

Background: Heparan sulfate (HS) plays a role in renal fibrosis.
Results: 6-O-sulfation of HS was increased after kidney transplantation, leading to the sequestration and activation of FGF2.
Conclusion: Increased 6-O-sulfation of HS regulates the fibrotic response associated with chronic renal allograft failure.
Significance: Blockade of the interaction between FGF2 and 6-O-sulfated HS might mitigate the development of renal fibrosis.
Keywords: Fibroblast Growth Factor (FGF), Fibrosis, Heparan Sulfate, Sulfotransferase, Transplantation
Abstract
Heparan sulfate (HS) plays a crucial role in the fibrosis associated with chronic allograft dysfunction by binding and presenting cytokines and growth factors to their receptors. These interactions critically depend on the distribution of 6-O-sulfated glucosamine residues, which is generated by glucosaminyl-6-O-sulfotransferases (HS6STs) and selectively removed by cell surface HS-6-O-endosulfatases (SULFs). Using human renal allografts we found increased expression of 6-O-sulfated HS domains in tubular epithelial cells during chronic rejection as compared with the controls. Stimulation of renal epithelial cells with TGF-β induced SULF2 expression. To examine the role of 6-O-sulfated HS in the development of fibrosis, we generated stable HS6ST1 and SULF2 overexpressing renal epithelial cells. Compared with mock transfectants, the HS6ST1 transfectants showed significantly increased binding of FGF2 (p = 0.0086) and pERK activation. HS6ST1 transfectants displayed a relative increase in mono-6-O-sulfated disaccharides accompanied by a decrease in iduronic acid 2-O-sulfated disaccharide structures. In contrast, SULF2 transfectants showed significantly reduced FGF2 binding and phosphorylation of ERK. Structural analysis of HS showed about 40% down-regulation in 6-O-sulfation with a parallel increase in iduronic acid mono-2-O-sulfated disaccharides. To assess the relevance of these data in vivo we established a murine model of fibrosis (unilateral ureteric obstruction (UUO)). HS-specific phage display antibodies (HS3A8 and RB4EA12) showed significant increase in 6-O-sulfation in fibrotic kidney compared with the control. These results suggest an important role of 6-O-sulfation in the pathogenesis of fibrosis associated with chronic rejection.
Introduction
Kidney transplantation is the optimal treatment for patients with end stage renal disease. Due to better immunosuppressive drugs, transplant loss to acute rejection is now rare. However, after the first year, ∼5% of grafts are lost each year. The number and severity of immune-mediated acute allograft rejection episodes are major risk factors for the development of chronic allograft dysfunction characterized by interstitial fibrosis and tubular atrophy (IFTA), which can lead to graft loss (1).
Recruitment of immune cells guided into the grafts by proinflammatory cytokines is considered as a definitive feature of rejection. The sequestration of growth factors/cytokines by heparan sulfate (HS)3 molecules within the extracellular matrix is thought to be essential for these molecules to be fully functional. HS is the glycosaminoglycan moiety of proteoglycans, which are associated with the cell surface, basement membrane, and extracellular matrix. HS chains are synthesized in the Golgi apparatus and consist of repeating disaccharide units of uronic acid (iduronic or glucuronic acid) linked to N-acetylglucosamine (GlcNAc).
During synthesis the HS chains are selectively sulfated at the N, 6-O, and 3-O positions of the glucosamine (GlcNS) and at the 2-O position of the uronic acid residue by HS sulfotransferases and modified by C5-epimerization of glucuronic acid (GlcA) to iduronic acid (IdoA). The following enzymes are involved in the modification: four glucosaminyl N-deacetylase/N-sulfotransferases, three glucosaminyl 6-O-sulfotransferases (HS6STs), and seven glucosaminyl HS3STs (glucosaminyl 3-O-sulfotransferase) but only one GlcA C-5 epimerase and one hexuronic acid HS2ST (glucosaminyl 2-O-sulfotransferase) (2). The modification reactions are generally incomplete, such that the resultant polysaccharides vary in structure in a tissue-specific manner. It is believed that the fine structure of HS is determined through a regulated expression of the modifying enzymes.
The interaction between many of these growth factors/cytokines and HS is critically dependent on the amount and position of the O-sulfate groups, in particular 6-O-sulfated glucosamine residue that forms binding sites for proteins (3). The pattern of 6-O-sulfation is generated during HS biosynthesis by the three HS6STs whose expression patterns are dynamically regulated during embryonic development (4), aging, and carcinogenesis (5, 6). The sulfation pattern of the mature HS chains may be further modified at the cell surface by HS 6-O-endosulfatases (SULF1 and -2), that specifically remove 6-O sulfate groups (7). Dysregulated expression of SULF1 and -2 can influence the activity of HS binding growth factors such as vascular endothelial growth factor and fibroblast growth factor (FGF) (8). FGF signal transduction is dependent on the sulfation at the 2-O and 6-O positions of HS chains (9). The 2-O sulfation is required for FGF2 binding to heparin, and 6-O-sulfation is required for FGF2-dependent dimerization and activation of the FGFR1 receptor as revealed by both biochemical and the crystal structure studies of FGF-FGFR1-heparin ternary complexes (9, 10).
Expression of N-sulfated HS is specifically increased during acute rejection in peritubular capillary vessels, and the CCL5 binding basement membrane of tubular epithelial cells was shown to co-localize with HS expression (11). In chronic vascular allograft rejection, altered HSPG expression characterized by low sulfated HS was shown to be present in fibrotic and sclerotic lesions in blood vessels, interstitium, and mesangium (12, 13). Increased levels of extracellular matrix molecules including collagen XVIII and perlecan are also reported (14). Although the expression and tissue distribution of HS in healthy kidney has been investigated, little is known about the abundance and localization of various HS structural motifs during chronic renal allograft rejection.
This study was designed to examine the expression of sulfated HS during renal chronic allograft rejection. A series of experiments was then performed to define the mechanism by which 6-O-sulfation modifies FGF binding and activity using renal epithelial cells. Finally, relevance of HS 6-O-sulfation was examined in vivo in a murine model of fibrosis.
MATERIALS AND METHODS
Human Tissue
Normal human kidney (n = 5) and allograft biopsies (n = 10) were obtained as formalin-fixed paraffin-embedded blocks from the local transplant tissue archive in accordance with the Newcastle and North Tyneside Local Research Ethics Committee. Samples were anonymous and coded before they were made available for the study. Donor kidneys that were anatomically unsuitable for transplantation provided normal control tissue.
Immunofluorescence
Paraffin-embedded formalin-fixed tissue sections were dewaxed. Antigen retrieval for staining with HS3A8 antibody was carried with trypsin/calcium chloride solution, pH 7.8, for 12 min, whereas antigen retrieval for FGF-2 was carried out in pressure cooker for 1 min in citrate buffer, pH 6. Heparan sulfate was detected with anti-VSV-tagged phage display antibody HS3A8 kindly provided by Dr. Van Kuppevelt, University of Nijmegen, The Netherlands (Table 1). The staining was visualized with rabbit anti-VSV TRITC-conjugated antibody (1:200, Sigma). To visualize the nuclei, DAPI solution (Sigma, 2 μg/ml) was used. Slides were mounted in fluorescence mounting medium (DakoCytomation, DAKO). Controls consisted of an unstained population of cells and cells stained with the secondary antibody only.
TABLE 1.
Epitopes of heparan sulfate antibodies
N-Acel, N-acetylated; N-S, N-sulfated; 2-OS, 2-O-sulfated; 6-OS, 6-O-sulfated; C-5, sulfated iduronic acid; 3-OS, 3-O-sulfated; GAG, glycosaminoglycan.
A mouse on mouse kit (Vector) was used for staining murine tissue (unilateral ureteral obstruction (UUO) model) with murine antibodies. Frozen sections were fixed with cold acetone for 10 min followed by blocking with avidin-biotin each for 15 min. Sections were incubated with HS3A8 and RB4EA12 antibodies at a 1:20 dilution overnight at 4C. Staining was visualized as described above. However, staining with collagen-1 antibody (rabbit anti-mouse, Millipore) was detected with biotin-conjugated goat anti-rabbit Ig (Vector Laboratories) followed by the addition of avidin-biotin peroxidase (Vector Laboratories). Color development was performed using 3,3′-diaminobenzidine (brown).
Immunofluorescent samples were analyzed with the Leica TCS-SP2UV confocal laser scanning microscope (Leica Lasertechnik). The excitation and emission wavelengths were 488 and 530 nm for fluorescein isothiocyanate (FITC), 543 and 580 nm for TRITC, and 358 and 461 nm for DAPI. Quantitative information of the renal sections analyzed was obtained using Image J software applying histogram analysis as described earlier (44). Mean fluorescence was calculated by taking all fluorescent readings into account that were between 1 (threshold set manually) and 255 (maximum fluorescence).
Generation of Stable Transfectants
Stable transfectants were generated after transfection of renal epithelial cells line HK2 and HKC8 with pcDNA3.1/Zeo encoding HS6ST1 and pcDNA3.1/Myc-his-encoding human SULF2, respectively. Individual colonies were expanded and selected with either zeocin (400 μg/ml) or G418 (800 μg/ml). Expression of the gene was verified by real time PCR using GAPDH as a reference gene. Mock cells transfected with empty vector were used as control. All results are representative of data on two independent clones of the transfectants.
Quantitative Real-time PCR
RNA was isolated from immortalized human renal TEC cells (HK-2 and HKC8; American Type Culture Collection, Manassas, VA) using TRIzol reagent (Sigma) and transcribed to cDNA using Superscript II (Invitrogen). For some experiments cells were stimulated with cytokines for 24 h before RNA isolation.
Primers and probe as well as the PCR mastermix were obtained from Applied Biosystems. Primers were exon spanning, and the probe contained the 5′-fluorescence probe 6-carboxyfluorescein and the 3′-quencher 6-carboxytetramethylrhodamine. Comparative Ct method (ΔΔCT) for relative quantitation of gene expression was used. The level of expression of the gene of interest was normalized by including an endogenous control, such as GAPDH, and the reaction was performed in an ABI Prism 7000 (Applied Biosystems). For the analysis of the transfectants, the ΔCT of the negative control (mock-transfected cells) was subtracted from the ΔCT of the transfectants, known as ΔΔCT. The control was set to 1, and the other samples were normalized to the control. Results were analyzed using REST 2008 software.
Staining of Cell Surface Antigens by FACS
Adherent cells were detached using non-enzymatic cell dissociation media (Sigma). Cells in suspension were incubated with primary antibodies in 2% BSA/PBS for 45 min. After the washing steps, fluorescently labeled secondary antibodies were added and incubated for further 30 min. The phage display antibodies were visualized with anti-VSV cy3-conjugated antibody (1:200, Sigma). Cells were resuspended in 200 μl of 2% BSA/PBS and analyzed using appropriate laser and filter settings (488 nm emission, 530 nm extinction for FITC). All flow cytometry data are expressed as mean fluorescence intensity.
FGF2 Binding Assay
A Fluorokine FGF2 flow cytometry kit was used in this study (R&D Systems). In brief, cells were detached using cell dissociation media, and after 2 washes in PBS, 100,000 cells were mixed with FGF2-biotin at a final concentration of 570 ng/ml and incubated for 60 min at 4 °C. Avidin-FITC was added to the cells and incubated for 30 min at 4 °C; after two additional washes cells were resuspended in 200 μl of PBS and analyzed by flow cytometry. As a negative control, cells were incubated with an unrelated biotinylated soybean protein. FGF2 binding was analyzed with LSRII flow cytometer.
Western Blotting Analysis
20 μg of the prepared protein lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (5% stacking gel and 10% running gel). The proteins were transferred onto a Hybond-PVDF membrane (GE Healthcare). The membrane was subsequently incubated with primary anti-phosphospecific ERK (1:1000, Millipore) overnight; after washing, it was incubated with secondary HRP-conjugated antibody (1:5000). Bands were detected using enhanced chemiluminescence (Thermo Scientific). The immunoblots were stripped and re-probed with tubulin (Sigma) to demonstrate fidelity of loading.
Heparan Sulfate Structure Analysis
Renal epithelial cells transfected with HS6ST1 or SULF2, and appropriate mock transfectants were labeled with [35S]sulfate to determine the structure changes of HS. The cells were grown in 25-cm2 flasks until subconfluent, and 5 ml of fresh medium containing 200 μCi/ml Na235SO4 (PerkinElmer Life Sciences) was added at 37 °C for 24 h. The cells were washed twice with cold PBS and treated with 3 ml of solubilization buffer (1% Triton X-100, 50 mm Tris, 0.15 m NaCl, pH 7.4, and protease inhibitor (Complete Mini Mixture, Roche Applied Science)) at 4 °C for 60 min in a shaking incubator. Lysates were centrifuged at 500 × g for 15 min, and the supernatant was used for purification of labeled glycosaminoglycans.
Glycosaminoglycans were isolated by treating supernatants from 35S-labeled lysates with diethylaminoethyl (DEAE) Sephacel (Sigma). After desalting by gel chromatography using PD-10 columns (Sephadex G-25, Amersham Biosciences) in 10% ethanol, the polysaccharide chains were released from the core protein by treatment with 0.5 m NaOH overnight at 4 °C. After completing β-elimination, the samples were neutralized with 4 m HCl and desalted by gel chromatography using PD-10 columns eluted with H2O followed by lyophilization. HS chains were recovered after gel chromatography on a column (1 × 180 cm) of Sephadex G-25 superfine in 0.2 m NH4HCO3 and desalted by lyophilization.
The samples were depolymerized with nitrous acid at pH 1.5 (which cleaves the glucosaminidic linkage at GlcNS units) followed by reduction with NaBH4. Disaccharides were isolated and analyzed as described (15).
Mouse UUO
Wild type C57BL/6N male inbred mice (Charles River Laboratories) age 8–10 weeks were used in accordance with the UK Home Office Animals (Scientific Procedures) Act of 1986. Mice received water and standard mouse chow. These mice were subjected to an abdominal incision where the left ureter was completely double-ligated under general anesthesia with isoflurane and oxygen. The right (unoperated) kidney served as the control. Animals were killed after 7 days of obstruction, and both kidneys were removed and immediately frozen in OCT embedding compound in liquid-nitrogen cooled isopentane.
Statistical Analysis
Student's t test was used to compare data that followed the Gaussian distribution. A one-way analysis of variance with Tukey's post test was used to compare 3 or more independent groups, and differences with p < 0.05 were considered to be significant. Error bars represent the S.E. of the mean.
RESULTS
Expression of Heparan Sulfate Domains during Renal Allograft Rejection
Immunofluorescence laser scanning confocal microscopy allowed precise localization and semiquantitative comparison of the abundance of distinct HS domains within normal renal tissue and a range of sections taken from rejection biopsies. The reactivity of HS3A8 phage display antibody, which recognizes highly sulfated domains containing iduronic acid and 6-O and 2-O-sulfated structures within HS chains (16), was examined within the renal tissue. Most prominent staining was found for HS associated with blood vessel walls (peritubular capillaries) and tubular basement membrane and to a lesser extent within the interstitium and glomerulus. Fig. 1A shows increased expression of the HS3A8 epitope during acute rejection. Interestingly, however, an even more prominent increase was observed in the tubular basement membrane and interstitium in tissues with features of interstitial fibrosis/tubular atrophy (p < 0.05). The mean fluorescence intensity of HS3A8 staining was significantly increased during chronic rejection (Fig. 1B). Human kidney showing normal histology expressed low levels of FGF2. However, FGF2 had widespread and increased distribution within the tubular epithelium during chronic rejection (Fig. 1C).
FIGURE 1.

Changes in HS3A8 epitope and FGF expression during renal rejection. A, immunohistochemistry of HS in kidney sections. Heavily sulfated HS was visualized with HS3A8 antibody and a Cy3-labeled secondary antibody. DAPI was used for nuclear counterstain. Kidney sections are representative for the indicated stages of rejection (acute rejection 1a, acute rejection 2a, chronic rejection) with normal tissue as control. The bottom two panels show representative stainings with FGF2 antibody and visualized with FITC-conjugated secondary antibody. B, quantification of the mean fluorescence intensity associated with the expression of HS3A8 epitope during various stages of renal rejection. C, quantification of the mean fluorescence intensity associated with the expression of FGF of normal (clear bar) and chronically rejected kidneys (filled). Five random fields per biopsy were analyzed using Image-J software. Results represent data from 15 patients in total (5 healthy, 3 acute rejection 1a, 2 acute rejection 2a, and 5 chronic rejection).
Role of 6-O-Sulfation in Fibrosis
It is known that TGF-β promotes renal fibrosis (17), but the mechanisms that regulate profibrotic genes remain unclear. We hypothesized that stimulation of renal epithelial cells by TGF-β may mediate fibrosis by modulating the expression of HS modifying enzymes.
Real-time PCR demonstrated a significant up-regulation of SULF2 (4.9-fold) 24 h after stimulation with TGF-β (Fig. 2). Stimulation with other factors e.g. phorbol 12-myristate 13-acetate, IL-17, and IFN-γ, did not significantly change the expression of SULF2. Analysis of the expression levels of HS6ST isoforms 1, 2, and 3 in renal epithelial cells revealed constitutive expression of HS6ST1 only (data not shown). No significant changes in expression levels of SULF1, heparanase, and HS6ST1 were observed after cytokine stimulation (phorbol 12-myristate 13-acetate, TNF-α, IFN-γ, and TGF-β; Fig. 2). To understand the biological significance of 6-O-sulfation during fibrosis, we generated HS6ST1- and SULF2-overexpressing renal epithelial cells.
FIGURE 2.

Gene expression levels of HS modifying enzymes after cytokine stimulation. Renal epithelial cells were incubated with IFN-γ (25 ng/ml), TGF-β (10 ng/ml), IL-17 (100 ng/ml), and phorbol 12-myristate 13-acetate (PMA; 50 ng/ml) for 24 h, and RNA was extracted and converted to cDNA, which was used in quantitative PCR. Relative transcript abundance was normalized to endogenous control GAPDH Three independent experiments (n = 3) were performed in duplicate (n = 2).
Pattern of HS Sulfation by HS6ST1 and SULF2 Transfectants
Overexpression of HS6ST1 and SULF2 was verified by quantitative real time-PCR. Approximately 10 individual clones were analyzed, and transfectants T7 (HS6ST1) and S11 (SULF2) expressing a significantly high level of respective genes were chosen for further studies (data not shown).
Phage display antibodies were used for investigation of HS structure after transfection. RB4EA12, HS3A8, and HS4C3 have been shown to target 6-O sulfate groups within differently sulfated HS structures (Table 1). HS6ST1 overexpression resulted in increased binding of RB4EA12, HS3A8, and HS4C3 to the overexpressing cells as compared with mock transfectants (Fig. 3A). In contrast, these antibodies showed significantly reduced binding to SULF2 transfectants, ∼50% lower binding compared with the mock transfected cells (p < 0.01) (Fig. 3B).
FIGURE 3.

HS-specific phage display antibody binding to stable transfectants. Mock and HS6ST1/SULF2 transfectants (A and B, respectively) were incubated on ice with HS3A8, HS4C3, or RB4EA12 antibody at 1:10 dilution followed by Cy3-labeled anti-VSV secondary antibodies. Fluorescence was measured by flow cytometry, and experiments were performed in duplicate (n = 2 and n = 3, *, p < 0.05; **, p < 0.01).
To determine the role of increased or reduced 6-O-sulfation on the organization of N-sulfated domains, the transfectants were stained with the 10E4 antibody. The ability of 10E4 to bind cell surface HS was significantly reduced in HS6ST1 transfectants, whereas SULF2 transfectants showed increased binding ability (Fig. 4). Specificity of this interaction was verified by using isotype control.
FIGURE 4.

Changes in 10E4 epitopes in stable transfectants. A, flow cytometric analysis of 10E4 antibody binding to HS6ST1 transfected cells (black), mock transfectants (gray), and isotype incubated cells (shaded gray). FL1-H, fluorescence 1-height. B, data were quantified using mean fluorescence intensity measured by flow cytometry, and statistics were performed using t tests for unpaired values. C, immunofluorescence staining of SULF2 transfectants with 10E4 antibody. Cells were grown on chamber slides and stained with 10E4 antibody, and expression was visualized by staining with a FITC-conjugated secondary antibody followed by scanning laser confocal microscopy. D, SULF2 transfectant binding to 10E4 antibody was analyzed using Image J software and Prism 4. (**, p < 0.01; ***, p < 0.001). The results are representative of three independent experiments.
Role of 6-O-Sulfation on FGF2 Binding
Next, we examined the effects of 6-O-sulfation on FGF2 binding. HS6ST1 and mock transfectants were incubated with biotinylated FGF2. Mean fluorescent intensity values for transfectants were approximately two times higher than that of the mock transfectants (p < 0.05, Fig. 5A). In contrast, median fluorescence intensity values for SULF2 transfectants were significantly reduced compared with the mock transfectants (p < 0.05, Fig. 5B). Specificity of the test was verified using soya bean protein (non-HS binding) biotinylated to the same level as FGF2 and blocking antibodies against FGF2. Heparitinase III treatment of the HS6ST1 transfectants significantly decreased FGF2 binding (∼75%, p < 0.05; Fig. 5C).
FIGURE 5.

Stable transfectants interaction with FGF2. Representative histograms of flow cytometry of FGF2 binding to HS6ST1 (A) and SULF2 (B) transfectants. Mock, HS6ST1, and SULF2 transfectants were incubated with biotinylated FGF2 (570 ng/ml) for 60 min at 4 °C. Binding was visualized by adding FITC-avidin and analyzed by flow cytometry. The specificity of binding was verified by incubating cells with unrelated biotinylated soya bean protein (negative control) and an FGF2 blocking antibody. The shaded gray histogram represents control cells with avidin-FITC conjugated substrate; the gray histogram represents mock transfectants, whereas black histograms represent HS6ST1 (A) and SULF2 (B) transfectants. The interaction was investigated by flow cytometry and expressed as mean fluorescence intensity. *, p < 0.05, n = 3 error bars represent the S.E. C, FGF2 binding after incubation with heparitinase III. Mock (filled bar) and HS6ST1 transfectants (patterned bar) were incubated with heparitinase III (10 units/ml) for 60 min at 37 C. Cells were subsequently incubated with biotinylated FGF2. Controls include untreated cells. D, FGF2 binding in the presence of various HS derivatives (500 μg/ml), which were added to Mock and HS6ST1 transfectants cells in the presence of biotinylated FGF2.(n = 3). FL1-H, fluorescence 1-height.
FGF2 binding was further examined in the presence of competitors such as heparin, HS, and de-N- and de-O-sulfated heparins (lacking sulfate groups at the N- and O- position, respectively). Incubation of FGF2 with de-N- or de-O-sulfated heparins produced no significant change in the median fluorescence intensity values for HS6ST1 transfectants compared with the control. In contrast, the addition of soluble HS or heparin inhibited FGF2 binding to both HS6ST1 transfectants and control (Fig. 5).
HS Structural Analysis
To investigate the changes in HS sulfation after overexpression of HS6ST1 and SULF2, cells were metabolically labeled with [35S]sulfate, and the fine structure of HS was determined after cleavage of HS into mono-O- and di-O-sulfated disaccharides.
Disaccharide analysis of N-sulfated HS sequences of the HS6ST1-overexpressing cells revealed a significant increase in HS 6-O-sulfation. The levels of each of the mono-O-sulfated disaccharides, GlcA-GlcNS6S (GMS) and IdoA-GlcNS6S (IMS), increased from ∼12% in control HS to ∼22% in the HS6ST1 transfectant, whereas IdoA2S-GlcNS and IdoA2S-GlcNS6S (ISM and ISMS, respectively) decreased (Fig. 6A).
FIGURE 6.
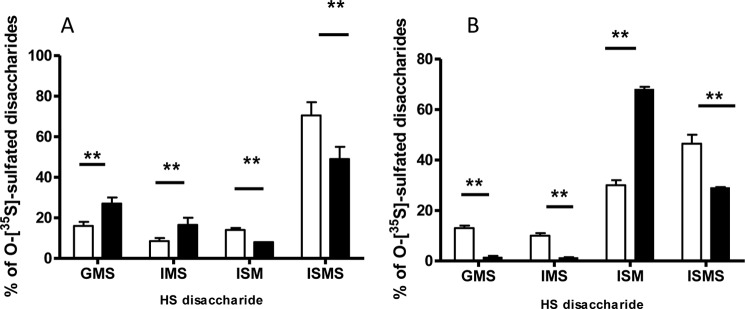
HS disaccharide composition in HS6ST1 and SULF2 transfectants. Mock, HS6ST1 (A), and SULF2 (B) transfectants were incubated with 200 μCi/ml [35S]sulfate for 24 h, and disaccharides, isolated as described under “Materials and Methods,” were separated using a strong anion-exchange HPLC Partisil 10 SAX column. GMS, IMS, ISM, and ISMS correspond to GlcA-GlcNS6S, IdoA-GlcNS6S, IdoA2S-GlcNS, and IdoA2S-GlcNS6S, respectively, in the intact HS chain. Clear bars represent mock cells, whereas black bars represent HS6ST1 or SULF2 transfectants.
The Sulf enzymes are known to preferentially release 6-O-sulfate groups from the trisulfated -IdoA2S-GlcNS6S- (ISMS) units with less pronounced activity on -IdoA-GlcNS6S- sequences and -GlcA-GlcNS6S- units. Interestingly, SULF2 transfectants removed 6-O sulfate groups not only from the highly sulfated disaccharides IdoA2S-GlcNS6S but also from mono-O-sulfated disaccharides of GlcA-GlcNS6S and IdoA-GlcNS6S units, whereas IdoA2S-GlcNS (ISM) increased reciprocally from 30% in control to 68% in the SULF transfectants (Fig. 6B).
Role of 6-O-Sulfation on FGF2 Signaling
To examine the potential role of HS6ST1 overexpression on cell proliferation, a series of experiments was set up with mock and HS6ST1 transfectants. HS6ST1 transfectants had a significantly higher proliferation by days 6 and 7 compared with the mock transfectants (Fig. 7A).
FIGURE 7.

Effect of HS6ST1 and SULF2 transfectants on FGF2 signaling. A, representative figure showing proliferation data for HS6ST1 transfectants (T-7). 50,000 cells were seeded in triplicate in 6-well plates with complete media. B, representative figure showing proliferation data for SULF2 transfectants (S-11). 100,000 cells were seeded in triplicate in 6-well plates with complete media. Viable cells (using trypan blue) were counted with Neubauer hemocytometer at the indicated time points (n = 3). Shown is a Western blot with anti-phospho ERK1/2 or α-tubulin of HS6ST1 (C) and SULF2 (D) after induction with 10 ng/ml FGF2 for 0 (3), 5, and 10 min. Quantitation by densitometry of pERK1/2 bands normalized to α-tubulin of HS6ST1 (E) and SULF2 (F) transfectants. Clear bars represent mock transfectants, whereas filled bars represent HS6ST1 or SULF2 transfectants. The analysis were performed using Image J and Prism 4 (n = 3; *, p < 0.05; ***, p < 0.001).
We further assessed whether the increased FGF2 binding by the HS6ST1 transfectants exerted its effect via activation of the ERK pathway. HS6ST1 overexpression resulted in a significant increase in the basal level of ERK activation compared with mock transfectants (Fig. 7C). Stimulation with FGF2 produced an increase in pERK activation in mock transfectants that increased with time; however, there was no significant change in the level of pERK in HS6ST1 transfectants. These results suggest that HS6ST1 overexpression increases basal FGF2-FGFR signaling by sequestering the endogenous FGF2 on low affinity HS receptors and facilitating the binding to high affinity receptors. Furthermore, perhaps these cells have reached their maximal level of activation so no further effect is seen after the addition of exogenous FGF2.
Earlier studies have shown that the SULF1or SULF2 inactivation results in increased FGF2-mediated signaling (18, 19). In agreement with published data we found that stimulation of SULF2 transfectants with FGF2 resulted in reduced ERK activation (at 3, 5, and 10 min, respectively) (Fig. 7D). Furthermore, SULF2 transfectants show significantly reduced proliferation compared with the mock transfectants (day 5, p = 0.002; day 6, p = 0.0052; day 7, p = 0.0014; Fig. 7B). These data are consistent with the reduced ERK activation observed.
Role of HS 6-O-Sulfation in Murine Model of Fibrosis
The murine model of UUO was used to examine the role of 6-O-sulfation in vivo. Fibrosis is usually accompanied by pathological changes including deposition of collagen. To verify the incidence of fibrosis after UUO, day 7 kidneys were stained; increased collagen deposition was seen in the basement membrane, and interstitial spaces of the operated kidneys were compared with the unoperated control kidneys. Furthermore, UUO sections showed that the HS3A8 antibody bound predominantly to tubular basement membrane and interstitial structures. Additionally, RB4EA12 antibody strongly stained the cortical tubules in the kidney and to a lesser extent interstitium and glomerulus (Fig. 8A). The changes in HS sulfation in UUO kidneys were shown to be statistically significant (p < 0.01) by measuring mean fluorescence intensity of selective tubular and interstitial structures (Fig. 8B).
FIGURE 8.

Changes in HS sulfation in murine model of fibrosis. UUO mice were generated. On day 7 the animals were sacrificed, and the kidneys were retrieved. A, frozen section of UUO and unoperated control kidneys were stained with antibodies against collagen-I and HS (HS3A8 and RB4EA12), respectively. Sections stained with collagen antibody (1:50) were visualized by incubating with biotinylated secondary antibody, and color was developed with 3,3-diaminobenzidine solution (brown). HS sulfation was examined using phage display antibodies HS3A8 and RB4EA12 followed by staining with Cy3-conjugated anti-VSV antibody. B, quantitative analysis was performed by measuring the density of tubular and interstitial areas randomly by Image J software (3–5 areas/kidney). Three animals were investigated. ***, p < 0.001; **, p < 0.01.
DISCUSSION
To explore the relationship between HS-6-O-sulfation and the sequestration of growth factors in vivo, a series of normal kidney and renal allograft biopsy sections was examined. Immunofluorescence allowed precise localization and semiquantitative comparison of the abundance of the HS3A8 epitope that recognizes highly sulfated domains within HS. Immunofluorescence labeling suggested that the HS3A8 epitopes were more abundant during chronic rejection compared with acute rejection. HS3A8 epitope was predominantly localized within basolateral region of the renal tubules and was also concentrated in the tubular basement membrane, where it is known that HS is a major component of the normal tubular basement membrane (20). Increased production of HS core proteins perlecan in glomerular basement membranes and collagen XVIII in cortical tubular basement membrane has already been demonstrated in a model of experimental renal transplantation (13). Further changes in HS-modifying enzymes were obtained after stimulation of human epithelial kidney cells with proinflammatory cytokines. TGF-β-induced up-regulation of SULF2 expression suggests its role during chronic rejection. Earlier reports showed that stimulation with TGF-β could also induce the expression of SULF1 in lung fibroblasts in vitro and in vivo; this enzyme has similar function as SULF2 with different tissue expression and substrate specificity (21).
To understand the biological significance of 6-O-sulfation during fibrosis, we generated HS6ST1- and SULF2-overexpressing renal epithelial cells. The 6-O-sulfate plays a significant role in the binding of a variety of cytokines and growth factors to HS (22, 23). HS6ST1 is a highly expressed isoform of the HS6ST enzyme (24, 25). HS6ST1 transfectants exhibited a significantly increased ability to bind phage display antibodies of HS3A8, HS4C3, and RB4EA12 compared with mock transfectants. These antibodies target HS epitopes consisting of trisulfated disaccharides, IdoA2S-GlcNS6S (16, 26), GlcNS3S6S, and IdoA-GlcNS6S, respectively, (Table 1). In contrast, SULF2 transfection significantly reduced HS3A8, HS4C3, and RB4EA12 antibody binding. SULF knock-out mice show increased expression of 6-O-sulfate groups and simultaneous increased binding of RB4EA12 antibody (27). Interestingly, increased binding of HS4C3 and RB4EA12 has been noticed in human chronic liver disease (28). These observations strongly suggest a possible role for HS 6-O-sulfation in the etiology and development of chronic diseases.
Furthermore, the increase in 10E4 epitopes in SULF2 transfectants and the decrease in HS6ST1 explains additional and non-substrate-specific changes in HS sulfation after the overexpression of these enzymes. Previous studies demonstrate similar changes in 10E4 epitopes accompanied by reduced binding to HS3A8 and HS4C3 in glomerular endothelial cells after stimulation by TNF-α (29). This is further supported by data showing TGF-β-induced overexpression of SULF1 causes changes in HS sulfation, including a reduction in the 6-O-sulfation and an increase in the N and 2-sulfate groups in the human lung fibroblasts (21).
FGF2 plays a key role in the pathology of renal fibrosis and epithelial mesenchymal transformation (30). This factor stimulates the proliferation of fibroblasts and mesangial cells and induces the secretion of TGF-β, which stimulates the epithelial mesenchymal transformation process (31). Overexpression of HS6ST1 significantly increases the binding of FGF2 to renal epithelial cells. Despite several studies on the heparin/HS binding motif for FGF2, no unique binding sequence has been described (3). The location of N- and 2-O-sulfate groups along with the chain length and overall O-sulfation seems more important for HS interaction than the specific distribution of sulfate groups (32). However, others have reported that the functional activation of FGF signaling demonstrates a higher degree of selectivity than just ligand or receptor binding (33). Nevertheless, SULF2 transfectants demonstrated reduced binding of FGF2, which indicates fundamental changes in the HS-FGF2 binding motif after the removal of 6-O sulfate group. In conclusion, this result supports the finding that HS-FGF2 binding does not depend completely on any single unique sequence but on the combination of several components (34).
Stimulation of HS6ST1 transfectants with FGF2 did not have any additive effect on ERK activation. It can be hypothesized that increased HS-6-O-sulfation may result in enhanced binding of endogenous growth factors to low affinity HS receptors, thus facilitating the binding and signaling through FGFR. This is consistent with the increased proliferation ratio observed with the HS6ST1 transfectants compared with the mock cells in the absence of exogenous FGF. Removal of 6-O sulfate groups is expected to affect FGF-FGFR signaling and, consequently, ERK activation (35, 36). Similarly, SULF2 transfectants showed a reduced proliferation rate compared with mock transfectants (data not shown). Previous work has also demonstrated that 6-O-endosulfatase enzymes have a negative effect on proliferation and mitogenic activity (35, 37).
Overexpression of HS6ST1 had a significant effect on HS disaccharide structure and distribution. This included increased mono-O-sulfated disaccharides (GlcA-GlcNS6S and IdoA-Glc-NS6S) and a corresponding decrease in IdoA2S-GlcNS and IdoA2S-GlcNS6S. Similar changes in 6-O-sulfation have been observed after HS6ST overexpression in human kidney 293 cells 38). Based on 10E4 staining, the changes observed in HS structure also included an increase in N-acetylated disaccharide/N-sulfated disaccharide domains after HS6ST1 overexpression. Interestingly, no significant change in the overall 6-O-sulfation was found. This may be related to the endogenous high level of sulfated HS produced by wild-type kidney epithelial cells; hence, the additive effect is not apparent.
Overexpression of SULF2 resulted in removal of 6-O-sulfate groups from trisulfated disaccharides, which is consistent with previous literature (7, 39). Interestingly, SULF2 overexpression had a significant effect not only on 6-O-sulfation but also on the relative overall 2-O and N-sulfation (data not shown), which is consistent with previous study (39). However, no significant changes in N- or 2-O-sulfation were found in TGF-β-induced SULF1 overexpression in vitro and in vivo in lung fibroblasts (21). Therefore, the observed effect of 6-O-desulfation on N- and 2-O-sulfation in our results might be related to the level of expression of the gene.
The model of UUO in mice generates progressive renal fibrosis (40). Expression of collagen-I confirmed that fibrosis is induced by day 7 post UUO. Phage display antibodies were used to examine the changes in HS sulfation. The expression of HS epitopes targeted by HS3A8 and RB4EA12 were significantly increased. Increased expression of RB4EA12 has been observed in human fibrogenic diseases including liver cirrhosis and focal nodular hyperplasia (28). Thus increased expression of HS3A8 and RB4EA12 is consistent with the suggested role of 6-O-sulfation changes in HS regulation during fibrogenic renal disease.
Our findings are of relevance to renal pathology. In the situation of chronic allograft rejection, HS 6-O-sulfation regulates FGF2 signaling by acting as a co-receptor and enabling the growth factor to bind to its cognate tyrosine kinase FGF2 receptor. This can lead to further production of profibrotic cytokines (e.g. TGF-β), MMP2, and growth factors. Thus, blockade of this interaction may provide a therapeutic option in shifting the balance toward allograft function rather than fibrosis.
Acknowledgment
Phage display antibodies were a generous gift from Toin H. van Kuppevelt (Nijmegen Centre for Molecular Life Sciences, Nijmegen, The Netherlands).
This work was supported by a grant from the Ministry of Higher Education, