4 dimer was performed using the HADDOCK Web server (31, 32). The residues on the convex side of CTX A4 (Lys-5, Lys-12, Lys-18, and Lys-35) were defined as the active residues, and those residues around the active residues were automatically selected as the passive residues. 1000 complex structures were generated from rigid body docking, and 200 lowest energy structures were obtained after semiflexible and water refinements. A model from the best scoring cluster based on the HADDOCK score was chosen as the putative docking model (shown in Fig. 2C).
FIGURE 2.

Structural comparisons of CTX A2, A3, and A4. A, three different views of electrostatic surfaces of the CTX homologues based on the crystal structures of CTX A2 (PDB code 4OM4 (this work)), CTX A3 (PDB code 1XT3), and CTX A4 (PDB code 4OM5 (this work)). Protein surfaces shown in blue, red, and white indicate positively charged, negatively charged, and hydrophobic regions, respectively. B, hydrophobic interface in the crystal structures of A4 dimers. Ribbon models of the CTX A4 dimer colored in cyan and green are superimposed with the CTX A2 dimer in gray. The N-terminal R1 and hydrophobic residues at the dimer interface of A4 are shown as sticks. C, the electrostatic surface of CTX A4 dimer showing a continuous patch of positively charged residues (Lys-5, Lys-12, Lys-18, and Lys-35) suitable for heparin binding. The locations of the R1 residues are indicated by arrows. de2OS, de-2-O-sulfation; de6OS, de-6-O-sulfation; deNS, de-N-sulfation.
RESULTS
Binding Targets and Three-dimensional Structure of CTX Homologues
We previously classified Taiwanese cobra CTX homologues into P-type (Pro-30-containing) and S-type (Ser-28-containing) based on the CTX-induced fusion activity of sphingomyelin vesicles, distinguishable by its hydrophobicity near the tip of loop II (18). A subsequent x-ray crystal structure determination of CTX A3 (See Fig. 1A for the sequence and Fig. 2A for three-dimensional structure; PDB code 1XT3) supports the idea that a more hydrophobic loop II would bridge hydrophobic loops I and III to form a continuous, long stretch of the hydrophobic domain and facilitate P-type CTX binding with penetration into lipid bilayers. The same conclusion can also be reached by a similar observation of the relative CTX A2-, A3-, and A4-induced membrane leakage activity toward negatively charged POPS vesicles (Fig. 1B). CTX A3 exhibited 4–5-fold higher membrane leakage activity than CTX A2 and A4 in 50% POPS-containing vesicles.
FIGURE 1.
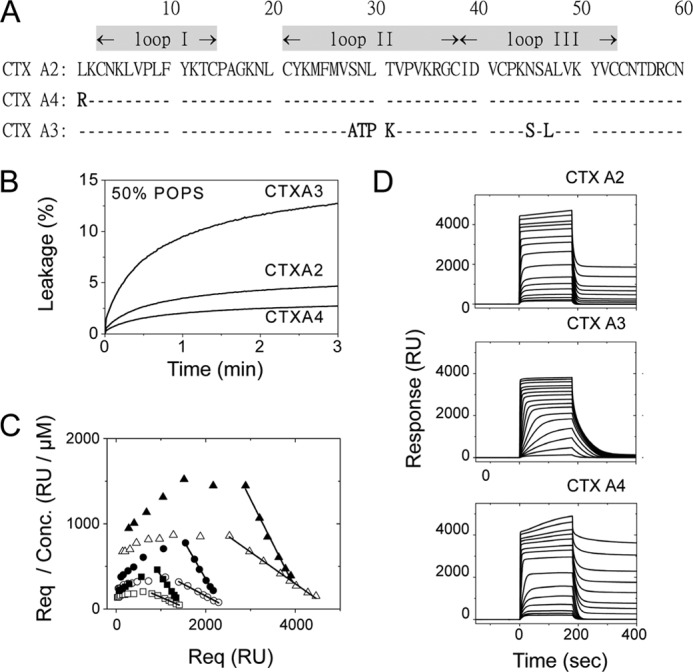
Heparin and negatively charged lipid as binding targets for three CTX homologues with a well defined spatial distribution of the positively charged domain. A, primary sequences of CTX homologues show high homologies but vary in several positively charged residues at the N-terminal and loop II regions. B, vesicle leakage assay on different CTX homologues shows differences in specificity toward negatively charged lipids. Time-dependent leakage of 6-CF dyes from 50% POPS vesicles was induced by CTX (0.16 μm). The higher affinity of CTX A3 toward the negatively charged lipids, POPS, was indicated by a higher leakage content, but this was not seen for CTX A2 or A4 under the same conditions. C, Scatchard plots (Req/concentration (Conc.) versus Req) for CTX A2 (open symbols) or CTX A4 (closed symbols) in an SPR binding assay (0.15–8 μm) on immobilized heparins under different surface densities (triangle, 700 RU; circle, 350 RU; square, 200 RU) were shown. D, SPR binding of CTX homologues (0.15–20 μm) onto immobilized heparin surfaces (700 RU as representative plots) showed different retention capabilities. Significant CTX retention on immobilized heparin surfaces was observed for CTX A2 and A4 at higher concentrations but not for CTX A3.
