

Background: Base J regulates Pol II transcription in trypanosomatids and is synthesized in two steps involving DNA hydroxylation and glucosylation.
Results: JGT catalyzes the transfer of glucose from uridine diphosphoglucose to hmU in DNA.
Conclusion: JGT is the glucosyltransferase involved in base J synthesis.
Significance: JGT is the first known glucosyltransferase that modifies eukaryotic DNA.
Keywords: DNA Enzyme, Epigenetics, Glycosyltransferase, Parasite, Transcription Regulation, Trypanosoma brucei, Base J, Trypanosomatid
Abstract
O-linked glucosylation of thymine in DNA (base J) is an important regulatory epigenetic mark in trypanosomatids. β-d-glucopyranosyloxymethyluracil (base J) synthesis is initiated by the JBP1/2 enzymes that hydroxylate thymine, forming 5-hydroxymethyluracil (hmU). hmU is then glucosylated by a previously unknown glucosyltransferase. A recent computational screen identified a possible candidate for the base J-associated glucosyltransferase (JGT) in trypanosomatid genomes. We demonstrate that recombinant JGT utilizes uridine diphosphoglucose to transfer glucose to hmU in the context of dsDNA. Mutation of conserved residues typically involved in glucosyltransferase catalysis impairs DNA glucosylation in vitro. The deletion of both alleles of JGT from the genome of Trypanosoma brucei generates a cell line that completely lacks base J. Reintroduction of JGT in the JGT KO restores J synthesis. Ablation of JGT mRNA levels by RNAi leads to the sequential reduction in base J and increased levels of hmU that dissipate rapidly. The analysis of JGT function confirms the two-step J synthesis model and demonstrates that JGT is the only glucosyltransferase enzyme required for the second step of the pathway. Similar to the activity of the related Ten-Eleven Translocation (TET) family of dioxygenases on 5mC, our studies also suggest the ability of the base J-binding protein enzymes to catalyze iterative oxidation of thymine in trypanosome DNA. Here we discuss the regulation of hmU and base J formation in the trypanosome genome by JGT and base J-binding protein.
Introduction
Trypanosomatids, including the human pathogens Trypanosoma brucei, Trypanosoma cruzi, and Leishmania species, possess a unique DNA modification within their genome known as β-d-glucopyranosyloxymethyluracil (base J)3 (1, 2). Base J is a hypermodified thymine residue that accounts for about 1% of thymines in the genome and is predominately present in repetitive sequences, such as telomeric repeats (2, 3). In addition, base J is also found at chromosome-internal regions that coincide with sites of RNA polymerase II (Pol II) transcription initiation and termination (4). The loss of base J from these chromosome-internal regions in T. cruzi leads to more open chromatin and increased Pol II occupancy, ultimately leading to changes in gene expression and parasite virulence (5, 6). Loss of base J in Leishmania tarentolae leads to transcription termination defects and generation of antisense RNAs corresponding to downstream genes (7). These data strongly support that base J represents a novel epigenetic marker involved in regulating Pol II transcription and gene expression. Understanding the enzymes involved in base J synthesis and their regulation is critical in furthering our understanding of the role base J plays in the regulation of gene expression in these parasites.
Indirect evidence, reviewed in Ref. 8, indicates a two-step pathway for the synthesis of base J (Fig. 1A). The first step of the pathway involves the hydroxylation of thymine in DNA by a thymidine hydroxylase enzyme, forming 5-hydroxymethyluracil (hmU). A glucose moiety is then attached to this intermediate, presumably by a glucosyltransferase enzyme, to form base J. Although the GT has not been identified, two thymidine hydroxylase enzymes (JBP1 and JBP2) involved in the first step of base J synthesis have been characterized (9, 10). Both JBP1 and JBP2 contain a thymidine hydroxylase domain at the N terminus that has led to the designation of these enzymes as belonging to the TET/base J-binding protein (JBP) subfamily of dioxygenases that require Fe2+ and 2-oxoglutarate for activity (11–15). JBP1 has a J-DNA binding domain in the C-terminal half of the protein (10, 16–18). JBP2 does not bind the modified base directly, but is able to bind chromatin in a base J-independent manner, presumably via the C-terminal SWI2/SNF2 domain (9). Although both JBP1 and JBP2 stimulate de novo thymidine hydroxylation in vivo, the ability of JBP1 to bind J-DNA is thought to play a role in J propagation/maintenance (9, 14, 19). Deletion of either JBP1 or JBP2 from the bloodstream form T. brucei results in a 20- and 8-fold reduction in the levels of base J, respectively (10, 14, 20). The simultaneous deletion of both JBP1 and JBP2 yields a cell line that is unable to synthesize base J unless cells are fed hmU (14). Finally, using recombinant protein produced in Escherichia coli, we demonstrated that JBP1 stimulates the hydroxylation of thymidine in the context of dsDNA in a Fe2+, 2-oxoglutarate-, and O2-dependent manner (15). These data confirm the identity of JBP1/2 as the thymidine hydroxylases catalyzing the first step of base J synthesis and that this step is independent from the subsequent step of glucose conjugation.
FIGURE 1.

JGT is an hmU-DNA glucosyltransferase. A, proposed mechanism of base J synthesis. As described in the text, base J synthesis is proposed as a two-step process in which thymines in DNA are first hydroxylated by the thymidine hydroxylases JBP1 and JBP2 to form hmU. JBP1 and JBP2 are members of the Fe2+/2-oxoglutarate dioxygenase family that utilize oxygen and 2-oxoglutarate as cosubstrates and release CO2 and succinate as byproducts. The intermediate hmU is then glycosylated by a glucosyltransferase, forming base J. Presumably, the glucose donor in the second step is UDP-glucose. B, schematic of the T. brucei (Tb) JGT protein depicting the GT domain, with the general topology shown above. Conserved residues characteristic of GT-A fold are shown below (adapted from Ref. 21). C, SDS-PAGE of purified JGT. Coomassie-stained gel (left panel) and anti-His Western blot analysis (right panel) are shown. LC-MS indicates that affinity purification using Talon resin resulted in copurification of JGT and two E. coli molecular chaperones (DnaK and GroEL). GroEL and DnaK are common contaminants of GT enzymes following expression in E. coli (48). Because DnaK runs at the same location as JGT on the SDS-PAGE gel, protein concentration was measured using a combination of SDS-PAGE and a bovine serum albumin standards and anti-His Western blot analysis as described under “Experimental Procedures.” The Western blot analysis indicates that the lower molecular weight proteins are degradation products of JGT. D, in vitro GTase reaction. Recombinant JGT and UDP-[3H]glucose were incubated with 20-nt-long, double-stranded DNA substrate containing four hmU molecules (ds hmU) or unmodified thymidines (ds T), as described under “Experimental Procedures.” CPM, counts per million, indicative of the transfer of glucose to DNA, were read for each sample. All experiments were performed in triplicate, and error bars are representative of mean ± S.D. E, equal amounts of recombinant WT JGT and r80a, d241a, and d243a point mutant JGT were assayed for transfer of [3H]glucose from UDP-glucose to hmU-DNA as described above. The results are mean ± S.D. of triplicates and expressed as percent activity relative to the WT control reaction. Bottom panel, anti-His Western blot analysis demonstrating that equal amounts of WT and mutant JGT enzyme were included in each assay.
As reviewed in Borst and Sabatini (8), attempts to identify the GT involved in the synthesis of base J have been unsuccessful. However, a recent bioinformatic study identified a possible candidate GT in trypanosomatids (21). Examining biochemical pathways for DNA modifications, Iyer et al. (21) identified a GT-A-like glucosyltransferase with an operonic association to a JBP-related gene within several phage genomes. The authors postulate that because operons contain functionally related genes, these TET/JBP-associated glycosyltransferases glycosylate substrates generated by the JBP-like enzymes. Multiple sequence alignment showed that the TET/JBP-associated glycosyltransferases, including the kinetoplastid homologs, possess many of the structural elements and catalytic residues characteristic of the Rossmannoid nucleotide-diphospho-sugar binding fold typical of the GT-A/fringe superfamily (21).
We now show the first functional analysis of the T. brucei TET/JBP-associated glycosyltransferase homolog and refer to it as base J-specific GT (JGT). Our in vitro assays show that recombinant JGT utilizes UDP-Glc to transfer glucose to dsDNA substrates containing hmU. Mutation of conserved residues in the catalytic domain of the GT-A fold in JGT impairs DNA glucosylation. In vivo, deletion of both JGT alleles in T. brucei results in the complete loss of base J. Reintroduction of an ectopically expressed, HA-tagged JGT in the JGT KO restores base J synthesis. RNAi knockdown of JGT led to a sequential reduction in base J and increased levels of hmU and 5-formyluracil (fU) in the T. brucei genome in a time-dependent manner. The analyses of JGT function in vivo not only confirm its identity and the two-step base J synthesis model but also indicate that it is the only GT catalyzing the second step of base J synthesis. These studies also suggest the ability of the JBP enzymes to convert hmU to fU. Here we discuss the regulation and consequence of the oxidation and glucosylation of hmU in the trypanosome genome by JBP and JGT.
EXPERIMENTAL PROCEDURES
Trypanosome Cell Culture and Generation of T. brucei Transfectants
The bloodstream form T. brucei cell line strain 427 and the 90–13 RNAi cell line were cultured in HMI9 medium supplemented with 10% heat-inactivated fetal bovine serum and 10% serum plus as described previously (9). 90-13 cells were cultured in the presence of 2.5 μg/ml neomycin and 5 μg/ml hygromycin to maintain the intergrated genes for T7 RNA polymerase and the tetracycline repressor, respectively. Transfections of bloodstream form T. brucei were essentially carried out as described previously (14).
Generation of the JGT Knockout
Constructs used to generate JGT knockout lines contain either a hygromycin phosphotransferase (Hyg) gene (pTub-Hygro) or a blasticidin S deaminase (BSR) gene (pTub-Blast). The T. brucei JGT-flanking regions used to target each allele were PCR-amplified from T. brucei 427 genomic DNA. The 611-bp fragment corresponding to the 5′ flank was PCR-amplified using the sense primer 5′-GCGGCCGCCGGCACTGACGATCTTACAT-3′ (the NotI site is underlined) and the antisense primer 5′-GGATCCCACATAATATAGCGCCACACATTC-3′ (the BamHI site is underlined). The resulting PCR products were cloned into the pTub-Hygro and pTub-Blast vectors digested with NotI and BamHI. The 595-bp fragment corresponding to the 3′ flank was PCR-amplified using the sense primer 5′-AAGCTTTGCAGATGGCGTGTTTCT-3′ (the HindIII site is underlined) and the antisense primer: 5′-CTCGAGACGTGTGCCTAATACACTTACC-3′ (the XhoI site is underlined) and cloned into the pTub-Hygro construct. For the pTub-Blast construct, the 3′ flank was generated using the sense primer 5′-GGGCCCTGCAGATGGCGTGTTTCT-3′ (the ApaI site is underlined) and the antisense primer 5′- GGGCCCAAGCTTACGTGTGCCTAATACACTTACC-3′ (the ApaI site is underlined). The resulting PCR product was cloned into the ApaI-digested pTub-Blast vector. The pJGT-Tub-Hygro knockout construct was digested with NotI and XhoI, and the pJGT-Tub-Blast knockout construct was digested with NotI and HindIII prior to transfection. Transfectants were selected for resistance at 5 μg/ml hygromycin and 5 μg/ml blasticidin. Two rounds of transfection were used to replace both JGT alleles. The correct integration of the KO constructs and deletion of the JGT ORF was confirmed by PCR.
Re-expression of HA-tagged JGT
To allow ectopic expression of HA-JGT fusion, we utilized a modified ptub-phleo construct (22) that contains the enhanced green fluorescent protein ORF cloned between the tubulin flanking sequences. A 2012-bp PCR product corresponding to the T. brucei JGT ORF was PCR-amplified using the sense primer CCTGCAGGATGGCTTACCCATATGATGTTCCAGATTACGCTGGAGGTCCAAGTGAGGGGAAG (the SbfI site is underlined and sequence coding for the HA tag is shown in boldface) and the antisense primer GGCGCGCCTTAGTCTGCCTGCGACCCTCC (the AscI site is underlined) and cloned into the ptub-GFP vector digested with SbfI and AscI. Insertion of the HA-JGT PCR product in the place of EGFP now allows expression of HA-JGT N terminus fusion protein after integration into the tubulin array. The final construct (HA-JGT-tub-phleo) was digested with XbaI and XhoI before electroporation, and transformants were selected for resistance to 2.5 μg/ml phleomycin.
JGT RNAi
To allow Tet-inducible ablation of the T. brucei JGT mRNA, the pZJM-JGT RNA interference construct was generated by inserting a 524-bp portion of the JGT ORF into the XhoI and HindIII sites of the RNAi vector pZJM (23). A 524-bp fragment of the coding sequence of the JGT gene (tb427.10.6900) was amplified from T. brucei 427 genomic DNA using the sense primer CTCGAGGTGAATGTGTATGCCAACGC (the XhoI site is underlined) and the antisense primer AAGCTTTTGTTCTGCTGGCAGATGTC (the HindIII site is underlined) and cloned into the XhoI- and HindIII-digested pZJM vector. The resulting construct, pZJM-JGT, was digested with NotI prior to transfection. Transfectants were selected for resistance at 2.5 μg/ml phleomycin.
Determination of the Genomic Level of J
To quantify the genomic J levels, DNA was isolated and utilized in the anti-J DNA immunoblot assay as described previously (24). Briefly, serially diluted genomic DNA was blotted to nitrocellulose, followed by incubation with anti-J antisera. Bound antibodies were detected by a secondary goat anti-rabbit antibody conjugated to HRP and visualized by ECL. The membrane was stripped and hybridized with a probe for the β-tubulin gene to correct for DNA loading.
Quantitative Reverse Transcription PCR
Total RNA was obtained using Qiagen RNeasy kits according to the instructions of the manufacturers. First-strand cDNA was synthesized from 1 μg of total RNA using an iScript cDNA synthesis kit (Bio-Rad) according to the instructions of the manufacturer. Heat-inactivated cDNA reaction mixtures were finally treated with RNase H at 37 °C for 45 min. Quantification of selected genes was performed on an iCycler with an iQ5 multicolor real-time PCR detection system (Bio-Rad). The reaction mixture contained 5 pmol of forward and reverse primer, 2× iQ SYBR Green Super Mix (Bio-Rad), and 2 μl of template cDNA. Standard curves were prepared for each gene using 5-fold dilutions of a known quantity (250 ng/μl) of genomic DNA from wild-type T. brucei DNA. The quantities were calculated using iQ5 optical detection system software. Each sample was normalized to enolase mRNA. The primer sequences utilized in this analysis were as follows: JGT, 5′-CCTGACTGAGAACCCTTACTTC-3′ (sense) and 5′-GGCACGTGTGACCATATACA-3′ (antisense); JBP1, 5′-GTGTCCTAGCTGTGCTCAAA-3′ (sense) and 5′-CAGGTGCGTATCGAAGAGTAAG-3′ (antisense); JBP2, 5′-CCTTCCACCTTTGTGTATTCCT-3′ sense) and 5′-CAACCGTCTCCTTCCTTGATAC-3′ (antisense); enolase, 5′-GGCCTGCAACTCTCTTCTAC-3′ (sense) and 5′-CATCACTGACCAGCCATTCT-3′ (antisense).
Preparation of Recombinant JGT
The open reading frame of T. brucei JGT (Tb927.10.6900 TriTrypDB) was codon-optimized for E. coli expression and cloned into pET16b expression vector by GeneArt. The codon-optimized JGT nucleotide sequence is available upon request. The final construct was Tb-JGT-pET16b. Expression and purification of the N-terminal His10-tagged Tb-JGT was performed with BL21-CodonPlus(DE3)- RIL-competent cells (Agilent). Briefly, freshly transformed bacterial cell cultures were induced at A600 nm 0.4–0.6 with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 16 h at 16 °C. Cell pellets were washed in 50 mm Hepes (pH 7.5), 300 mm NaCl, 10 mm β-mercaptoethanol, and 100 mm PMSFand then flash-frozen and stored at −70 °C until purification. Cells were lysed and sonicated in lysis buffer (50 mm Hepes-NaOH (pH 7.5), 300 mm NaCl, 10 mm β-mercaptoethanol, and 10% glycerol) with protease inhibitors (aprotinin, leupeptin, pepstatin, PMSF, and EDTA-free complete protease inhibitor; Roche). Cell lysates were clarified at 31,000 × g for 20 min at 4 °C. Recombinant JGT was affinity-purified with Talon resin (CloneTech) at 4 °C for 1 h, washed with 600 mm NaCl lysis buffer in a batch, and eluted by gravity column with 150 mm imidazole lysis buffer. Purified protein was concentrated by Centricon (Millipore) and visualized by colloidal blue-stained SDS-PAGE and anti-His Western blot analysis. JGT concentration was estimated on the basis of mass spec analysis indicating the ratio of JGT and DnaK in the purified sample and comparison to BSA standards on Coomassie-stained SDS-PAGE.
Mutagenesis of the T. brucei JGT was achieved using the Q5 site-directed mutagenesis kit (New England Biolabs). As a template for the PCR Tb-JGT-pET16b construct, the following primer pairs were used: r(80)a, 5′-GAGCAAAGGTgcaTTTTATCATGAACGTGGC-3′ (forward) and 5′-GGAACAAAAA TCGGAACAG-3′ (reverse); d241a, 5′-TGGGTTATGGcTGATGATATCG-3′ (forward) and 5′-ATACCACTGTGCTGCATG-3′ (reverse); and d243a, 5′ATGGATGATGGcTATCGCCAAATTTTTC-5′ (forward) and 5′-AACCCAATACCACTGTGC-3′ (reverse). Nucleotides shown in lowercase indicate altered amino acids to yield a point mutation. The mutated construct was verified by DNA sequencing and utilized as described above for expression and purification.
In Vitro Glucosylation Assay
A standard glucosylation assay consisted of 88 μm UDP-[3H]glucose (specific activity 45Ci/mmol), ∼0.05 μm recombinant JGT, and 100 μm DNA containing four hmU residues in buffer (50 mm potassium acetate, 20 mm Tris acetate, 10 mm manganese acetate, and 1 mm DTT (pH 7.9)). Reaction mixtures were incubated at 37 °C for 30 min, and the reaction was stopped by addition of 10 μl of 400 μm cold UDP-glucose and flash-freezing in liquid nitrogen for processing. The reaction mixtures were thawed and applied immediately to a 2.5-cm DE81 membrane (GE Healthcare, catalog no. 3658-325) under air pressure using a vacuum manifold. The filters where then washed in 3 × 2 ml of 0.2 m ammonium bicarbonate, 3 × 2 ml of water, and 3 × 2 ml of 100% ethanol. Membranes were air-dried and placed in scintillation vials containing 5 ml of scintillation fluid. The solution was mixed, and tritium incorporation was measured for 1 min. All glucosylation reaction values were corrected for nonspecific binding of UDP-[3H]glucose to the filters. Background values were determined using reactions performed in the absence of enzyme but in the presence of UDP-[3H]glucose.
Oligos modified with hmU (H) (ACCCHAACCCHAACCCHAACCCHA) and an unmodified (thymidine) control (ACCCTAACCCTAACCCTAACCCTA) were hybridized to complementary oligo (TAGGGTTAGGGTTAGGGTTAGGGT), synthesized by Integrated DNA Technologies to generate hmU modified and unmodified dsDNA substrates, respectively. DNA duplexes were made by boiling complementary oligos for 10 min and allowing them to cool overnight.
Western Blotting
Proteins from 3 × 107 cell equivalents were separated by SDS-PAGE 8% gel, transferred to nitrocellulose, and probed with anti-HA antibody (Santa Cruz Biotechnology, catalog no. SC-805) (1:1000 dilution), anti-histone H3 (Abcam, catalog no. ab8580) (1:2500 dilution) or anti-JBP2 antibody, as described previously (19). Bound antibodies were detected by a secondary goat anti-rabbit antibody conjugated to HRP and visualized by ECL.
Detection of Base J, hmdU, and fdU by Mass Spectrometry
To minimize artificial DNA oxidation, a modified procedure was used to isolate total genomic DNA for LC/MS analysis. Briefly, T. brucei cells were pelleted, washed in 1× PBS, and then resuspended in lysis buffer (1% SDS, 25 mm EDTA, 0.4 m NaCl, 50 mm Tris-HCl (pH 7.9), and 400 μg/ml proteinase K) and incubated at 37 °C overnight. 5 m NaCl was then added and centrifuged to remove protein and cell debris, and DNA was precipitated with cold 100% ethanol. Purified genomic DNA from the indicated trypanosome cell lines was digested to nucleosides and subjected to LC/MS analysis.4 Briefly, DNA was digested to nucleosides as described previously, and isotopically labeled 5-hydroxymethyl-2′-deoxyuridine (hmdU) was added to the final mixture (25). Prior to digestion, single-stranded oligodeoxynucleotides housing 5-((β-d-glucopyranosyloxy)-methyl)-cytidine (glc-hmdC) was added to the sample to provide a surrogate internal standard for LC-MS/MS quantification of 5-((β-d-glucopyranosyloxy)-methyl)-uridine (dJ). The target nucleosides, dJ, glc-hmdC, and hmdU, were enriched via offline HPLC and collected separately for subsequent LC-MS/MS measurement. LC-MS/MS analysis of dJ and glc-hmdC was conducted on an LTQ XL linear ion trap mass spectrometer equipped with a nanoelectrospray ionization source coupled to an EASY-nLC II system (Thermo Fisher Scientific, San Jose, CA). The enriched fraction containing hmdU was separated on an Agilent 1200 capillary HPLC, and the eluent was directed to an LTQ linear ion-trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) following procedures reported previously (25).
For LC-MS/MS/MS quantification of fdU in the genome, 250 fmol of [1,3-15N2-2′-D]-5-fdU was added to the enzymatic digestion mixture of 1 μg of genomic DNA. The chemical synthesis of the labeled 5-hmdU and 5fdU standards is described in Ref. 26. To improve the detection limit of fdU in positive-ion electrospray ionization (ESI)-MS, the digested DNA was then derivatized with Girard reagent T to form a hydrazone conjugate harboring a precharged quaternary ammonium moiety, as reported previously (27). The nucleoside mixture containing unlabeled and isotope-labeled fdU-GirT hydrazone was subsequently extracted with chloroform to remove the enzymes, and the aqueous layer was subjected to LC-MS/MS/MS analysis.
Microscopy
The detection of HA-JGT expressed in the T. brucei JGT−/− was performed by anti-HA immunofluorescence analysis. Cells were fixed in 1% paraformaldehyde for 5 min on ice and then washed in HMI9 medium (without serum additives). Samples were applied to slides and allowed to air-dry. Slides were then blocked in 20% FBS in PBS for 30 min. Slides were incubated with anti-HA antibody (Covance, catalog no. MMS-101R) (1:100 dilution), followed by Alexa Fluor 594 goat anti-mouse antibody (Invitrogen, catalog no. A21125) (1:500 dilution). Images were acquired using an Axioobserver Z1 equipped with an Axiocam MRm camera controlled by Axiovision version 4.6 software.
RESULTS
JGT Is a Glucosyltransferase Utilizing UDP-Glucose to Transfer Glucose to hmU
The putative base J-specific GT encoded in the trypanosomatid genome contains many of the structural elements and catalytic residues characteristic of the Rossmannoid nucleotide-diphospho-sugar binding fold typical of the GT-A/fringe superfamily (21) (Fig. 1B). Common features of the GT-A domain that are found in JGT include a positively charged residue (Arg-80) and a “DXD” (here DDD) motif that are typically involved in nucleotide sugar binding (28–30). To test whether the JGT protein possessed glucosyltransferase activity, the T. brucei JGT was cloned and expressed in E. coli to produce recombinant protein with an N-terminal histidine tag (Fig. 1C). Using the purified recombinant enzyme fraction, we are able to show that JGT is able to utilize UDP-glucose to transfer glucose onto dsDNA substrates containing hmU (Fig. 1D). We see little transfer using the same DNA substrate with unmodified Thr residues instead of hmU. Alanine substitution of Arg-80, Asp-241, and Asp-243 (within the DXD motif) caused a significant reduction in in vitro GT activity (Fig. 1E). Alanine substitution of equivalent conserved (Asp) residues in other GTs resulted in similar reductions in activity (31, 32). Thus, elimination of one critical residue may not necessarily result in the complete loss of substrate binding. These data not only demonstrates that the JGT possesses glucosyltransferase activity but that UDP-glucose is the sugar donor and that hmU-modified DNA is a substrate for the enzyme. JGT is also predicted to be an inverting enzyme consistent with the structure of base J.
JGT Is the Glucosyltransferase Involved in Base J Synthesis in Vivo
To investigate whether JGT is involved in the synthesis of base J, we deleted both alleles of JGT from bloodstream form T. brucei. Loss of JGT mRNA was confirmed using quantitative reverse transcription PCR analysis (Fig. 2A). J levels in the genomic DNA of JGT−/+ and JGT−/− cells were examined using an anti-base J dot blot. Although the deletion of one allele decreased the levels of J in the genome, the deletion of both alleles of JGT resulted in the complete loss of J synthesis (Fig. 2B), as confirmed by LC-MS/MS analysis (Fig. 2C and Fig. 3). In this regard, we observed a peak in the selected ion chromatograms for monitoring the m/z 420 → 304 transition for glc-hmdC and the m/z 421 → 125, 143 transition for dJ (Fig. 3). The former transition is due to the cleavage of the N-glycosidic linkage in the [M+H]+ ion of glc-hmdC (m/z 420). The collisional activation of the [M+H]+ ion (m/z 421) of dJ again leads to the facile cleavage of the N-glycosidic bond to yield the ion of m/z 305, which can further eliminate a glucose or part of the glucose (C6H10O5) to give fragment ions of m/z 125 and 143, respectively. To confirm that the reduction and loss of base J was caused by the loss of the JGT protein, an HA-tagged version of JGT was re-expressed in the JGT−/− cell line from the tubulin locus (Fig. 2, A and D). Upon re-expression, JGT localizes to the nucleus (Fig. 2E) and restores J synthesis (Fig. 2B). Taken together, these results clearly identify that JGT is the GT involved in the synthesis of base J in T. brucei.
