

Background: There is a need for alternative HIV-1 inhibitors.
Results: An engineered antibody fragment (Fab) against the essential HIV-1 protein Rev significantly reduces reverse transcriptase activity in human cell culture, and synthetic antigen-binding peptides from the Fab block Rev polymerization.
Conclusion: The Fab inhibits viral replication by blocking Rev function.
Significance: The Fab and its antigen-binding peptides represent a new class of anti-HIV-1 agents.
Keywords: AIDS, Cell-penetrating peptide (CPP), Cyclic Peptide, Monoclonal Antibody, Phage Display
Abstract
The HIV-1 protein Rev oligomerizes on viral transcripts and directs their nuclear export. Previously, a Fab against Rev generated by phage display was used to crystallize and solve the structure of the Rev oligomerization domain. Here we have investigated the capability of this Fab to block Rev oligomerization and inhibit HIV-1 replication. The Fab itself did not have antiviral activity, but when a Tat-derived cell-penetrating peptide was appended, the resulting molecule (FabRev1-Tat) was strongly inhibitory of three different CCR5-tropic HIV-1 isolates (IC50 = 0.09–0.44 μg/ml), as assessed by suppression of reverse transcriptase activity in infected peripheral blood mononuclear cells, and had low cell toxicity (TC50 > 100 μg/ml). FabRev1-Tat was taken up by both peripheral blood mononuclear and HEK293T cells, appearing in both the cytoplasm and nucleus, as shown by immunofluorescence confocal laser scanning microscopy. Computational alanine scanning was used to identify key residues in the complementarity-determining regions to guide mutagenesis experiments. Residues in the light chain CDR3 (LCDR3) were assessed to be important. Residues in LCDR3 were mutated, and LCDR3-Tyr92 was found to be critical for binding to Rev, as judged by surface plasmon resonance and electron microscopy. Peptides corresponding to all six CDR regions were synthesized and tested for Rev binding. None of the linear peptides had significant affinity for Rev, but four of the amide-cyclic forms did. Especially cyclic-LCDR3 (LGGYPAASYRTA) had high affinity for Rev and was able to effectively depolymerize Rev filaments, as shown by both surface plasmon resonance and electron microscopy.
Introduction
With the advent of combination antiretroviral therapy, the long term survival of HIV-1-positive patients has increased to the point that the disease can now often be considered a chronic condition (1). Currently approved small molecule inhibitors of the viral reverse transcriptase, integrase, and protease can, in the right combinations, drive viral load below the limits of detection, and new inhibitors are continually being developed. Monoclonal antibodies and their derivatives, such as antigen binding fragments (Fab)2 and the corresponding complementarity determining region (CDR) peptides, have been described as plausible agents for inhibiting HIV-1. These have been directed either against gp120, the envelope glycoprotein, or more commonly, the CD4 receptor (2–8). All have shown promise. Nevertheless, there remains a demonstrable need for antiviral agents with alternative modes of action for patients with drug-resistant isolates. One currently unrealized target is the essential viral regulatory protein Rev.
HIV-1 Rev enables the nuclear export of partially spliced and non-spliced viral transcripts used for the production of the late-stage proteins and as genomic RNA for progeny virions, and as such is indispensible for viral replication. Rev binds to a highly structured region on viral RNAs, the Rev response element (RRE), initially at a high affinity site and subsequently at 7–9 subsidiary sites. Binding is a cooperative process mediated in part by the amino-terminal oligomerization domains of the Rev molecules (9). The Rev molecules then interact via their carboxyl-terminal activation regions with Crm1 and Ran-GTP to facilitate export of the RNA from the nucleus (10–13).
Rev has not been a target for intervention, in large part due to a lack of structural information about both Rev and the RRE. In the case of Rev, the difficulty was due to the low solubility and a strong tendency of the protein to polymerize into filaments and then aggregate (14). A low resolution structure of the filaments was determined by cryo-electron microscopy and helical reconstruction (15), and the solution structure of a short helical peptide from the oligomerization domain of Rev bound to stem loop IIB of the RRE was solved by nuclear magnetic resonance (16).
We have recently used a chimeric human/rabbit Fab generated by phage display as a crystallization chaperone to solve the structure of Rev (17). We found that the oligomerization domain of Rev, constituting approximately one-half of the molecule, consists of two coplanar α-helices arranged like a hairpin, and that these dimerize in a chevron-like arrangement with the open ends of the hairpins located toward the middle of the dimer. A Fab molecule is bound on either side of the paired oligomerization domains, blocking association between Rev dimers. The carboxyl-terminal region of Rev is unstructured in the crystals (18). A very similar structure for Rev was found by using a soluble mutant form of the protein (19). More recently we have also determined a low resolution structure for the RRE, derived in part from small-angle x-ray scattering data (20). Here we show that the Fab, with a Tat cell-penetrating peptide (21) attached (FabRev1-Tat), effectively enters human peripheral blood mononuclear cells (PBMC) and has high anti-HIV-1 activity. We further show that the light chain CDR3 (LCDR3) is a critical component of the Fab paratope, and that a cyclic form of LCDR3 (cLCDR3) effectively disrupts Rev polymers in vitro.
EXPERIMENTAL PROCEDURES
Preparation of Proteins
The expression and purification of full-length Rev1–116 have been described in detail (14). Two truncated forms of the protein, Rev1–93 and Rev1–69, were prepared by standard procedures and purified in essentially the same way as Rev1–116.
The selection, expression, and purification of a Rev amino-terminal domain-specific chimeric rabbit/human antibody fragment (Fab) using phage display have also been described in detail, as have the expression and purification of the corresponding single chain variable domain fragment (scFv) (17). In the present study, two additional antibody fragments were prepared. The first corresponds to the Fab described above, but with a His6 tag appended to the carboxyl-terminal end of the heavy chain followed by the Tat cell-penetrating peptide (RKKRRQRRR) (21), and is herein referred to as FabRev1-Tat. The second was an scFv directed against a linear epitope located near the carboxyl terminus of Rev. This protein was isolated from the same phage display library by the methods described previously, and is herein referred to as scFvRev2.
Peptide Synthesis
The peptide Rev94–116 (TQGVGSPQILVESPTVLESGAKE) was synthesized by United Biosystems. Peptides corresponding to the Fab CDR loops were synthesized in both the linear and amide-cyclic forms by LifeTein. All peptides were characterized by HPLC and MS analysis.
Thermal and Serum Stability Assays
For the analysis of thermal stability, FabRev1-Tat (100 μg/ml) in PBS was heated to 65 °C for times ranging from 0.5 to 3 h, and then chilled on ice. For the analysis of serum stability, FabRev1-Tat (100 μg/ml) was incubated in DMEM supplemented with 20% fetal bovine serum at 37 °C for 2, 4, or 6 days. The ability to bind Rev1–116 after treatment was determined by ELISA. 96-Well plates were coated with 50 μg/ml of Rev1–116 for 1 h at 37 °C, blocked with 5% nonfat milk/PBS for 1 h, and washed with PBS. The Fab samples were diluted serially and then added to the wells and incubated for 1 h at room temperature. Three replicates were included for each sample. Bound FabRev1-Tat was detected with anti-rabbit IgG followed by peroxidase-conjugated anti-human IgG. The color reaction was initiated by adding horseradish peroxidase (HRP) substrate and read at 405 nm. Standard curves were generated using the original protein stocks.