In addition to the lipid binding activity, CTX homologues had been shown to bind glycosaminoglycans with certain specificity (33). Heparan sulfate (HS), one type of GAG with alternating high and low sulfated domains, presented near the cell surface through membrane-associated proteoglycans. Heparin was a structural mimic of heparan sulfate and presented as a highly sulfated structure at the N-sulfate, 2-O-sulfate, and 6-O-sulfate positions. The affinity of CTX A3 binding to immobilized heparin was consistent with previous similar studies (19) showing binding affinities of ∼0.42 ± 0.1 μm under different heparin surface densities (data not shown). CTX A2 and A4 exhibited a micromolar binding affinity to the heparin surface in an SPR binding measurement using three different surface densities (Fig. 1C). By using Scatchard plots within the CTX concentration range of 0.15 to 8 μm, the Kd value toward immobilized heparin was determined to be around 3.6 ± 0.9 μm for CTX A2 and 1.1 ± 0.2 μm for CTX A4 under different heparin surface densities. Positive slopes were observed at low CTX A2/A4 concentration, indicating positive cooperativities of CTX-heparin binding with Hill coefficients of ∼1.3 and ∼1.5 for CTX A2 and A4, respectively.
Interestingly, a significant difference could be seen for the amount of CTX A2/A4 retention on surfaces during the washing-out time period, between 200 and 400 s (Fig. 1D). A similar retention behavior was observed in citrate-induced CTX A3 dimer formation as studied by kinetic binding of CTX A3 to the heparin surface in the presence of anionic citrate (19). In addition, under different surface densities, the percentage of CTX A2/A4 retained on immobilized heparin (amount of CTX retention/equilibrium response) increased with reduced surface density (data not shown). These observations suggest that CTX A2 and A4 exhibit significant dimer/oligomer formation and become significantly retained, mainly on heparin surface under low density condition.
To understand the structural basis of the aforementioned observation, we crystallized CTX A2 and A4 and determined their x-ray structures at 2.74 and 2.55 Å resolution (Table 1), respectively, as there were no available x-ray structures for CTX A2 and A4 to allow a direct comparison with CTX A3. Fig. 2A shows the ribbon models and surface diagrams with the spatial distribution of positively charged domains for the CTX homologues studied. Basically, the depicted structures exhibit a similarly positively charged distribution, with the exception that CTX A4 consists of a more extended, positively charged region to the convex site due to the substitution of the N-terminal residue Leu-1 into Arg-1 (Fig. 2A). Interestingly, both CTX A2 and A4 form a similar dimer interface consisting of exclusively hydrophobic residues such as Leu-6, Val-7, Pro-8, and Leu-9 and Val-32, Pro-33, and Val-34 from the loop I and II regions, respectively (Fig. 2B). A positively charged region formed by Lys-5, Lys-12, Lys-18, and Lys-35 on the convex side of CTX allowed heparin binding. An electrostatic potential map of the CTX A4 dimer showed a connection between two convex sites on two CTX monomers and generated a long stretch of positively charged region, linking the convex sites from two CTX monomers for possible heparin binding (Fig. 2C). Accordingly, the extended, positively charged region of a CTX dimer could bind to a heparin chain with 10 monosaccharides (Fig. 2C), as shown by a molecular docking experiment using the HADDOCK Web server (31, 32).
As we had shown previously that dimer formation of the CTX A3 molecules requires the binding of anionic citrate to interact with Lys-31 near the tip of loop II (19), the much higher surface retention of CTX A2 and A4 in the SPR binding measurement could be interpreted as a CTX binding-induced cross-linking effect on the heparin surface. Similarly, we could also attribute the significantly larger amount of surface retention of CTX A4 than of CTX A2 to the more extended positively charged domain of the former near the N-terminal region (Fig. 1D). Indeed, Arg-1 of CTX A4 is located right at the extended position of the heparin molecule to allow CTX A4 binding to heparin with a longer chain length (see the two arrows indicated by R1 in Fig. 2C).
Sulfate Groups Involved in the Binding to CTX
Quantifying the importance of sulfate groups for CTX A2 and A4 binding to heparin, we first performed SPR competition experiments in the presence and absence of heparin mimetics with a different degree of desulfation (22) to determine the effects of desulfation on the Ki value (concentration of heparin competitor to exhibit, 50% inhibition). As shown in Fig. 3A, the relative responses to the binding were reduced upon increasing the heparin concentrations. The Ki values for CTX A2 and A4 were similar for native heparin (0.40 versus 0.32 μm), de-N-sulfated heparin (16 versus 19 μm), de-2-O-sulfate heparin (1.7 versus 2.0 μm), and de-N-acetylated heparin (2.1 versus 2.0 μm). Nevertheless, reduction by about a factor of 2 was observed for de-6-O-sulfate heparin (0.55 versus 1.0 μm), as clearly indicated by the arrows on Fig. 3A.
Two conclusions can be made based on these observations. First, the binding of heparin mimetics of de-N-sulfated heparin to both CTX A2 and A4 was similarly reduced by almost 2 orders of magnitude as compared with the native heparin, indicating an important role for the N-sulfate group for binding to both CTX A2 and A4. Second, the presence of 6-O-sulfate gave a slight preference for binding to CTX A4 as compared with CTX A2, suggesting a possible involvement of the N-terminal Arg-1 of CTX A4 for its electrostatic interaction with 6-O-sulfate.
As a large amount of CTX A4 was observed to exhibit significant retention on SPR surfaces during a 200–400-s washing out, we also examined the effect of the sulfation of heparin molecules such as N-sulfate, 2-O-sulfate, and 6-O-sulfate on the retention behavior of CTX A4 as compared with that of CTX A2. We performed competition experiments by injecting heparin or heparin derivatives over the CTX-retained SPR chips to monitor the dissociation of CTX from the heparin surface. The competition-mediated dissociation by the injected heparin was seen to be concentration-dependent (Fig. 3B, inset). Moreover, as seen in Fig. 3C, native heparin was more competitive in eluting the retained CTX than the other heparin derivatives with different levels of desulfation or deacetylation. No dissociation could be observed by injecting de-N-sulfated heparin, consistent with our previous conclusion on the important role of the N-sulfate group in its binding to both CTX A2 and A4. The relative potency of different sulfated groups in CTX binding/retention can be determined as N-sulfate > 2-O-sulfate > 6-O-sulfate for both CTX A2 and A4 (Fig. 3C). A similar conclusion was also reached via an SPR competition experiment (Fig. 3A).