FIGURE 2.

JGT is involved in the synthesis of base J. A, relative expression of JGT mRNA levels. RNA was isolated from WT, JGT−/+, JGT−/−, and JGT−/− expressing an HA-tagged version of JGT (+JGT), and levels of JGT were measured by quantitative reverse transcription PCR as described under “Experimental Procedures.” B, anti-base J dot blot. DNA isolated from the indicated cell lines was serially diluted 2-fold and analyzed by anti-base J dot blot. The membrane was then hybridized with a radioactive tubulin probe to control for DNA loading. JBP−/− refers to the J null cell line that has all four alleles for JBP1 and JBP2 deleted from the genome (14). C, quantitation of the levels of nucleosides dJ and hmdU by nanoLC-MS/MS and LC-MS/MS/MS. Genomic DNA of the WT, JBP−/−, and JGT−/− cell lines was analyzed by tandem mass spectrometry as described under “Experimental Procedures.” D, anti-HA Western blot analysis of cell lysates from WT, JGT−/−, and JGT−/− cell lines expressing an HA-tagged version of JGT. E, anti-HA IFA of +JGT cells. The analysis of the untransfected JGT−/− cells is provided as a negative control.
FIGURE 3.
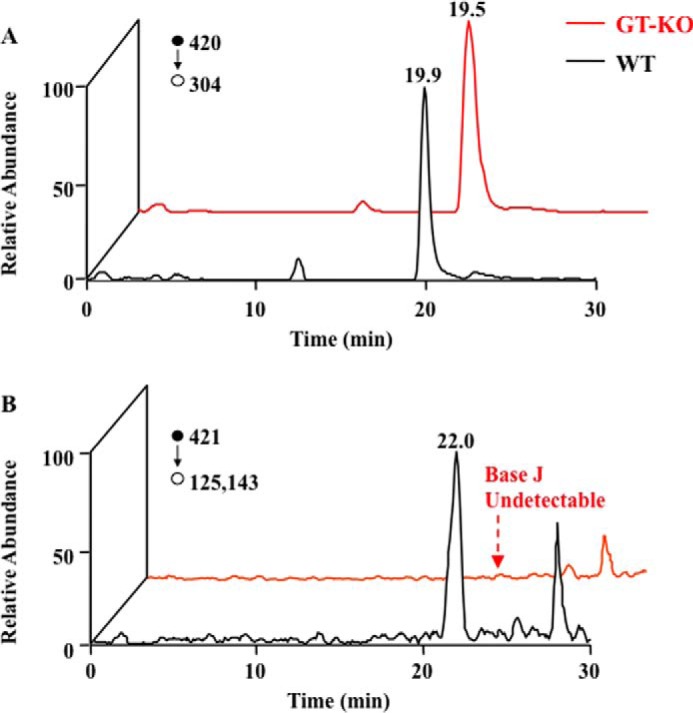
Representative LC-MS/MS results for the quantification of dJ. Shown are the selected ion chromatograms for monitoring the indicated transitions for the surrogate internal standard (A, glc-hmdC) and the analyte (B, dJ). The red and black lines represent samples from JGT KO and wild-type T. brucei cells, respectively.
According to the two-step model of base J synthesis (Fig. 1A), the GT modifies hmU generated by the thymidine hydroxylase (JBP1/2). We have shown here that JGT can utilize hmU-DNA in vitro (Fig. 1D). Therefore, we might expect an increase in hmU levels in the genome upon loss of JGT in vivo. However, unexpectedly, we did not detect an increase but, rather, a slight decrease in hmU levels in JGT−/− compared with WT T. brucei (Fig. 2C). One possible explanation is that JGT loss leads to altered levels or activity of JBP1/2. Although we lack antisera against JBP1, we detected no change in JBP2 levels upon deletion of JGT by Western blot analysis (data not shown). Although it is possible that JGT somehow stimulates JBP hydroxylase activity, a more feasible explanation for the lack of hmU accumulation upon the loss of GT is that the intermediate is subsequently shunted to the formation of additional base analogs. The mammalian JBP/TET enzymes (TET1–3) function in the demethylation pathway in which 5-methylcytosine can be hydroxylated iteratively to 5-hydroxymethylcytosine (hmC), 5-formylcytosine, and 5-carboxycytosine (33, 34). Potentially, JBP1/2 function in a similar manner, and accumulation of hmU in trypanosome DNA is prevented via conversion to fU and 5-carboxyluracil. To test this hypothesis and more closely investigate the role of the JGT in base J biosynthesis, we took advantage of the inducible RNAi system in T. brucei. As expected, RNAi-mediated ablation of JGT results in decreasing levels of base J over an ∼3-day time period (Fig. 4, A–C). LC-MS/MS/MS analyses indicated an initial increase in hmdU during the RNAi time course, followed by decreased levels by day 3.5 (Fig. 4C). These results indicate that hmU does increase following the loss of JGT but that it is rapidly lost. The detection of a slight increase in fdU is consistent with the loss of hmdU occurring via conversion to fU (and other analogs).
FIGURE 4.

RNAi knockdown of JGT leads to a decrease of base J and changes in hmU and fU. A, relative expression of JGT mRNA levels using quantitative RT-PCR in JGT RNAi cells with our without tetracycline 36 h after induction. B, anti-base J dot blot. DNA isolated from the indicated cell lines was serially diluted by 2-fold and spotted onto a nitrocellulose membrane to measure base J levels as described under “Experimental Procedures.” The membrane was then hybridized with a radioactive tubulin probe to control for DNA loading. 90-13, RNAi cell line untransfected with the JGT-pZJM construct; −Tet, uninduced JGT RNAi cells; +Tet, induced with tetracycline for the indicated days. C, genomic DNA isolated from JGT RNAi cells uninduced (−Tet) and at the indicated days following induction with tetracycline was analyzed by LC-MS/MS as described under “Experimental Procedures.” We detected a slight increase in levels of fdU in the tetracycline-induced samples compared with the uninduced samples (day 1.5, p = 0.5751; day 3.5, p = 0.1872).
DISCUSSION
Functional studies have established base J as an important regulatory epigenetic mark in trypanosomatids. Therefore, characterization of the enzymes and mechanisms regulating the modification of specific thymidine residues within key regulatory regions along the chromosome is critical for understanding the control of trypanosome gene expression.