Culture of Cells for Transduction
PBMC were isolated from fresh human blood provided by the blood bank of the National Institutes of Heath. Diluted blood cells were layered over Ficoll-Plaque (GE Healthcare) and centrifuged. The mononuclear cell layer was washed twice with PBS. After stimulation overnight at 37 °C with 5 μg/ml of phytohemagglutinin (Invitrogen) the isolated PBMC were cultured in complete RPMI 1640 (Invitrogen) supplemented with 100 μg/ml of penicillin/streptomycin and 5% recombinant IL-2 (Roche Applied Science). HEK293T cells were obtained from the American Type Culture Collection (ATCC) and maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Transduction of Fab into Cells
PBMC were plated in 6-well Falcon plates in complete medium overnight. HEK293T cells were grown on poly-d-lysine-coated glass coverslips (BD Biosciences) overnight. After washing, the cells were incubated with serum-free medium containing either 1 μm FabRev1 or FabRev1-Tat protein for 1 h. The cells were then washed and cultured in serum-free medium for an additional hour. The cells were fixed with 4% formaldehyde in PBS for 20 min at 4 °C, permeabilized with 0.2% Triton X-100 for 10 min, and blocked with 20% fetal bovine serum in PBS for 1 h. Anti-His tag antibody (Santa Cruz Biotechnology) was diluted 1/1000 with blocking buffer and applied for 1 h. After three washes (10 min each) the samples were incubated in Alexa 488 goat anti-mouse secondary antibody. After another three washes, the samples were embedded in Prolong Gold anti-fade mounting reagent with DAPI (Invitrogen) and viewed with a Zeiss 780 confocal microscope.
Antiviral Activity Assays
All antiviral and cytotoxicity testing was done under contract (Southern Research) by standard methods. Six HIV-1 virus isolates from the NIAID AIDS Research and Reference Reagent Program were employed; three were CCR5 tropic and three were CXCR4 tropic. Briefly, human PBMC were isolated from screened donors known to be seronegative for HIV and hepatitis B virus. Infections were done in a microplate format using serial dilutions of viral inhibitor. Negative (buffer) and positive (azidothymidine) controls were also done. Anti-HIV-1 activity was assessed by a microtiter plate-based reverse transcriptase assay of the cell-free culture supernatants. Cytotoxicity was assessed on identically prepared uninfected cells by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (Promega) staining assay. All experiments were done in triplicate.
Computational Alanine Scanning
Paratope residues likely to mediate antigen binding were identified with the following web servers: Robetta (22), DrugScorePPI (23), HotPoint (24), ANCHOR (25), PCRPi-W (26), and KFC2 (27). For DrugScorePPI the cutoff criteria were set as for Robetta, i.e. a residue was considered “hot” if ΔΔG was >1 kcal/mol, and “warm” if ΔΔG was <1 but buriedness was >7.0 or if a salt bridge was formed. For ANCHOR, a residue was considered hot if the binding energy was < −5 kcal/mol, and warm if the binding energy was < −0.5 kcal/mol and the ΔSASA was >0.5 Å2. For the remainder the default settings were used.
Site-directed Mutagenesis
A FabRev1-pET11a expression construct was used as a template for mutant construction using standard procedures. All mutations on the final constructs were verified by DNA sequencing.
Circular Dichroism
Spectra were collected using a Jasco J-715 spectrometer as described (14). For each sample, four accumulations were collected between 190 and 240 nm using a 0.01-cm path length cell. Scanning was done at a speed of 20 nm/min with a 0.1-nm data pitch. After baseline subtraction the raw data were converted to molar ellipticities and smoothed with Jasco software. The results were analyzed using the online software DichroWeb.
Surface Plasmon Resonance
All experiments were performed on a BIAcore X100 (GE Healthcare) instrument at 25 °C. HBS-EP+ (10 mm Hepes, pH 7.4, 150 mm sodium chloride, 3 mm EDTA, 0.05% Polysorbate 20) was used as the running buffer and data were analyzed using Biacore X100 evaluation software (GE Healthcare). Cell 1 was left untreated to serve as a reference surface and cell 2 was used as the experimental surface. The full-length and truncated Rev proteins were diluted in HBS-EP+ buffer and immobilized on CM5 sensor chips by the standard amine coupling method (Amine Coupling kit, GE Healthcare) at a flow rate of 5 μl/min. The immobilization levels of the proteins on the sensor chip surfaces were as follows: 750–1250 response units of Rev1–116 for Fab binding, 2500–3000 response units of Rev1–116 for peptide binding, and 500–1000 response units of Rev1–93 or Rev1–69 for Fab binding. For kinetic analysis, analytes were prepared by serial dilution with HBS-EP+ buffer over a range of greater than 100-fold and injected over both the reference and experimental surfaces at a flow rate of 30 μl/min. Sensor chips were regenerated by a 60-s injection of 50 mm sodium hydroxide. Signals from the reference surface and an ensemble of buffer blank injections were subtracted to correct for nonspecific binding and injection artifacts. The corrected results were globally fitted to a 1:1 binding model and the association rate constant (ka), dissociation rate constant (kd), and equilibrium dissociation constant (KD) were extracted.
Electron Microscopy
Rev (as filaments) and Fab (or scFv) were diluted in cold 50 mm Tris (pH 7.5), 150 mm sodium chloride, and 25 mm sodium citrate and typically mixed to give a 1:2 (Rev dimer:Fab) molar ratio and a final Rev concentration of 0.1 mg/ml. The CDR-derived peptides were used in 5–40-fold molar excess over Rev dimer. The mixtures were incubated overnight at 4 °C (conditions under which Rev filaments alone remain polymerized), applied to 400-mesh carbon-coated copper grids made hydrophilic by glow discharge in a 25:75 oxygen:argon atmosphere, and negatively stained with 1% uranyl acetate. Images were recorded with a Philips CM120 electron microscope on a CCD at ×35,000 magnification.
Western Blotting
Reduced Rev1–116 protein was run on a 4–12% SDS-polyacrylamide gel and electrotransferred to a PVDF membrane (Invitrogen). After blocking with 5% nonfat milk solution for 1 h, the membrane was incubated with either scFvRev1 or scFvRev2, which was then detected with peroxidase-conjugated anti-rabbit IgG (KPL). Signals were detected with a chemiluminescence reagent (Thermo) and exposure on x-ray film (Kodak).