Distinct HS Domains and Endocytotic Routes for CTX A2/A4 Retention and Internalization
To understand the biological significance of the distinct sulfate group of HS in its binding to CTX, we first examined whether CTX A2 and A4 targeted different types of HS domains on cell membranes for their biological actions. It has been well established that heparinase I recognizes and degrades fully sulfated HS domains, whereas heparinase III recognizes and degrades the low sulfated domain (34). As 2-O-sulfate and 6-O-sulfate are located mainly in the fully sulfated HS domain, we could then examine the effect of different heparinase treatments on the cellular activities of CTX A2 and A4.
Previously we had designed a novel capillary retention assay to quantify the cell surface retention capability of CTX on immobilized mammalian cells such as CHO-K1 or its GAG-deficient pgsA745 cell lines (19). Using a similar approach, we show in Fig. 4A that cell surface retention of CTX A2/A4 was indeed GAG-sensitive. It gave evidence of less CTX retention on the surface of immobilized pgsA745 cells, i.e. GAG-deficient CHO cells, than on the surface of parental CHO-K1 cells. Interestingly, CTX A2 and A4 exhibited opposite sensitivities to heparinase I and heparinase III treatment of CHO-K1 cells. For CTX A2, pretreatment by heparinase I did not affect its retention on CHO-K1 cells, whereas heparinase III pretreatment eliminated CTX A2 retention as revealed by shortened strips of blots (Fig. 4A). This indicates that low-sulfated domains were responsible for CTX A2 binding (or retention) on cell surfaces. However, heparinase I pretreatment, but not heparinase III pretreatment, decreased the CTX A4 retention on CHO-K1 cell surfaces. This implies that the fully sulfated HS domains were involved in CTX A4 binding (or retention) onto CHO-K1 cell surfaces.
FIGURE 4.

Distinct heparinase I and III sensitivity to CTX A2/A4 retention and endocytosis indicating the involvement of different HS domains. A, retentions of different CTX on immobilized cell surfaces were dependent on surface GAG and were sensitive to different heparinase treatments as revealed by the reduced length of the blots (three individual repeats to show retained CTX). B, endocytotic CTX A2 and CTX A4 inside the H9C2 cells was located at the lysosome (as indicated by LysoTracker) but not at the mitochondria (as indicated by MitoTracker). Bar = 20 μm. C, endocytosis of different CTX into H9C2 or CHO cells was dependent on surface GAG and was sensitive to different heparinase treatments. Bar = 5 μm. D, CTX A2 and A4 induced lysosomal membrane permeabilization. CTX A2 or A4 decreased the red fluorescence of acridine orange (upper panels; green fluorescence was from LysoTracker Green DND-26) and the green fluorescence of LysoSensor DND-189 (middle panels) as compared with naphthazarin (lysosomal destabilizer, 5 μm). Treatment with naphthazarin released cathepsin D into the cytosol, but this was not observed for CTX A2 orA4 (intact spots remained). Data were mean ± S.E. (n > 10); **, p < 0.01.
One should note that CTX was internalized after binding to the cell surface and exhibited cell toxicity. CTX A3 internalization into mitochondria was through the sulfatide-mediated membrane insertion, internalization, and toxicity (3, 5). Further experiments may determine how the HS-mediated retention of CTX A2 and A4 on cell surfaces would be different from the sulfatide lipid-mediated CTX A3 internalization and the eventual location of the intracellular compartments. As shown in Fig. 4B, colocalization of rhodamine-labeled CTX A2 and A4 with the organelle markers of lysosome and mitochondria by confocal imaging revealed that CTX A2 and A4 were located in the lysosome rather than the mitochondria. This was not surprising, because most of the endocytotic cargo was distributed mainly in the lysosome of living cells. However, the distinct distribution of CTX A2/A4 (in the lysosomes) compared with that of CTX A3 (in the mitochondria) was still intriguing because they shared similar three-dimensional structures but slightly different spatial distributions of positive charges.
Next, we examined whether distinct types of HS domain were responsible for CTX A2/A4 endocytosis. As shown in Fig. 4C, the pretreatment of heparinase III abolished CTX A2 internalization into CHO-K1 or H9C2 cells, whereas heparinase I pretreatment decreased CTX A4 internalization. This suggests that different HS domains were responsible for CTX A2 and A4 binding and internalization to CHO-K1 or H9C2 cells. A follow-up question would be whether their respective toxicity could be through the mechanism of lysosome damage. As seen in Fig. 4D, treatment with CTX A2 and A4 and naphthazarin (a lysosome destabilizer) decreased the red fluorescence of acridine orange and the green fluorescence of LysoSensor DND-189. It implies the loss of lysosome integrity and acidification by CTX. Surprisingly, CTX A2 and A4 treatment did not induce the release of cathepsin D, a lysosomal enzyme (35, 36), within 30 min. Although the reasons behind the lack of release of cathepsin are unknown, these results suggest that CTX A2 and A4 may induce lysosomal membrane permeabilization at least partially to exert their toxicity.