Several lines of evidence support the hypothesis that base J is synthesized in DNA in two separate steps, as depicted in Fig. 1A. The specific localization of J within the kinetoplastid genome provides the strongest evidence that thymidine residues are modified in DNA rather than being synthesized and then incorporated during DNA replication (2, 4). In contrast, when trypanosomes are grown in the presence of hmU, it is incorporated randomly into DNA and then converted to J. Furthermore, the expression of the mammalian base excision DNA repair enzyme SMUG1 leads to T. brucei cell death by excessive DNA repair, indicating that hmU is a freely accessible intermediate in J biosynthesis (35). The hmU intermediate is also detectable in trypanosome DNA by thin layer chromatography (15) and mass spectrometry.4 We have recently shown the ability of JBP1 to oxidize Thr residues in dsDNA in vitro and regulate J synthesis in a DNA replication-independent manner in vivo (14, 15). We now provide conclusive evidence for the two-step base J biosynthesis mechanism by identifying the base J-associated GT and demonstrating its ability to specifically modify hmU residues in dsDNA substrate in vitro and regulate base J synthesis in vivo. In contrast with the first step involving two distinct hydroxylases, JGT is the only glucosyltransferase involved in the second step of base J synthesis.
Glycosyltransferases catalyze the transfer of monosaccharide primarily from an activated sugar donor (UDP sugars) to various substrates, including carbohydrates, proteins, and glycoproteins (36). Because they are central to all biosynthetic processes involving sugars, GTs are important targets for the development of novel drugs to help combat cancer as well as microbial infections (including parasitic diseases). JGT represents a unique glucosyltransferase in that it targets chromatin-bound DNA and is localized to the nucleus. To date, no other enzyme with similar substrate (DNA) specificity has been described in eukaryotes. Interestingly, a mammalian homologue of JGT called GREB1 has also been identified in the aforementioned bioinformatic study (21). GREB1 is an estrogen-responsive gene that has a largely unknown function but has been implicated in the proliferation of estrogen receptor-positive breast cancer cells (37, 38). GREB1 localizes to the nucleus and has been found bound to chromatin, presumably functioning as a transcriptional coactivator of estrogen receptor-mediated transcription (39). On the basis of the characterization of JGT presented here, an interesting possibility is that hmC present in the mammalian genome, generated by the TET enzymes, can be glucosylated by GREB1.
Glucosyltransferases that modify hmC have been described in other organisms, namely the β-glucosyltransferase present in T-even bacteriophages. T4 β-glucosyltransferase catalyzes the transfer of a glucose residue from UDP-glucose onto hmC, converting it to β-glycosyl-5-hydroxymethylcytosine (40, 41). The resulting hypermodified base prevents the phage DNA from being degraded by bacterial restriction enzymes (42, 43). T4 β-glucosyltransferase was the first GT x-ray crystal structure to be solved, in 1994, and is a founding member of the GT-B structural fold family of GTs (44). Despite the broad similarity in substrate, JGT is predicted to have a GT-A structural fold, suggesting that there is no ancestral kinship between these two enzymes. A better understanding of GT enzymes, including JGT, and their mechanism of action in vivo and in vitro is essential for rational drug design as well as increasing our knowledge regarding overall glycosylation machinery. The identification of a novel nuclearly localized GT in early-branching eukaryotes will provide a critical boost in this direction. Structure-function studies are underway to fully characterize the JGT enzyme. Of particular interest is clarifying the function of regions of JGT outside of the GT-A domain (see Fig. 1B).
Metazoan TET proteins serially oxidize 5-methylcytosine to hmC, 5-formylcytosine, and 5-carboxycytosine, which function as stable epigenetic marks or as potential intermediates in the DNA demethylation pathway (33, 34). In a recent study, we reported a reverse-phase HPLC coupled with tandem mass spectrometry (LC-MS/MS) method for the accurate detection of the nucleosides dJ and hmdU in T. brucei DNA.4 As shown here, this method has indicated that a low but detectable level of fdU is present along with hmdU in the genome of T. brucei. This provided the first indication that JBP enzymes may catalyze three similar, iterative catalytic/oxidation cycles of thymine in trypanosome DNA. The in vivo analysis of JGT function in T. brucei further supports this hypothesis. We have demonstrated previously that both JBP1 and JBP2 can stimulate de novo synthesis of hmU, which is subsequently converted to base J (9, 14, 24). Upon loss of JGT activity, we would expect de novo activities of JBP1/2 to modify the same sites as in WT cells but without having the hmU converted to J. Trypanosomatids lack DNA glycosylases (i.e. SMUG1) that recognize 5-hmU in DNA, and no activity against 5-hmU was detected in T. brucei extracts (35, 45). Therefore, even in the absence of any propagation activity of JBP1 (because of the loss of base J), we would expect an increased level of hmU in the JGT KO genome. However, we were able to observe the transient nature of thymine modifications because base J synthesis was inhibited in a time-dependent manner. During the time course of the RNAi ablation of JGT, the levels of hmU rise initially but then decrease concomitantly with an apparent small rise in fU levels. Although we have not ruled out the possibility that JGT somehow directly regulates JBP function, the results support the idea that, in the absence of JGT, sustained hmU accumulation is prevented by subsequent conversion to fU (and possibly 5-carboxyluracil) by JBP1/2.
In vitro studies of JBP1 function support the hypothesis that JBP enzymes act as TET enzymes by performing iterative hydroxylation reactions on modified substrates. Not only is JBP1 able to bind hmU-modified DNA (17), but detection of hmU formation by JBP1 in vitro peaks early during the reaction but then decreases over time.5 Although we acknowledge that further work is needed to conclusively demonstrate JBP-stimulated conversion of hmU to fU and 5-carboxyluracil, the data thus far allow us to propose a model where JBP and JGT compete for hmU substrate in the trypanosome genome for conversion to base J or additional base analogs (fU and 5-carboxyluracil).
The ability to genetically segment the base J synthesis pathway at the hydroxylation step and the glucosylation step provides the tools to specifically study these epigenetic markers. Interestingly, 5-formylcytosine and 5-carboxycytosine can significantly reduce the kinetics of yeast RNA polymerase II transcription, suggesting that these modifications play a role in splicing and termination (46). The coregulation of base J and hmU synthesis by JGT and JBP enzymes in trypanosomatids is intriguing because the studies demonstrating the role of base J in Pol II transcription were performed by halting hmU synthesis via altering JBP function (5–7). Future experiments utilizing the JGT KO cell line will allow us to address the specific role of hmU, fU, and base J in regulating Pol II kinetics and trypanosomatid gene expression.
Finally, the ability to delete JGT or both JBP thymidine hydroxylases from T. brucei is consistent with the non-essential nature of base J in this organism (4, 14, 15). Although deletion of either JBP1 or JBP2 in T. cruzi and JBP2 in L. tarentolae results in similar reductions in J levels as seen in the T. brucei mutants, attempts to delete both JBPs have been unsuccessful, leading to the idea that base J is essential in these organisms (5, 20, 47). However, chemical inhibition of JBP enzymes via a specific inhibitor (dimethyloxoglycine) reduces base J to extremely low levels without affecting cell growth (15). Therefore, it is possible that JBP1/2 have additional functions outside of base J synthesis, explaining the apparent essential nature of these genes. With the identity of JGT, we can now directly address this conundrum in Leishmania major and T. cruzi.