RESULTS
Antibody Fragments Binding to the Amino-terminal Region of Rev
We have previously used phage display to generate a chimeric rabbit/human anti-Rev Fab (17), which binds to a conformational epitope involving the two α-helices in the amino-terminal domain of Rev (Fig. 1A) (18). To determine whether there are any effects on the binding from the carboxyl-terminal region of Rev, we prepared the full-length protein Rev1–116 and two deletion mutants: Rev1–69 and Rev1–93 (Fig. 1A). The Rev1–69 construct was chosen to correspond to the structured portion of the molecule observed in crystals (18), whereas Rev1–93 was chosen on the basis of the observation that deletions beyond residue 92 do not affect Rev function (28). The proteins were purified by ion-exchange and gel filtration chromatography (Fig. 1B) as previously described (14). Far UV-CD analysis indicated that the deletion mutants were folded with a predominantly helical secondary structure (Fig. 1C, data for Rev1–93 not shown). The near UV-CD spectrum, considered a conformational fingerprint for tertiary folding, of Rev1–93 is very similar to that of the full-length protein (14). This indicates that the carboxyl-terminal domain, which does not contain any aromatic residues and is therefore invisible in the near-UV CD, has no major influence on the folded structure of the amino-terminal domain, as would be expected if it were intrinsically unstructured.
FIGURE 1.

Rev-related proteins used in this study. A, linear organization of Rev1–116, Rev1–93, Rev1–69, and the peptide Rev94–116. Shown are the two α-helices in the amino-terminal oligomerization domain and the probably unstructured carboxyl-terminal activation region, as well as the nuclear localization signal (NLS) and nuclear export signal (NES). The conformational epitope of FabRev1 and the linear epitope of FabRev2 are indicated by the dashed boxes. B, size exclusion chromatography, and inset, SDS-PAGE analysis of Rev1–116, Rev1–93, and Rev1–69. C, far-UV circular dichroic spectra of Rev1–116, Rev1–69, and Rev94–116. The spectrum for Rev1–93 is not shown.
The kinetics of FabRev1 binding to Rev1–116, Rev1–93, and Rev1–69 were studied by surface plasmon resonance. The kinetic constants are shown in the upper half of Table 1. The overall equilibrium constants for binding to the truncated forms (Kd = 1.6 or 1.8 × 10−10 m) were slightly lower than that for the full-length protein (Kd = 6.8 × 10−10 m). The higher association rate of Rev1–69 suggested that the carboxyl-terminal region interferes somewhat with Fab binding. We also performed a similar kinetic analysis of FabRev1 engineered as a single chain variable fragment (scFvRev1). The kinetic constants are shown in the lower half of Table 1. In all three cases, the equilibrium constants were ∼10-fold lower than for the FabRev1, largely due to lower association rates. As with FabRev1, the scFv1 had lower binding affinity to Rev1–116 (Kd = 5.9 × 10−9 m) than the truncated forms (Kd = 2.0 × 10−9 m). These results suggest that the carboxyl-terminal region of Rev interferes, to a limited extent, with FabRev1 binding to the epitope of Rev protein.
TABLE 1.
Kinetic constants of binding between Fab and Rev proteins
Values are the mean ± S.E. from three separate experiments
| Fab | Rev | ka | kd | KD |
|---|---|---|---|---|
| m−1s−1 | s−1 | m | ||
| FabRev1 | Rev1–116 | 1.74 ± 1.06 × 105 | 9.46 ± 3.63 × 10−5 | 6.80 ± 0.44 × 10−10 |
| Rev1–93 | 1.84 ± 1.29 × 105 | 3.15 ± 2.66 × 10−5 | 1.61 ± 0.28 × 10−10 | |
| Rev1–69 | 4.19 ± 2.21 × 105 | 7.10 ± 3.07 × 10−5 | 1.77 ± 0.22 × 10−10 | |
| scFvRev1 | Rev1–116 | 1.64 ± 0.08 × 104 | 9.64 ± 1.84 × 10−5 | 5.89 ± 0.10 × 10−9 |
| Rev1–93 | 4.52 ± 0.07 × 104 | 9.24 ± 0.20 × 10−5 | 2.04 ± 0.03 × 10−9 | |
| Rev1–69 | 2.74 ± 0.54 × 105 | 5.20 ± 1.45 × 10−5 | 1.97 ± 0.91 × 10−9 |
Antiviral Activity of FabRev1-Tat
As the oligomerization of Rev on the RRE is required for nuclear export of viral transcripts, we reasoned that blocking this oligomerization might inhibit viral replication. Initial immunofluorescence experiments showed that FabRev1 did not enter PBMC (not shown). To facilitate cell entry, the Tat cell penetration peptide was appended to the carboxyl-terminal end of the Fab. The resulting protein (FabRev1-Tat) was purified by nickel affinity- and size-exclusion chromatography (Fig. 2A). Kinetic analysis of surface plasmon resonance experiments (Fig. 2B) showed that the affinity of FabRev1-Tat (Kd = 5.0 × 10−10 m) was similar to that of the untagged protein (Kd = 6.8 × 10−10 m). Like FabRev1, FabRev1-Tat depolymerized Rev filaments (Fig. 2, C and D), showing that the presence of the Tat peptide did not interfere with binding to Rev.
FIGURE 2.

FabRev1-Tat preparation and Rev binding. A, size exclusion chromatography analysis of FabRev1-Tat, and inset, SDS-PAGE analysis of purified FabRev1 and FabRev1-Tat. B, sensograms of the binding between FabRev1-Tat and Rev1–116. Electron micrographs of negatively stained Rev1–116 filaments (C) and complexes of Rev1–116 and FabRev1-Tat (D). Bar = 50 nm (C and D).
The thermal stability of FabRev1-Tat was tested prior to assaying its antiviral activity. The binding affinity of FabRev1-Tat was not affected by incubation at 65 °C for up to 3 h (Fig. 3A). Serum stability was tested by incubation with 20% serum at 37 °C, which mimicked the conditions during transduction. No detectable loss of binding affinity was observed after being cultured under these conditions for up to 6 days (Fig. 3B).
FIGURE 3.

FabRev1-Tat inhibits HIV-1. A, thermal stability and B, serum stability of FabRev1-Tat. The stability of FabRev1-Tat in both assays was assessed by the binding of peroxidase-conjugated IgG, measured as the absorbance at 405 nm of a color reaction in ELISA. C, inhibition of replication of HIV-1 Ba-L; and D, PBMC cell toxicity of FabRev1 and FabRev1-Tat. Replication was measured as reverse transcriptase activity in cell culture supernatants. Cell toxicity was determined from residual cell viability measured with a commercial colorimetric assay. Values plotted are the mean ± S.D. of triplicate experiments. Complete IC90 and IC50 data for all the tested virus strains are shown in Table 2.
FabRev1-Tat uptake was first tested in PBMC, the same cell type as was used for the viral inhibition assays, where it appeared in both the cytoplasm and nucleus (Fig. 4, A–F). In PBMC the distribution of fluorescence between the cytoplasm and nucleus is difficult to ascertain due to the relatively small volume of the cytoplasm relative to that of the nucleus. Uptake was therefore also assessed in HEK293T cells, which afford a better view of the cytoplasm. In HEK293T cells, as in PBMC, FabRev1-Tat was observed in both the cytoplasm and nucleus, however, immunofluorescence had a more punctate appearance (Fig. 4, G–L). Alternative locations and spacings for the Tat peptide were tested but did not appreciably affect uptake (not shown). In control experiments, HIV-Tat fluorescence did not appear in the nucleus (not shown).