We had previously shown that CTX A3 internalization is through a novel sulfatide-induced membrane insertion rather than adopting an endocytotic process. The endocytotic routes for CTX A2/A4 and CTX A2/A4-induced cell toxicity were investigated. Dynamin2 is a crucial protein for clathrin-mediated or caveolae-mediated endocytosis (11, 12). Dynasore functions as a specific dynamin2 inhibitor by blocking its GTPase activity (37). As shown in Fig. 5A, dynasore significantly inhibited the endocytosis of CTX A2 and A4 but not that of CTX A3. This was further strengthened by the observation that dynasore rescued CTX A2/A4-induced cell toxicity (Fig. 5B). No effect of dynasore could be observed in CTX A3-induced cell toxicity. We concluded that CTX A2/A4 internalization was mediated by HS-dependent endocytosis in a dynamin2-dependent manner.
FIGURE 5.

Dynasore effect on the internalization of CTX homologues. A, CTX A2 and A4 showed dynamin-dependent endocytosis as evidenced by dynasore treatment, whereas CTX A3 endocytosis was dynamin-independent. Bar = 20 μm. B, CTX-induced cytotoxicities were performed in the absence and presence of dynasore, a specific blocker of dynamin-dependent endocytosis. Dynasore inhibited CTX A2/A4-induced cytotoxicity but had no effect on that of CTX A3. Data were mean ± S.E. (n = 5); **, p < 0.01. C, confocal images show partial colocalization of rhodamine-labeled CTX A2 and fluorescin-labeled CTX A4 in H9C2 cells. The percentage of colocalization was calculated as 67.0% (n = 5). Bar = 20 μm. D, colocalization experiments show possible endocytotic mechanisms of CTX A2 and A4. Fluorescence-labeled albumin (Alb), transferrin (Tfn), and dextran (Dex) were indicators for caveolae-mediated, clathrin-mediated, and marcopinocytosis pathways, respectively. The percentage of colocalization with transferrin, albumin, and dextran distinguished possible mechanisms of endocytosis (n ≦ 5). Bar = 20 μm. Data were mean ± S.E. (n = 5); **, p < 0.01.
Finally, when different fluorescence-labeled A2 and A4 CTX, i.e. Rho-A2 and FITC-A4, were treated and internalized into H9C2 cells, they were partially colocalized (∼60–70% (Fig. 5C)), suggesting that different endocytotic routes might be in operation for these two CTX. Other experiments were also performed on the colocalization of CTX A2/A4 with clathrin-mediated pathway markers (transferrin) or caveolae-mediated pathway markers (albumin). As seen in Fig. 5D, the colocalization of CTX A2 with albumin was more significant than with transferrin and dextran, whereas CTX A4 appeared to be colocalized with both transferrin and albumin. The data suggest that both CTX A2 and A4 became internalized through caveolae-mediated pathways but that additional clathrin-mediated pathways are in operation for CTX A4. Future research could examine the process by which different endocytotic routes mediate CTX A2/A4 endocytosis through distinct HS domains.
Opposite Cholesterol Sensitivity for the Internalization of CTX Homologues
We showed in Fig. 2A that both the three-dimensional structure and the spatial charge distribution were similar for the CTX A2 and A3 molecules. The major difference was that the hydrophobic region in CTX A2 contributed to amino acid residues located near the tip of a three-fingered loop II. The reduced hydrophobic domain (or expansion of the positive-charged domain) by the presence of Lys-31 in CTX A3, resulted in a change in their molecular target from water soluble GAGs to the other type of sulfated glycoconjugates., i.e. membrane sulfatides (38).
As the membrane sulfatides tended to be colocalized with the cholesterol-rich lipid raft domain, we examined the effect of cholesterol concentration on the internalization of CTX A2 and A3. It should be pointed out that cholesterol concentration varies significantly both in intracellular organelles and plasma membranes. As shown in Fig. 6, A and B, upon cholesterol depletion, endocytosis of CTX A2 and A4 was slightly inhibited, but that of CTX A3 was significantly enhanced. In contrast, when we increased the membrane cholesterol concentration, endocytosis of both CTX A2 and A4 was significantly enhanced, but that of CTX A3 was inhibited. There was an apparent correlation between CTX A3-induced cell toxicity and cholesterol-sensitive CTX A3 internalization (Fig. 6C). With the increase in MβCD concentration, which resulted in a decreased cholesterol level, both the cytotoxicity and the internalization of CTX A3 increased. In contrast, both the CTX A3-induced cell toxicity and level of internalization decreased upon cholesterol incorporation into the plasma membranes.
The biological roles and detailed mechanisms of why structurally similar CTX homologues exhibited opposite cholesterol sensitivity for their internalization are not clear. However, based on the model membrane studies of CTX A3-induced vesicle leakage shown in Fig. 6D, the introduction of cholesterol into sulfatide-containing vesicles inhibited its activity. This indicates that the presence of cholesterol in membrane vesicles prohibited the insertion of CTX A3 into lipid bilayers for its follow-up activity. In contrast, the presence of cholesterol on cell membranes appeared to trigger the endocytosis process responsible for CTX A2 and A4 internalization. Despite this detailed mechanism, the presence of distinct types of CTX homologues for the opposite cholesterol sensitivity should be of evolutionary advantage for CTX to target different cell types or intracellular membrane organelles.
Finally, the literature on pore-forming proteins suggests that membrane repair mechanisms could be easily triggered along with the internalization of pore-forming proteins under Ca2+/energy-dependent conditions (39). As shown in Fig. 7A, extracellular Ca2+ influx into Fluo-3-loaded NIH3T3 cells was induced within 1 min by CTX A3-specific pore formation; but such an effect was not observed with CTX A2 and A4. The presence of extracellular Ca2+ could significantly reduce CTX A3 internalization at 37 °C (Fig. 7B) as a result of the membrane repair mechanism. Similarly, the presence of extracellular Ca2+ could also suppress the propidium iodide influx (Fig. 7C) and even the internalization of FITC-dextran (4 kDa) (Fig. 7D) through the same membrane repair mechanism.
FIGURE 7.
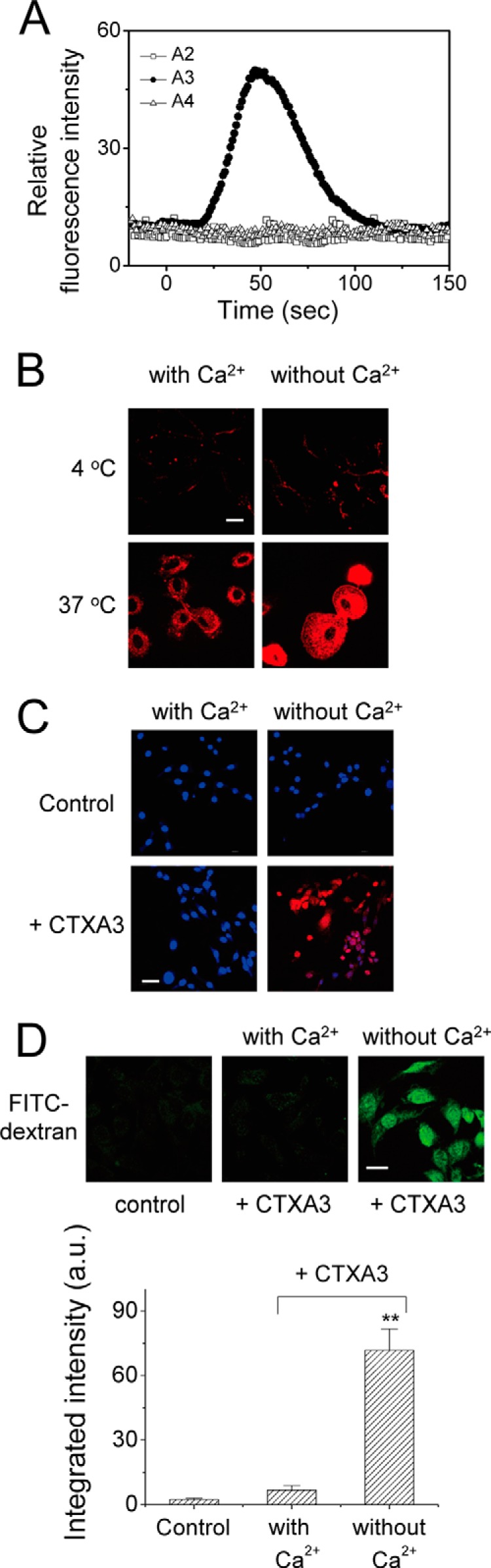
Extracellular Ca2+-mediated membrane repair mechanism to hinder CTX A3-induced cytotoxicity. A, extracellular Ca2+ influx into Fluo-3-loaded NIH3T3 cells was induced by CTX A3-specific (but not by CTX A2 and A4) pore formations within 1 min. B, depletion of extracellular Ca2+ enhanced CTX A3 internalization. C, plasma membrane repair of CTX A3-permeabilized NIH3T3 cells was Ca2+-dependent. The blue color for Hoechst staining indicates the nuclear location; propidium iodide influx, stained red, indicates that the cells failed to reseal. D, endocytosis of fluorescence-labeled dextran (4 kDa) was significantly enhanced by CTX A3 under Ca2+-free condition. Bar = 20 μm. Data were mean ± S.E. (n = 5); **, p < 0.01.
DISCUSSION
Evolutionary Advantage of Toxin Homologues
In this article, we have demonstrated that two CTX homologues from Taiwanese cobra venom, i.e. CTX A2 and A4, with only one amino acid difference at the N-terminal residue, could target lysosome and increase lysosomal membrane permeability through endocytosis. However, there appears to be a difference in their internalization pathways by engaging distinct sulfated glycosaminoglycans on the cell surface (Fig. 4). This was evidenced by the distinct heparinase I and III sensitivity to cell surface GAG binding and the endocytosis following binding.
There are about 7–10 CTX homologues within each cobra species, and it is generally believed that there must be an evolutionary advantage for capturing the prey in the arms race between the snake and its prey. Recent proteomic and genomic investigations of snake venom toxins have indicated that toxin genes are important for capturing prey and have been massively expanded by variable levels of gene duplication, resulting in protein neofunctionalization with directional selection (14). However, the exact molecular interactions involved in these fast evolving toxin homologues at the cellular level have not been addressed. Our study has shed some light on the advantage of the existence of multiple toxin homologues, because their plasma membranes and intracellular targets appear to be highly sensitive to the toxin molecules. For instance, If CTX A3 was the only CTX homologue present in cobra venom, the cytotoxicity induced by the CTX could have been stopped easily via a membrane-repairing mechanism. Under this condition, the existence of CTX A2 and A4 would have the ability to get into the cell through alternative HS-mediated endocytosis processes and induce cytotoxicity by increasing lysosome membrane permeability. A massive expansion of toxin homologues through variable levels of gene duplication may well be the best evolutionary mechanism for the biological arms race of snake venom toxins.
Structural Basis of GAG-dependent Surface Retention
The binding affinities of CTX homologues such as CTX A2, A3, and A4 have been determined to be in the range of ∼3.6, 0.42, and 1.1 μm, respectively, based on the Scatchard analysis of the sensograms measured in SPR binding experiments. However, the apparent amount of surface retention of CTX A3 on the heparin-coated SPR chip surface is minimal unless citrate-induced CTX A3 dimerization occurs during the binding process (19). Interestingly, both CTX A2 and A4 are found to form a dimer in the determined crystal structures via exclusive hydrophobic interaction involving the loop I and loop II regions (Fig. 2B), and both CTX A2 and A4 dimers consist of long stretches of positively charged regions for heparin binding (Fig. 2C). CTX A2 and A4 are structurally similar, with a single amino acid difference at the N terminus; therefore, both the hydrophobic interaction to promote toxin oligomerization and the electrostatic interaction to promote protein-carbohydrate interaction may account for the variation of toxin retention on the cell surface. Alternatively, the CTX A2 and A4 dimers may also bind to other membrane protein receptors to trigger the related internalization processes.