Acknowledgments
We thank Dr. Lance Wells and Peng Zhao for performing MS analyses of the purified recombinant JGT preparation. We also thank members of the Steve Hajduk laboratory for discussions, Tony Szempruch for help with microscopy, and Laura Cliffe and David Reynolds for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants 2R56AI063523-07A1 (to R. S.), R01 CA101864 (to Y. W.), and P01GM107012 and P4GM103490 (in support of spectrometry analysis). This work was also supported by the Georgia Research Alliance in support of mass spectrometry analysis.
S. Liu, D. Ji, L. Cliffe, R. Sabatini, and Y. Wang, submitted for publication.
L. Cliffe, J. Wang, Y. Wang, and R. Sabatini, unpublished observations.
- base J
- β-d-glucopyranosyloxymethyluracil
- Pol II
- polymerase II
- hmU
- 5-hydroxymethyluracil
- GT
- glucosyltransferase
- JGT
- base J-associated glucosyltransferase
- JBP
- base J-binding protein
- fU
- 5-formyluracil
- hmdU
- 5-hydroxymethyl-2′-deoxyuridine
- fdU
- 5-formyl-2′-deoxyuridine
- dJ
- 5-((β-d-glucopyranosyloxy)-methyl)-uridine
- hmC
- 5-hydroxymethylcytosine
- TET
- Ten-Eleven Translocation
- glc-hmdC
- β-d-glucopyranosyloxy-methyl-cytidine.
REFERENCES
- 1. Gommers-Ampt J. H., Van Leeuwen F., de Beer A. L., Vliegenthart J. F., Dizdaroglu M., Kowalak J. A., Crain P. F., Borst P. (1993) β-d-Glucosyl-hydroxymethyluracil: a novel modified base present in the DNA of the parasitic protozoan T. brucei. Cell 75, 1129–1136 [DOI] [PubMed] [Google Scholar]
- 2. van Leeuwen F., Taylor M. C., Mondragon A., Moreau H., Gibson W., Kieft R., Borst P. (1998) β-d-Glucosyl-hydroxymethyluracil is a conserved DNA modification in kinetoplastid protozoans and is abundant in their telomeres. Proc. Natl. Acad. Sci. 95, 2366–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Leeuwen F., de Kort M., van der Marel G. A., van Boom J. H., Borst P. (1998) The modified DNA base β-d-glucosyl-hydroxymethyluracil confers resistance to micrococcal nuclease and is incompletely recovered by 32P-postlabeling. Anal. Biochem. 258, 223–229 [DOI] [PubMed] [Google Scholar]
- 4. Cliffe L. J., Siegel T. N., Marshall M., Cross G. A., Sabatini R. (2010) Two thymidine hydroxylases differentially regulate the formation of glucosylated DNA at regions flanking polymerase II polycistronic transcription units throughout the genome of Trypanosoma brucei. Nucleic Acids Res. 38, 3923–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ekanayake D. K., Minning T., Weatherly B., Gunasekera K., Nilsson D., Tarleton R., Ochsenreiter T., Sabatini R. (2011) Epigenetic regulation of transcription and virulence in Trypanosoma cruzi by O-linked thymine glucosylation of DNA. Mol. Cell Biol. 31, 1690–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ekanayake D., Sabatini R. (2011) Epigenetic regulation of polymerase II transcription initiation in Trypanosoma cruzi: modulation of nucleosome abundance, histone modification, and polymerase occupancy by O-linked thymine DNA glucosylation. Eukaryot. Cell 10, 1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Luenen H. G., Farris C., Jan S., Genest P. A., Tripathi P., Velds A., Kerkhoven R. M., Nieuwland M., Haydock A., Ramasamy G., Vainio S., Heidebrecht T., Perrakis A., Pagie L., van Steensel B., Myler P. J., Borst P. (2012) Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional readthrough in Leishmania. Cell 150, 909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borst P., Sabatini R. (2008) Base J: discovery, biosynthesis, and possible functions. Annu. Rev. Microbiol. 62, 235–251 [DOI] [PubMed] [Google Scholar]
- 9. DiPaolo C., Kieft R., Cross M., Sabatini R. (2005) Regulation of trypanosome DNA glycosylation by a SWI2/SNF2-like protein. Mol. Cell 17, 441–451 [DOI] [PubMed] [Google Scholar]
- 10. Cross M., Kieft R., Sabatini R., Wilm M., de Kort M., van der Marel G. A., van Boom J. H., van Leeuwen F., Borst P. (1999) The modified base J is the target for a novel DNA-binding protein in kinetoplastid protozoans. EMBO J. 18, 6573–6581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu Z., Genest P. A., ter Riet B., Sweeney K., DiPaolo C., Kieft R., Christodoulou E., Perrakis A., Simmons J. M., Hausinger R. P., van Luenen H. G., Rigden D. J., Sabatini R., Borst P. (2007) The protein that binds to DNA base J in trypanosomatids has features of a thymidine hydroxylase. Nucleic Acids Res. 35, 2107–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iyer L. M., Tahiliani M., Rao A., Aravind L. (2009) Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 8, 1698–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cliffe L. J., Kieft R., Southern T., Birkeland S. R., Marshall M., Sweeney K., Sabatini R. (2009) JBP1 and JBP2 are two distinct thymidine hydroxylases involved in J biosynthesis in genomic DNA of African trypanosomes. Nucleic Acids Res. 37, 1452–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cliffe L. J., Hirsch G., Wang J., Ekanayake D., Bullard W., Hu M., Wang Y., Sabatini R. (2012) JBP1 and JBP2 proteins are Fe2+/2-oxoglutarate-dependent dioxygenases regulating hydroxylation of thymidine residues in trypanosome DNA. J. Biol. Chem. 287, 19886–19895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toaldo C. B., Kieft R., Dirks-Mulder A., Sabatini R., van Luenen H. G., Borst P. (2005) A minor fraction of base J in kinetoplastid nuclear DNA is bound by the J-binding protein 1. Mol. Biochem. Parasitol. 143, 111–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sabatini R., Meeuwenoord N., van Boom J. H., Borst P. (2002) Recognition of base J in duplex DNA by J-binding protein. J. Biol. Chem. 277, 958–966 [DOI] [PubMed] [Google Scholar]
- 18. Heidebrecht T., Christodoulou E., Chalmers M. J., Jan S., Ter Riet B., Grover R. K., Joosten R. P., Littler D., van Luenen H., Griffin P. R., Wentworth P., Jr., Borst P., Perrakis A. (2011) The structural basis for recognition of base J containing DNA by a novel DNA binding domain in JBP1. Nucleic Acids Res. 39, 5715–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kieft R., Brand V., Ekanayake D. K., Sweeney K., DiPaolo C., Reznikoff W. S., Sabatini R. (2007) JBP2, a SWI2/SNF2-like protein, regulates de novo telomeric DNA glycosylation in bloodstream form. Trypanosoma brucei. Mol. Biochem. Parasitol. 156, 24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vainio S., Genest P. A., ter Riet B., van Luenen H., Borst P. (2009) Evidence that J-binding protein 2 is a thymidine hydroxylase catalyzing the first step in the biosynthesis of DNA base. J. Mol. Biochem. Parasitol. 164, 157–161 [DOI] [PubMed] [Google Scholar]
- 21. Iyer L. M., Zhang D., Burroughs A. M., Aravind L. (2013) Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 41, 7635–7655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rudenko G., Blundell P. A., Taylor M. C., Kieft R., Borst P. (1994) VSG gene expression site control in insect form Trypanosoma brucei. EMBO J. 13, 5470–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Z., Morris J. C., Drew M. E., Englund P. T. (2000) Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 275, 40174–40179 [DOI] [PubMed] [Google Scholar]
- 24. van Leeuwen F., Wijsman E. R., Kieft R., van der Marel G. A., van Boom J. H., Borst P. (1997) Localization of the modified base J in telomeric VSG gene expression sites of Trypanosoma brucei. Genes Dev. 11, 3232–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J., Cao H., You C., Yuan B., Bahde R., Gupta S., Nishigori C., Niedernhofer L. J., Brooks P. J., Wang Y. (2012) Endogenous formation and repair of oxidatively induced G[8–5m]T intrastrand cross-link lesion. Nucleic Acids Res. 40, 7368–7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hong H., Cao H., Wang Y., Wang Y. (2006) Identification and quantification of a guanine-thymine intrastrand crosslink lesion induced by Cu(II)/H2O2/ascorbate. Chem. Res. Toxicol. 19, 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hong H., Wang Y. (2007) Derivatization with Girard reagent T combined with LC-MS/MS for the sensitive detection of 5-formyl-2′-deoxyuridine in cellular DNA. Anal. Chem. 79, 322–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 29. Liu J., Mushegian A. (2003) Three monopyletic superfamilies account for the majority of the known glycosyltransferases. Protein Sci. 12, 1418–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Breton C., Snajdrova L., Jeanneau C., Koca J., Imberty A. (2006) Structures and mechanisms of glycosyltransferases. Glycobiology 16, 29R–37R [DOI] [PubMed] [Google Scholar]
- 31. Keenleyside W. J., Clarke A. J., Whitfield C. (2001) Identification of residues involved in catalytic activity of the inverting glycosyl transferase WbbE from Salmonella enterica serovar Borreze. J. Bacteriol. 183, 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garinot-Schneider C., Lellouch A. C., Geremia R. A. (2000) Identification of essential amino acid residues in the Sinorhizobium meliloti glucosyltransferase ExoM. J. Biol. Chem. 275, 31407–31413 [DOI] [PubMed] [Google Scholar]
- 33. Wu H., Zhang Y. (2011) Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes and Dev. 25, 2436–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A, He C., Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carbosylcytosine. Science 333, 1300–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ulbert S., Cross M., Boorstein R. J., Teebor G. W., Borst P. (2002) Expression of the human DNA glycosylase hSMUG1 in Trypanosoma brucei causes DNA damage and interferes with J biosynthesis. Nucleic Acids Res. 30, 3919–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roseman S. (2001) Reflections on glycobiology. J. Biol. Chem. 276, 41527–41542 [DOI] [PubMed] [Google Scholar]
- 37. Liu M., Wang G., Gomez-Fernandez C. R., Guo S. (2012) GREB1 functions as a growth promoter and is modulated by IL6/STAT3 in breast cancer. PLoS One 7, e46410. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38. Ghosh M. G., Thompson D. A., Weigel R. J. (2000) PDZK1 and GREB1 are estrogen-regulated genes expressed in hormone-responsive breast cancer. Cancer Res. 60, 6367–6375 [PubMed] [Google Scholar]
- 39. Mohammed H., D'Santos C., Serandour A. A., Ali R., Brown G. D., Atkin A., Rueda O., Holmes K. A., Theodorou V., Robinson J., Zwart W., Saadi A., Ross-Innes C. S., Chin S. F., Menon S., Stingl J., Palmieri C., Caldas C., Carroll J. S. (2013) Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 3, 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Volkin E. (1954) The linkage of glucose in coliphage nucleic acids. J. Am. Chem. Soc. 76, 5892–5893 [Google Scholar]
- 41. Jesaitis M. A. (1956) Differences in the chemical composition of the phage nucleic acids. Nature 178, 637–641 [DOI] [PubMed] [Google Scholar]
- 42. Kornberg S. R., Zimmerman S. B., Kornberg A. (1961) Glucosylation of deoxyribonucleic acid by enzymes from bacteriophage- infected Escherichia coli. J. Biol. Chem. 236, 1487–1493 [PubMed] [Google Scholar]
- 43. Josse J., Kornberg A. (1962) Glucosylation of deoxyribonucleic acid. III: α- and β-glucosyltransferases from T4- infected Escherichia coli. J. Biol. Chem. 237, 1968–1976 [PubMed] [Google Scholar]
- 44. Vrielink A., Rüger W., Driessen H. P., Freemont P. S. (1994) Crystal structure of the DNA modifying enzyme β-glycosyltransferase in the presence and absence of the substrate uridine diphosphoglucose. EMBO J. 13, 3413–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ulbert S., Eide L., Seeberg E., Borst P. (2004) Base J, found in nuclear DNA of Trypanosoma brucei, is not a target for DNA glycosylases. DNA Repair 3, 145–154 [DOI] [PubMed] [Google Scholar]
- 46. Kellinger M. W., Song C. X., Chong J., Lu X. Y., He C., Wang D. (2012) 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 19, 831–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Genest PA., ter Riet B., Dumas C., Papadopoulou B., van Luenen H. G., Borst P. (2005) Formation of linear inverted repeat amplicons following targeting of an essential gene in Leishmania. Nucleic Acids Res. 33, 1699–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Der Wel H., Morris H. R., Panico M., Paxton T., Dell A., Kaplan L., West C. M. (2002) Molecular cloning and expression of a UDP-N-acetylglucosamine (GlcNAc):hydroxyproline polypeptide GlcNAc-transferase that modifies Skp1 in the cytoplasm of Dictyostelium. J. Biol. Chem. 277, 46328–46337 [DOI] [PubMed] [Google Scholar]