FIGURE 4.

Transduction of FabRev1 and FabRev1-Tat. Immunofluorescence confocal laser scanning microscopy was used to assess the transduction of FabRev1 (A–C and G–I) and FabRev1-Tat (D–F and J–L) into PBMC (A–F) and HEK293T cells (G–L). 2-Fold enlargements of cells are shown in the bottom right-hand corner of the panels. Green is Alexa 488 fluorescence arising from an anti-Fab secondary antibody (A, D, G, and J); blue is DAPI fluorescence that represents nucleic acid (B, E, H, and K); the corresponding merged images are in C, F, I, and L.
The antiviral activity of FabRev1 and FabRev1-Tat was assessed as inhibition of reverse transcriptase activity in HIV-1-infected PBMC cell culture supernatants. Representative results are shown in Fig. 3, C and D, and the complete results are shown in Table 2. FabRev1-Tat exhibited antiviral activity against three different CCR5 isolates, but not against the CXCR4 isolates. The IC50 values are in the range of 0.09–0.44 μg/ml and cytotoxicity was low (>100 μg/ml).
TABLE 2.
Inhibition of HIV-1 in PBMC by FabRev1-Tat
Values are the means from assays done in triplicate.
| Isolate | Tropism | IC90 | IC50 | TC50 |
|---|---|---|---|---|
| μg/ml | ||||
| Ba-L | CCR5 | 3.13 | 0.09 | >100 |
| 91US004 | CCR5 | 1.75 | 0.44 | >100 |
| JV1083 | CCR5 | 2.73 | 0.44 | >100 |
| NL4–3 | CXCR4 | 56.4 | 1.94 | >100 |
| 92HT599 | CXCR4 | >100 | >100 | >100 |
| 92UG021 | CXCR4 | >100 | >100 | >100 |
Mutational Analysis of the FabRev1 Paratope
To identify candidate residues for mutational analysis, and identify which CDR loop might serve best as an inhibitor of Rev oligomerization (Fig. 5), we analyzed the CDR sequences of FabRev1 by computational alanine scanning. Six web servers were employed for this: Robetta (22), DrugScorePPI (23), HotPoint (24), ANCHOR (25), PCRPi-W (26), and KFC2 (27). The results are shown in Table 3. The HCDR2 loop contained two of the strongest predicted hot spots, residues Tyr52 and Trp59. The LCDR3 loop had one strongly predicted hot spot, residue Tyr92, and five residues in this loop (Ala94, Ala95, Ser96, Tyr97, and Arg98) were also classed as hot spots by at least one prediction method, suggesting a critical role for this loop. To test these predictions experimentally, a number of conservative mutations were introduced individually into the LCDR3 of FabRev1, the mutant proteins were expressed in Escherichia coli, and the binding to Rev1–116 was analyzed by surface plasmon resonance. The results are shown in Table 4. All mutations decreased the binding affinity, but Y92S had a greater effect than the others, causing a 500-fold decrease in affinity.
FIGURE 5.
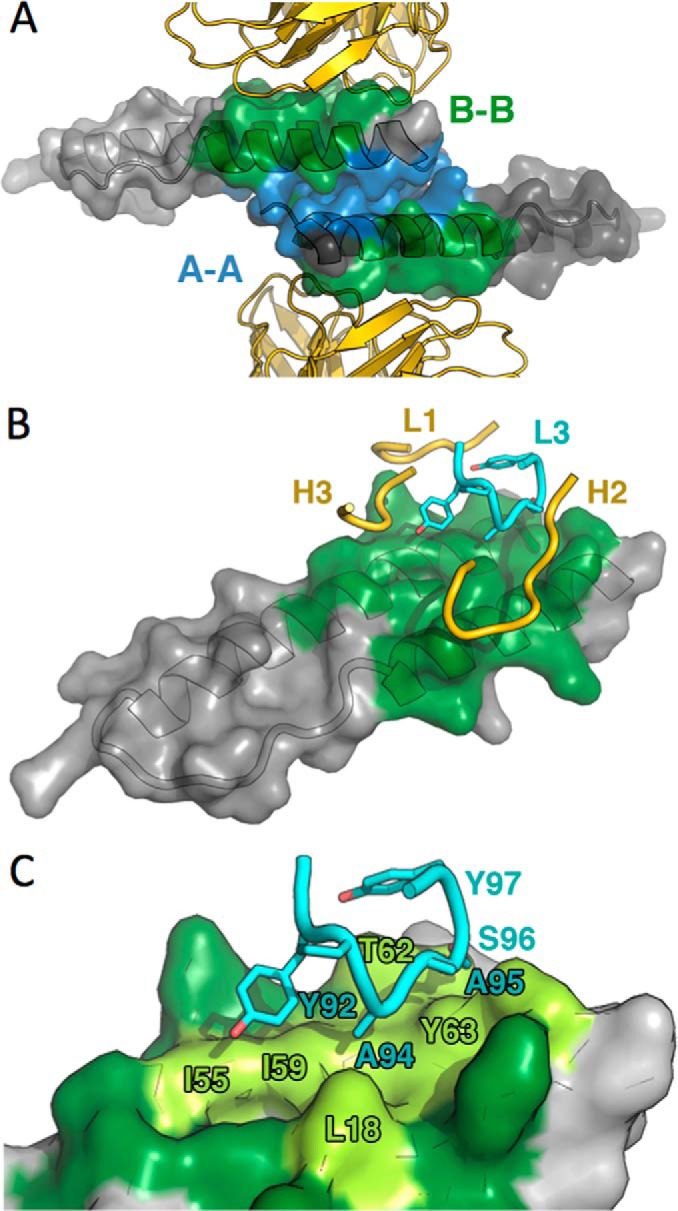
Model of the FabRev1 paratope. A, ribbon representation of FabRev1 molecules (gold) shown flanking a surface-rendered Rev dimer (translucent, gray) (PDB code 2X7L). The A-A and B-B Rev-Rev interfaces are colored blue and green, respectively (PDB codes 2X7L and 3LPH). B, one Rev monomer, rendered as in A, together with four of the CDR loops that engage it. For clarity, only subsets of the loops making contact are shown. LCDR3 is shown in cyan. C, close-up of LCDR3 engaging the epitope. The Rev surface colored dark green represents the B-B interface between Rev monomers; light green represents the region contacted by LCDR3.
TABLE 3.