The surface retention of CTX A3 is more complex than CTX A2/A4, as CTX A3 also binds to membrane lipids such as sulfatides and form pores through lipid-induced oligomerization processes. Nevertheless, even in this case, CTX oligomerization still plays an important role. It is interesting to note that the oligomerization of other HS-binding proteins such as chemokines also plays an important function in their cell signaling processes (40).
Plasma Membrane Microenvironment for Toxin Internalization
Sulfate group recognition contributes binding specificity for different heparin-binding proteins (41, 42). The sulfate specificity of CTX A2 and A4 binding to heparin was investigated by competition SPR. The N-sulfate is most critical in CTX A2 and A4 binding to heparin. The removal of 6-O-sulfate groups appears to have some differential effects between CTX A2 and A4, implying a possible interaction between the N-terminal arginine of CTX A4 with the 6-O-sulfate of heparin. Of note is the role of N-acetylation in the CTX A2/A4-heparin interaction, also indicated by competition SPR experiments, because the de-N-acetylation also resulted in an approximately 5-fold reduction in the apparent Ki for both CTX A2 and A4 (data not shown). It is known that the N-acetylated domain (a low or non-sulfated domain) is involved in protein dimer binding to heparin due to the characteristics of hydrophobic interaction between protein dimer and non-sulfated domains on heparin (43–46). Therefore, CTX A2 may shift its low-sulfated domain specificity (heparinase III-sensitive) into high-sulfated domain specificity (heparinase I-sensitive) if an additional positive charge is present at the N terminus for 6-O-sulfate recognition, as is the case for CTX A4.
Multiple mechanisms have been proposed for the internalization and/or penetration of cationic peptide into living cells, such as temperature/energy dependence (47), the involvement of proteoglycans (48, 49), hydrophobicity (50), amphiphilicity (51), and distinct endocytotic pathways (52–54). The CTX homologues in the cobra venom are cationic polypeptide with different ligand-binding specificities and cellular targets. CTX A3 differs from CTX A2 with characteristic positive charges and hydrophobic residues at the tip of loop II. The structural difference not only determined the sulfatide specificity for CTX A3 translocation into cells through cholesterol-sensitive/dynamin-independent mechanisms (Fig. 6) but also allowed the trafficking of CTX A3 into mitochondria (5). Plasma membrane microenvironments such as lipid rafts have been suggested to play a role in this internalization process (38, 55, 56).
CTX A4 differs from CTX A2 by a positively charged arginine at the N terminus of the CTX three-fingered molecule. However, it could shift heparan sulfate specificity from low-sulfated domain sensitivity into high-sulfated domain sensitivity for internalization through dynamin-sensitive routes. As the endocytotic routes between CTX A2 and A4 are also observed to be different, it will be interesting to see whether membrane microenvironments constituted by GAG through distinct proteoglycans or membrane receptors may account for the GAG-dependent endocytotic pathways.
Acknowledgments
We thank KunLin Ho, Yu-Chen Huang, JinLang Lin, Cheng-Ming Tasi, YiAn Liou, and JunYu Jen for assistance with cell studies and Stephen Sanders for editorial suggestions. The confocal microscopy was performed at the Nano Imaging Core Facility, National Synchrotron Radiation Research Center, Taiwan.
This work was supported by Grants NSC94-2311-B007-023 and NSC98-2311-B007-009-MY3 (to W.-G. W.) and NSC97-2311-B030-001 and NSC101-2311-B-030-002 (to S-C. L.) from the National Science Council, Taiwan.
The atomic coordinates and structure factors (codes 4OM4 and 4OM5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CTX
- cardiotoxin
- GAG
- glycosaminoglycan
- SPR
- surface plasmon resonance
- MβCD
- methyl-β-cyclodextran
- PDB
- Protein Data Bank
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- HS
- heparan sulfate
- RU
- response unit.