Computational alanine scanning of HCDR2 and LCDR3
| Chain | Position | Amino acid | Robetta | DSPPI | Hot point | Anchor |
PCR Pi |
KFC |
||
|---|---|---|---|---|---|---|---|---|---|---|
| SASA | E | Probability | 2a | 2b | ||||||
| kcal/m | ||||||||||
| H | 33 | Trp | 1.32 | 0.29 | H | 9.6 | −0.7 | 0.051 | Hot | |
| H | 52 | Tyr | 1.52 | 1.78 | H | 52.9 | −1.8 | 0.4324 | Hot | |
| H | 59 | Trp | 3.71 | 1.87 | H | 76.6 | 0 | 0.2969 | Hot | Hot |
| H | 100 | Asp | 0.72 | 0.64 | H | 33.9 | −6.0 | 0.1765 | ||
| L | 32 | Trp | 1.99 | 1.57 | H | 69.4 | −0.1 | 0.0879 | Hot | |
| L | 92 | Tyr | 1.92 | 1.15 | H | 29.5 | −1.4 | 0.1595 | Hot | |
| L | 94 | Ala | H | 26.3 | −1.5 | 0.9997 | Hot | |||
| L | 95 | Ala | NH | 8.6 | −1.3 | 0.4187 | ||||
| L | 96 | Ser | 0.83 | 0.35 | NH | 54.6 | −1.1 | 0.9251 | ||
| L | 97 | Tyr | 0.48 | 0.77 | H | 16.5 | −0.3 | 0.0082 | ||
| L | 98 | Arg | 2.95 | 0.33 | NH | 23.7 | 7.1 | 0.0998 | ||
TABLE 4.
Kinetic constants of binding between FabRev1 LCDR3 mutants and Rev1–116
Values are the mean ± S.E. from three separate experiments.
| Fab | ka | kd | KD |
|---|---|---|---|
| m−1s−1 | s−1 | m | |
| Fab Y92S | 6.16 ± 3.56 × 104 | 1.61 ± 0.71 × 10−2 | 2.91 ± 0.78 × 10−7 |
| Fab A94G | 5.50 ± 3.89 × 104 | 1.76 ± 0.22 × 10−3 | 4.39 ± 2.81 × 10−8 |
| Fab A95G | 1.32 ± 0.60 × 104 | 8.74 ± 0.30 × 10−4 | 7.38 ± 2.49 × 10−8 |
| Fab S96A | 1.47 ± 0.56 × 104 | 4.27 ± 0.25 × 10−4 | 3.18 ± 1.39 × 10−8 |
| Fab Y97S | 7.04 ± 1.22 × 104 | 2.38 ± 0.24 × 10−3 | 3.46 ± 0.94 × 10−8 |
| Fab R98A | 2.83 ± 1.98 × 105 | 7.94 ± 2.08 × 10−4 | 3.38 ± 1.63 × 10−9 |
Characterization of CDR-related Peptides
Linear peptides corresponding to the six FabRev1 CDR loops were synthesized and tested for binding to immobilized Rev1–116 using surface plasmon resonance but none were found to bind (not shown). The amide-cyclic (head-to-tail) forms of the peptides were then prepared and tested for binding to Rev (Fig. 6). Four peptides were found to bind, particularly cLCDR3 (Kd = 2.7 × 10−8 m) (Table 5). The ability of cyclic peptides cHCDR2 and cLCDR3 to disrupt Rev oligomerization was tested by mixing the peptides with Rev filaments and observing them by negative stain electron microscopy. The peptide cHCDR2 only perturbed the structure of the filaments (Fig. 6C), whereas cLCDR3 effectively depolymerized the filaments to a fine granular state (Fig. 6D) visually similar to that obtained with the parent Fab (Fig. 2D). This result together with mutational scanning of the Fab confirms the importance of LCDR3 in making contacts with Rev.
FIGURE 6.

Cyclization of Fab1 CDR-related peptides enhances their affinity for Rev. Sensograms of cHCDR2 (A) and cLCDR3 (B) binding to immobilized Rev1–116. The kinetic constants of the CDR-derived peptides are given in Table 5. Electron micrographs of negatively stained preparations of Rev filaments mixed with either cHCDR2 (C) or cLCDR3 (D). Bar = 50 nm (C and D).
TABLE 5.
Kinetic constants of binding between cyclic CDR-derived peptides and Rev1–116
Values are the mean ± S.E. from three separate experiments.
| Peptide | Sequence | ka | kd | KD |
|---|---|---|---|---|
| m−1s−1 | s−1 | m | ||
| cHCDR1 | GFWLNW | 1.27 ± 0.25 × 102 | 2.94 ± 0.79 × 10−3 | 2.44 ± 1.05 × 10−5 |
| cHCDR2 | AIYRGSGSEWYASW | 1.20 ± 0.50 × 102 | 2.60 ± 0.55 × 10−3 | 2.36 ± 0.68 × 10−5 |
| cHCDR3 | AADTTDNGYFTI | —a | ||
| cLCDR1 | QASQSISSWLS | 0 | 0 | 0 |
| cLCDR2 | YDASNLA | 2.28 ± 0.10 × 101 | 2.05 ± 0.15 × 10−3 | 9.04 ± 1.02 × 10−5 |
| cLCDR3 | LGGYPAASYRTA | 5.85 ± 1.82 × 105 | 1.51 ± 0.33 × 10−2 | 2.68 ± 0.41 × 10−8 |
a Could not be synthesized.
Antibody Fragment Binding to the Carboxyl-terminal Region of Rev
An antibody fragment targeting the carboxyl-terminal activation region of Rev was also isolated from the phage display library using Rev1–116 directly adsorbed to microtiter plates for screening. This method differed from that which led to FabRev1 where the carboxyl-terminal biotinylated Rev1–116 was immobilized on strepavidin-coated microtiter plates (17). The single chain form of this Fab (scFvRev2) bound to Rev1–116 with a mean equilibrium dissociation constant of 1.9 × 10−8 m, characterized by both a high association rate and a high dissociation rate (Fig. 7A). The scFvRev2 did not bind to Rev1–93 and Rev1–69, suggesting that the epitope was located in the carboxyl-terminal region beyond residue 93 (not shown). This was confirmed by a competition experiment wherein the carboxyl-terminal peptide Rev94–116 effectively interfered with scFvRev2 binding to immobilized Rev1–116 (Fig. 7B). As the carboxyl-terminal region of Rev is unstructured, as demonstrated by CD analysis (Fig. 1C), the epitope for this antibody was presumed to be linear. This was confirmed by Western blot analysis in which scFvRev2 recognized Rev immobilized on a PVDF membrane, whereas scFvRev1, which is known to have a conformational epitope, failed to do so (Fig. 7C). Negative stain electron microscopy showed that, unlike scFvRev1, which effectively depolymerizes Rev1–116 filaments to soluble complexes, scFvRev2 decorates the surface of the filaments and does not cause disruption (Fig. 7, D and E). The carboxyl terminally truncated forms of Rev do not polymerize as filaments.3
FIGURE 7.

scFvRev2 binding to the carboxyl-terminal region of Rev. A, sensograms of scFvRev2 binding to immobilized Rev1–116. The Kd (1.9 × 10−8 m) was determined by equilibrium analysis, i.e. by measuring the steady-state response (Req) over a range of scFvRev2 concentrations, as plotted in the inset. B, peptide Rev94–116 competes with scFvRev2 binding to immobilized Rev1–116. Rev1–116 was covalently immobilized on a CM5 sensor chip and analyzed with a mixture of scFvRev2 and varying amounts of the peptide Rev94–116. The analytes were mixtures of 0.19 μm scFvRev2 and 0, 1.25, 2.5, 5, 10, 20, 40, and 80 nm Rev94–116. C, Western blot of scFvRev1 and scFvRev2 binding to immobilized Rev1–116. Electron micrographs of negatively stained Rev1–116 (D) filaments and Rev1–116 (E) filaments decorated with scFvRev2. Bar = 50 nm (D and E).
DISCUSSION
HIV-1 Rev is required for viral replication, serving as an adaptor between viral transcripts and the transport factor Crm1, thereby allowing their nuclear export for the expression of late-stage proteins and to serve as genomes in progeny virions (10, 11, 13). Rev functions by oligomerizing on the RRE of viral RNA molecules through interactions between Rev monomers (29). Given the essential role of Rev in HIV-1 replication it is a potentially viable, but as yet largely untested, target for antiviral intervention (9, 30).
The Antiviral Activity of FabRev1-Tat
FabRev1 binds strongly to the amino-terminal oligomerization domain of Rev, although the unstructured carboxyl-terminal region of Rev sterically perturbs this binding slightly. Based on the crystal structure of the Rev-FabRev1 complex (18), the binding site targeted is one of the faces of the helical hairpin whereby Rev monomers oligomerize (Fig. 5). The 4.0 × 10−11 m affinity of FabRev1 for Rev (17) exceeds those reported for both the initial binding of Rev to the RRE and for the subsequent Rev oligomerization (29). It may be the capability of FabRev1-Tat to block, or even disrupt, Rev oligomerization on the RRE, and hence interfere with the nuclear export of viral RNA that is responsible for the inhibition that we have observed. Alternatively, FabRev1-Tat could conceivably inhibit nuclear import of Rev, or the association of Rev with numerous other cellular factors that have been identified as Rev interaction partners (9, 31). There is no indication that FabRev1-Tat interferes with Rev binding to RRE-containing RNA in vitro.3
FabRev1 did not have antiviral activity but FabRev1-Tat did, suggesting that it functions intracellularly rather than on the cell surface. FabRev1-Tat had an inhibitory affect on three CCR5-tropic HIV-1 isolates but not on three CXCR4-tropic isolates. We do not know the reason for this difference. There may be differences in cell penetration, in the interaction with Rev, or both. It has been shown that Tat protein is a CXCR4-specific antagonist (32), and that arginine-rich peptides (derived from Rev) are both CXCR4- and Rev-specific antagonists (33). However, that cLCDR3-Tat in the absence of cell penetration did not have antiviral activity (not shown) suggests that a Tat-peptide interaction with surface receptors is not the mechanism of action of FabRev1-Tat. The low cytotoxicity of FabRev1-Tat also suggests that the antiviral effect is not merely due to cellular dysfunction.
Dissection of the FabRev1-Tat Paratope and Identification of the Key Binding CDR
The FabRev1 LCDR3 was predicted by computational alanine scanning to be a major contributor to the binding affinity of Fab. Whereas the linear version of the peptide did not bind to Rev, the (presumably more rigid) cyclic version did, and with an affinity higher than any other CDR derived from FabRev1. Furthermore, cLCDR3 mimicked FabRev1 in its ability to depolymerize Rev filaments, suggesting that the peptide disrupts the same protein-protein interactions as the parent Fab.
Comparison of FabRev1-Tat with Other Agents
The antiviral activity of FabRev1-Tat was assessed as inhibition of reverse transcriptase activity in PBMC cell culture supernatants. The IC50 values were in the range of 0.09–0.44 μg/ml. This concentration (2–8 nm) is comparable with the activity typically exhibited in this assay by several small molecules such as azidothymidine (a nucleoside reverse transcriptase inhibitor), efavirenz (non-nucleoside reverse transcriptase inhibitor), darunavir (protease inhibitor), and raltegravir (integrase inhibitor), but an order of magnitude less potent than commonly exhibited by rilpivirine (non-nucleoside reverse transcriptase inhibitor) and elvitegravir (integrase inhibitor).4 The inhibitory and toxic properties of FabRev1-Tat (IC50 = 2–8 nm; TC50 > 2000 nm) also compare favorably with those of the recently reported myxobacterial metabolite ratjadone A that inhibits Rev/Crm1-mediated nuclear export (EC50 = 1.7 nm; CC50 = 4.6 nm) (34).
Considerations of Intracellular Distribution on the Activity of FabRev1-Tat
Proteins transduced into cells with Tat cell penetration peptides are known to accumulate in endosomes where they may become trapped and eventually degraded in lysosomes (35) (although our protein would be expected to benefit from the known resistance of Fab to acid proteolysis). For proteins to be targeted to the nucleus they must first escape from the endosome and enter the cytoplasm. A number of endosomal escape mechanisms have been identified, including pH buffering of the endosomal compartment, for example, by the imidazole ring of histidine residues in cargo proteins (the so-called “proton sponge” effect) (35). In at least one instance the addition of a His10 peptide to a Tat-tagged cargo resulted in a substantial (7000-fold) increase in nuclear localization (36). The His7 tag on FabRev1-Tat could be serving a similar function (whereas HIV-Tat, which did not have a His tag, remained in dense accumulations in the cytoplasm and did not enter nuclei well). Once released from the endosome the Tat peptide could target the Fab to the nucleus via an interaction with importin-β (37).
As mentioned above, alternative positioning and spacings of the Tat peptide were tested with no significant change in uptake. These constructs had the Tat peptide located at the carboxyl-terminal end. We were not able to express the proteins with the peptide in the amino-terminal position. There are reports for some other proteins that the Tat peptide position is not important (38) but this has not been our experience in this instance. Of the several other cell-penetrating peptides that have been described (21, 39), the most promising would be those that have been demonstrated to significantly enhance nuclear import (40, 41).