REFERENCES
- 1. Ownby C. L., Fletcher J. E., Colberg T. R. (1993) Cardiotoxin 1 from cobra (Naja naja atra) venom causes necrosis of skeletal muscle in vivo. Toxicon 31, 697–709 [DOI] [PubMed] [Google Scholar]
- 2. Su S. H., Su S. J., Lin S. R., Chang K. L. (2003) Cardiotoxin-III selectively enhances activation-induced apoptosis of human CD8+ T lymphocytes. Toxicol. Appl. Pharmacol. 193, 97–105 [DOI] [PubMed] [Google Scholar]
- 3. Wang C. H., Monette R., Lee S. C., Morley P., Wu W. G. (2005) Cobra cardiotoxin-induced cell death in fetal rat cardiomyocytes and cortical neurons: different pathway but similar cell surface target. Toxicon 46, 430–440 [DOI] [PubMed] [Google Scholar]
- 4. Chen T. S., Chung F. Y., Tjong S. C., Goh K. S., Huang W. N., Chien K. Y., Wu P. L., Lin H. C., Chen C. J., Wu W. G. (2005) Structural difference between group I and group II cobra cardiotoxins: X-ray, NMR, and CD analysis of the effect of cis-proline conformation on three-fingered toxins. Biochemistry 44, 7414–7426 [DOI] [PubMed] [Google Scholar]
- 5. Wang C. H., Wu W. G. (2005) Amphiphilic β-sheet cobra cardiotoxin targets mitochondria and disrupts its network. FEBS Lett. 579, 3169–3174 [DOI] [PubMed] [Google Scholar]
- 6. Feofanov A. V., Sharonov G. V., Astapova M. V., Rodionov D. I., Utkin Y. N., Arseniev A. S. (2005) Cancer cell injury by cytotoxins from cobra venom is mediated through lysosomal damage. Biochem. J. 390, 11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu M., Ming W., Tang Y., Zhou S., Kong T., Dong W. (2013) The anticancer effect of cytotoxin 1 from Naja atra Cantor venom is mediated by a lysosomal cell death pathway involving lysosomal membrane permeabilization and cathepsin B release. Am. J. Chin. Med. 41, 643–663 [DOI] [PubMed] [Google Scholar]
- 8. Vanhollebeke B., Uzureau P., Monteyne D., Pérez-Morga D., Pays E. (2010) Cellular and molecular remodeling of the endocytic pathway during differentiation of Trypanosoma brucei bloodstream forms. Eukaryot. Cell 9, 1272–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joffre C., Barrow R., Ménard L., Calleja V., Hart I. R., Kermorgant S. (2011) A direct role for Met endocytosis in tumorigenesis. Nat. Cell Biol. 13, 827–837 [DOI] [PubMed] [Google Scholar]
- 10. Kliemt S., Lange C., Otto W., Hintze V., Möller S., von Bergen M., Hempel U., Kalkhof S. (2013) Sulfated hyaluronan containing collagen matrices enhance cell-matrix interaction, endocytosis, and osteogenic differentiation of human mesenchymal stromal cells. J. Proteome Res. 12, 378–389 [DOI] [PubMed] [Google Scholar]
- 11. Schmid S. L., Frolov V. A. (2011) Dynamin: functional design of a membrane fission catalyst. Annu. Rev. Cell Dev. Biol. 27, 79–105 [DOI] [PubMed] [Google Scholar]
- 12. Ferguson S. M., De Camilli P. (2012) Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gonzalez-Jamett A. M., Momboisse F., Haro-Acuna V., Bevilacqua J. A., Caviedes P., Cardenas A. M. (2013) Dynamin-2 function and dysfunction along the secretory pathway. Front. Endocrinol. (Lausanne) 4, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vonk F. J., Casewell N. R., Henkel C. V., Heimberg A. M., Jansen H. J., McCleary R. J., Kerkkamp H. M., Vos R. A., Guerreiro I., Calvete J. J., Wüster W., Woods A. E., Logan J. M., Harrison R. A., Castoe T. A., de Koning A. P., Pollock D. D., Yandell M., Calderon D., Renjifo C., Currier R. B., Salgado D., Pla D., Sanz L., Hyder A. S., Ribeiro J. M., Arntzen J. W., van den Thillart G. E., Boetzer M., Pirovano W., Dirks R. P., Spaink H. P., Duboule D., McGlinn E., Kini R. M., Richardson M. K. (2013) The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. U.S.A. 110, 20651–20656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel L. N., Zaro J. L., Shen W. C. (2007) Cell-penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm. Res. 24, 1977–1992 [DOI] [PubMed] [Google Scholar]
- 16. Nakase I., Akita H., Kogure K., Gräslund A., Langel U., Harashima H., Futaki S. (2012) Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Acc. Chem. Res. 45, 1132–1139 [DOI] [PubMed] [Google Scholar]
- 17. Cleal K., He L., Watson P. D., Jones A. T. (2013) Endocytosis, intracellular traffic and fate of cell-penetrating peptide-based conjugates and nanoparticles. Curr. Pharm. Des. 19, 2878–2894 [DOI] [PubMed] [Google Scholar]
- 18. Chien K. Y., Chiang C. M., Hseu Y. C., Vyas A. A., Rule G. S., Wu W. (1994) Two distinct types of cardiotoxin as revealed by the structure and activity relationship of their interaction with zwitterionic phospholipid dispersions. J. Biol. Chem. 269, 14473–14483 [PubMed] [Google Scholar]
- 19. Lee S. C., Guan H. H., Wang C. H., Huang W. N., Tjong S. C., Chen C. J., Wu W. G. (2005) Structural basis of citrate-dependent and heparan sulfate-mediated cell surface retention of cobra cardiotoxin A3. J. Biol. Chem. 280, 9567–9577 [DOI] [PubMed] [Google Scholar]
- 20. Bjornsson T. D., Schneider D. E., Hecht A. R. (1988) Effects of N-deacetylation and N-desulfation of heparin on its anticoagulant activity and in vivo disposition. J. Pharmacol. Exp. Ther. 245, 804–808 [PubMed] [Google Scholar]
- 21. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nieba L., Krebber A., Plückthun A. (1996) Competition BIAcore for measuring true affinities: large differences from values determined from binding kinetics. Anal. Biochem. 234, 155–165 [DOI] [PubMed] [Google Scholar]
- 23. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 24. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Laskowski R. A., Macarthur M. W., Moss D. S., Thornton J. M. (1993) Procheck: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 28. Mulloy B., Forster M. J., Jones C., Davies D. B. (1993) n.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 293, 849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schüttelkopf A. W., van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 30. van Aalten D. M., Bywater R., Findlay J. B., Hendlich M., Hooft R. W., Vriend G. (1996) PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 10, 255–262 [DOI] [PubMed] [Google Scholar]
- 31. Karaca E., Melquiond A. S., de Vries S. J., Kastritis P. L., Bonvin A. M. (2010) Building macromolecular assemblies by information-driven docking: introducing the HADDOCK multibody docking server. Mol. Cell. Proteomics 9, 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Vries S. J., van Dijk M., Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 33. Patel H. V., Vyas A. A., Vyas K. A., Liu Y. S., Chiang C. M., Chi L. M., Wu W. (1997) Heparin and heparan sulfate bind to snake cardiotoxin: sulfated oligosaccharides as a potential target for cardiotoxin action. J. Biol. Chem. 272, 1484–1492 [DOI] [PubMed] [Google Scholar]
- 34. Linhardt R. J., Turnbull J. E., Wang H. M., Loganathan D., Gallagher J. T. (1990) Examination of the substrate specificity of heparin and heparan sulfate lyases. Biochemistry 29, 2611–2617 [DOI] [PubMed] [Google Scholar]
- 35. Minarowska A., Minarowski L., Karwowska A., Gacko M. (2007) Regulatory role of cathepsin D in apoptosis. Folia Histochem. Cytobiol. 45, 159–163 [PubMed] [Google Scholar]
- 36. Zaidi N., Maurer A., Nieke S., Kalbacher H. (2008) Cathepsin D: a cellular roadmap. Biochem. Biophys. Res. Commun. 376, 5–9 [DOI] [PubMed] [Google Scholar]
- 37. Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850 [DOI] [PubMed] [Google Scholar]
- 38. Wang C. H., Liu J. H., Lee S. C., Hsiao C. D., Wu W. G. (2006) Glycosphingolipid-facilitated membrane insertion and internalization of cobra cardiotoxin: the sulfatide.cardiotoxin complex structure in a membrane-like environment suggests a lipid-dependent cell-penetrating mechanism for membrane binding polypeptides. J. Biol. Chem. 281, 656–667 [DOI] [PubMed] [Google Scholar]
- 39. Idone V., Tam C., Goss J. W., Toomre D., Pypaert M., Andrews N. W. (2008) Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 180, 905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuo J. H., Chen Y. P., Liu J. S., Dubrac A., Quemener C., Prats H., Bikfalvi A., Wu W. G., Sue S. C. (2013) Alternative C-terminal helix orientation alters chemokine function: structure of the anti-angiogenic chemokine, CXCL4L1. J. Biol. Chem. 288, 13522–13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dreyfuss J. L., Regatieri C. V., Jarrouge T. R., Cavalheiro R. P., Sampaio L. O., Nader H. B. (2009) Heparan sulfate proteoglycans: structure, protein interactions, and cell signaling. An. Acad. Bras. Cienc. 81, 409–429 [DOI] [PubMed] [Google Scholar]
- 42. Nugent M. A., Zaia J., Spencer J. L. (2013) Heparan sulfate-protein binding specificity. Biochemistry 78, 726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lortat-Jacob H., Turnbull J. E., Grimaud J. A. (1995) Molecular organization of the interferon γ-binding domain in heparan sulphate. Biochem. J. 310, 497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stringer S. E., Gallagher J. T. (1997) Specific binding of the chemokine platelet factor 4 to heparan sulfate. J. Biol. Chem. 272, 20508–20514 [DOI] [PubMed] [Google Scholar]
- 45. Spillmann D., Witt D., Lindahl U. (1998) Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 273, 15487–15493 [DOI] [PubMed] [Google Scholar]
- 46. Stringer S. E., Forster M. J., Mulloy B., Bishop C. R., Graham G. J., Gallagher J. T. (2002) Characterization of the binding site on heparan sulfate for macrophage inflammatory protein 1α. Blood 100, 1543–1550 [PubMed] [Google Scholar]
- 47. Drin G., Cottin S., Blanc E., Rees A. R., Temsamani J. (2003) Studies on the internalization mechanism of cationic cell-penetrating peptides. J. Biol. Chem. 278, 31192–31201 [DOI] [PubMed] [Google Scholar]
- 48. Poon G. M., Gariépy J. (2007) Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem. Soc. Trans. 35, 788–793 [DOI] [PubMed] [Google Scholar]
- 49. Fang S. L., Fan T. C., Fu H. W., Chen C. J., Hwang C. S., Hung T. J., Lin L. Y., Chang M. D. (2013) A novel cell-penetrating peptide derived from human eosinophil cationic protein. PLoS One 8, e57318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gupta A., Mandal D., Ahmadibeni Y., Parang K., Bothun G. (2011) Hydrophobicity drives the cellular uptake of short cationic peptide ligands. Eur. Biophys. J. 40, 727–736 [DOI] [PubMed] [Google Scholar]
- 51. Tang Q., Cao B., Wu H., Cheng G. (2012) Selective gene delivery to cancer cells using an integrated cationic amphiphilic peptide. Langmuir 28, 16126–16132 [DOI] [PubMed] [Google Scholar]
- 52. Kaplan I. M., Wadia J. S., Dowdy S. F. (2005) Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 102, 247–253 [DOI] [PubMed] [Google Scholar]
- 53. Yamano S., Dai J., Yuvienco C., Khapli S., Moursi A. M., Montclare J. K. (2011) Modified Tat peptide with cationic lipids enhances gene transfection efficiency via temperature-dependent and caveolae-mediated endocytosis. J. Control. Release 152, 278–285 [DOI] [PubMed] [Google Scholar]
- 54. Ohara K., Kohno M., Hamada T., Kawakami K. (2013) Entry of a cationic lytic-type peptide into the cytoplasm via endocytosis-dependent and -independent pathways in human glioma U251 cells. Peptides 50, 28–35 [DOI] [PubMed] [Google Scholar]
- 55. Lajoie P., Nabi I. R. (2010) Lipid rafts, caveolae, and their endocytosis. Int. Rev. Cell Mol. Biol. 282, 135–163 [DOI] [PubMed] [Google Scholar]
- 56. Hanzal-Bayer M. F., Hancock J. F. (2007) Lipid rafts and membrane traffic. FEBS Lett. 581, 2098–2104 [DOI] [PubMed] [Google Scholar]