Fab Binding to the Carboxyl-terminal Region of Rev
Rev serves as an adaptor between viral transcripts and nuclear export machinery, binding to the RRE with its amino-terminal region and Crm1 with its carboxyl-terminal region. In principle, it is possible that a Fab binding to either region would interfere with Rev function. It has been found in HIV-1 patients that Rev mutants associated with increased activity (but that are silent in the overlapping Tat and Env proteins) are located near the carboxyl terminus (residues 102–111) (42). We therefore also attempted to produce Fabs that would bind to the carboxyl-terminal region. The molecule that we produced, scFvRev2, did bind near the carboxyl terminus of Rev (residues 93–116) but the epitope was non-conformational and it had low affinity, and was therefore judged to have little inhibitory potential. scFvRev2 may, however, be useful for structural studies. The carboxyl-terminal region of Rev is disordered in crystals (18). scFvRev2 decorates the outer surface of Rev filaments, thus localizing the carboxyl-terminal region in these structures. This will assist in ongoing studies aimed at determining the structure of Rev filaments that in turn may serve as a model for Rev oligomerization in vivo.5
Conclusions
We have shown that an anti-Rev Fab has high anti-HIV-1 activity and low cytotoxicity. The activity of the Fab is dependent on the presence of the Tat cell penetration peptide, and immunofluorescence results show that the tagged Fab enters cells even to the nucleus. From the structure of the Fab-Rev complex and the Fab-induced in vitro dissociation of Rev filaments, it is likely that the Fab inhibits HIV-1 by blocking Rev oligomerization, in the cytoplasm, nucleus, or both. A major determinant of the interaction of FabRev1-Tat with Rev resides in LCDR3. Cyclic forms of the peptide (cLGGYPAASYRTA) bind to Rev and effectively dissociate Rev polymers.
Acknowledgments
We thank Drs. Marc Nicklaus and Megan Peach (NCI/National Institutes of Health) for performing the computational alanine scanning, and Drs. Steven Turk (NIAID/National Institutes of Health) and Christoph Rader (Scripps) for much appreciated discussions. Drs. Evelyn Ralston and Kristina Zaal (NIAMS/National Institutes of Health) provided advice on the immunofluorescence microscopy. We also thank Joshua Kaufman and Ira Palmer (NIAMS/National Institutes of Health) for expert technical assistance. The anti-HIV testing was conducted by the Southern Research Institute using federal funds from the Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, under contract HHSN272200700041C entitled “Confirmatory In Vitro Evaluations of HIV Therapeutics.”
This work was supported by the NIAMS Intramural Research Program of the National Institutes of Health.
X. Zhuang, S. J. Stahl, N. R. Watts, M. A. DiMattia, A. C. Steven, and P. T. Wingfield, unpublished observations.
S. Turk, personal communication.
M. DiMattia, N. Watts, S. Stahl, C. Rader, P. Wingfield, D. Stuart, A. Steven, and J. Grimes, unpublished data.
- Fab
- antigen binding fragment
- CDR
- complementarity determining region
- cLCDR3
- cyclic light chain complementarity domain region 3
- RRE
- Rev response element
- scFv
- single chain variable domain fragment
- PBMC
- peripheral blood mononuclear cells.
REFERENCES
- 1. McManus H., O'Connor C. C., Boyd M., Broom J., Russell D., Watson K., Roth N., Read P. J., Petoumenos K., Law M. G., and Australian HIV Observational Database (2012) Long-term survival in HIV positive patients with up to 15 years of antiretroviral therapy. PLoS One 7, e48839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Levi M., Sällberg M., Rudén U., Herlyn D., Maruyama H., Wigzell H., Marks J., Wahren B. (1993) A complementarity-determining region synthetic peptide acts as a miniantibody and neutralizes human immunodeficiency virus type 1 in vitro. Proc. Natl. Acad. Sci. U.S.A. 90, 4374–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Levi M., Hinkula J., Wahren B. (2000) A retro-inverso miniantibody with anti-HIV activity. AIDS Res. Hum. Retroviruses 16, 59–65 [DOI] [PubMed] [Google Scholar]
- 4. Monnet C., Laune D., Laroche-Traineau J., Biard-Piechaczyk M., Briant L., Bès C., Pugnière M., Mani J. C., Pau B., Cerutti M., Devauchelle G., Devaux C., Granier C., Chardès T. (1999) Synthetic peptides derived from the variable regions of an anti-CD4 monoclonal antibody bind to CD4 and inhibit HIV-1 promoter activation in virus-infected cells. J. Biol. Chem. 274, 3789–3796 [DOI] [PubMed] [Google Scholar]
- 5. Bès C., Briant-Longuet L., Cerruti M., De Berardinis P., Devauchelle G., Devaux C., Granier C., Chardès T., DeBerardinis P. (2001) Efficient CD4 binding and immunosuppressive properties of the 13B8.2 monoclonal antibody are displayed by its CDR-H1-derived peptide CB1. FEBS Lett. 508, 67–74 [DOI] [PubMed] [Google Scholar]
- 6. Laune D., Molina F., Ferrières G., Villard S., Bès C., Rieunier F., Chardès T., Granier C. (2002) Application of the Spot method to the identification of peptides and amino acids from the antibody paratope that contribute to antigen binding. J. Immunol. Methods 267, 53–70 [DOI] [PubMed] [Google Scholar]
- 7. Casset F., Roux F., Mouchet P., Bes C., Chardes T., Granier C., Mani J. C., Pugnière M., Laune D., Pau B., Kaczorek M., Lahana R., Rees A. (2003) A peptide mimetic of an anti-CD4 monoclonal antibody by rational design. Biochem. Biophys. Res. Commun. 307, 198–205 [DOI] [PubMed] [Google Scholar]
- 8. Heap C. J., Wang Y., Pinheiro T. J., Reading S. A., Jennings K. R., Dimmock N. J. (2005) Analysis of a 17-amino acid residue, virus-neutralizing microantibody. J. Gen. Virol. 86, 1791–1800 [DOI] [PubMed] [Google Scholar]
- 9. Vercruysse T., Boons E., Venken T., Vanstreels E., Voet A., Steyaert J., De Maeyer M., Daelemans D. (2013) Mapping the binding interface between an HIV-1 inhibiting intrabody and the viral protein Rev. PLoS One 8, e60259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pollard V. W., Malim M. H. (1998) The HIV-1 Rev protein. Annu. Rev. Microbiol. 52, 491–532 [DOI] [PubMed] [Google Scholar]
- 11. Frankel A. D., Young J. A. (1998) HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 67, 1–25 [DOI] [PubMed] [Google Scholar]
- 12. Jain C., Belasco J. G. (2001) Structural model for the cooperative assembly of HIV-1 Rev multimers on the RRE as deduced from analysis of assembly-defective mutants. Mol. Cell 7, 603–614 [DOI] [PubMed] [Google Scholar]
- 13. Cullen B. R. (2003) Nuclear mRNA export: insights from virology. Trends Biochem. Sci. 28, 419–424 [DOI] [PubMed] [Google Scholar]
- 14. Wingfield P. T., Stahl S. J., Payton M. A., Venkatesan S., Misra M., Steven A. C. (1991) HIV-1 Rev expressed in recombinant Escherichia coli: purification, polymerization, and conformational properties. Biochemistry 30, 7527–7534 [DOI] [PubMed] [Google Scholar]
- 15. Watts N. R., Misra M., Wingfield P. T., Stahl S. J., Cheng N., Trus B. L., Steven A. C., Williams R. W. (1998) Three-dimensional structure of HIV-1 Rev protein filaments. J. Struct. Biol. 121, 41–52 [DOI] [PubMed] [Google Scholar]
- 16. Battiste J. L., Mao H., Rao N. S., Tan R., Muhandiram D. R., Kay L. E., Frankel A. D., Williamson J. R. (1996) α-Helix-RNA major groove recognition in an HIV-1 Rev peptide RRE RNA complex. Science 273, 1547–1551 [DOI] [PubMed] [Google Scholar]
- 17. Stahl S. J., Watts N. R., Rader C., DiMattia M. A., Mage R. G., Palmer I., Kaufman J. D., Grimes J. M., Stuart D. I., Steven A. C., Wingfield P. T. (2010) Generation and characterization of a chimeric rabbit/human Fab for co-crystallization of HIV-1 Rev. J. Mol. Biol. 397, 697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DiMattia M. A., Watts N. R., Stahl S. J., Rader C., Wingfield P. T., Stuart D. I., Steven A. C., Grimes J. M. (2010) Implications of the HIV-1 Rev dimer structure at 3.2 Å resolution for multimeric binding to the Rev response element. Proc. Natl. Acad. Sci. U.S.A. 107, 5810–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Daugherty M. D., Liu B., Frankel A. D. (2010) Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat. Struct. Mol. Biol. 17, 1337–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fang X., Wang J., O'Carroll I. P., Mitchell M., Zuo X., Wang Y., Yu P., Liu Y., Rausch J. W., Dyba M. A., Kjems J., Schwieters C. D., Seifert S., Winans R. E., Watts N. R., Stahl S. J., Wingfield P. T., Byrd R. A., Le Grice S. F., Rein A., Wang Y. X. (2013) An unusual topological structure of the HIV-1 Rev response element. Cell 155, 594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Madani F., Lindberg S., Langel U., Futaki S., Gräslund A. (2011) Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kortemme T., Kim D. E., Baker D. (2004) Computational alanine scanning of protein-protein interfaces. Science's STKE 2004, pl2. [DOI] [PubMed] [Google Scholar]
- 23. Krüger D. M., Gohlke H. (2010) DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 38, W480–W486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tuncbag N., Keskin O., Gursoy A. (2010) HotPoint: hot spot prediction server for protein interfaces. Nucleic Acids Res. 38, W402–W406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meireles L. M., Dömling A. S., Camacho C. J. (2010) ANCHOR: a web server and database for analysis of protein-protein interaction binding pockets for drug discovery. Nucleic Acids Res. 38, W407–W411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Segura Mora J., Assi S. A., Fernandez-Fuentes N. (2010) Presaging critical residues in protein interfaces-web server (PCRPi-W): a web server to chart hot spots in protein interfaces. PLoS One 5, e12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu X., Mitchell J. C. (2011) KFC2: a knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins 79, 2671–2683 [DOI] [PubMed] [Google Scholar]
- 28. Malim M. H., Böhnlein S., Hauber J., Cullen B. R. (1989) Functional dissection of the HIV-1 Rev trans-activator: derivation of a trans-dominant repressor of Rev function. Cell 58, 205–214 [DOI] [PubMed] [Google Scholar]
- 29. Pond S. J., Ridgeway W. K., Robertson R., Wang J., Millar D. P. (2009) HIV-1 Rev protein assembles on viral RNA one molecule at a time. Proc. Natl. Acad. Sci. U.S.A. 106, 1404–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wong R. W., Balachandran A., Haaland M., Stoilov P., Cochrane A. (2013) Characterization of novel inhibitors of HIV-1 replication that function via alteration of viral RNA processing and rev function. Nucleic Acids Res. 41, 9471–9483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jäger S., Cimermancic P., Gulbahce N., Johnson J. R., McGovern K. E., Clarke S. C., Shales M., Mercenne G., Pache L., Li K., Hernandez H., Jang G. M., Roth S. L., Akiva E., Marlett J., Stephens M., D'Orso I., Fernandes J., Fahey M., Mahon C., O'Donoghue A. J., Todorovic A., Morris J. H., Maltby D. A., Alber T., Cagney G., Bushman F. D., Young J. A., Chanda S. K., Sundquist W. I., Kortemme T., Hernandez R. D., Craik C. S., Burlingame A., Sali A., Frankel A. D., Krogan N. J. (2012) Global landscape of HIV-human protein complexes. Nature 481, 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiao H., Neuveut C., Tiffany H. L., Benkirane M., Rich E. A., Murphy P. M., Jeang K. T. (2000) Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. U.S.A. 97, 11466–11471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimane K., Kodama E. N., Nakase I., Futaki S., Sakurai Y., Sakagami Y., Li X., Hattori T., Sarafianos S. G., Matsuoka M. (2010) Rev-derived peptides inhibit HIV-1 replication by antagonism of Rev and a co-receptor, CXCR4. Int. J. Biochem. Cell Biol. 42, 1482–1488 [DOI] [PubMed] [Google Scholar]
- 34. Fleta-Soriano E., Martinez J. P., Hinkelmann B., Gerth K., Washausen P., Diez J., Frank R., Sasse F., Meyerhans A. (2014) The myxobacterial metabolite ratjadone A inhibits HIV infection by blocking the Rev/CRM1-mediated nuclear export pathway. Microb. Cell Fact. 13, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Varkouhi A. K., Scholte M., Storm G., Haisma H. J. (2011) Endosomal escape pathways for delivery of biologicals. J. Control. Release 151, 220–228 [DOI] [PubMed] [Google Scholar]
- 36. Lo S. L., Wang S. (2008) An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials 29, 2408–2414 [DOI] [PubMed] [Google Scholar]
- 37. Truant R., Cullen B. R. (1999) The arginine-rich domains present in human immunodeficiency virus type 1 Tat and Rev function as direct importin β-dependent nuclear localization signals. Mol. Cell. Biol. 19, 1210–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo Q., Zhao G., Hao F., Guan Y. (2012) Effects of the TAT peptide orientation and relative location on the protein transduction efficiency. Chem. Biol. Drug Des. 79, 683–690 [DOI] [PubMed] [Google Scholar]
- 39. Dupont E., Prochiantz A., Joliot A. (2011) Penetratin story: an overview. Methods Mol. Biol. 683, 21–29 [DOI] [PubMed] [Google Scholar]
- 40. Zaro J. L., Vekich J. E., Tran T., Shen W. C. (2009) Nuclear localization of cell-penetrating peptides is dependent on endocytosis rather than cytosolic delivery in CHO cells. Mol. Pharm. 6, 337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lewis H. D., Husain A., Donnelly R. J., Barlos D., Riaz S., Ginjupalli K., Shodeinde A., Barton B. E. (2010) Creation of a novel peptide with enhanced nuclear localization in prostate and pancreatic cancer cell lines. BMC Biotechnol. 10, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sloan E. A., Kearney M. F., Gray L. R., Anastos K., Daar E. S., Margolick J., Maldarelli F., Hammarskjold M. L., Rekosh D. (2013) Limited nucleotide changes in the Rev response element (RRE) during HIV-1 infection alter overall Rev-RRE activity and Rev multimerization. J. Virol. 87, 11173–11186 [DOI] [PMC free article] [PubMed] [Google Scholar]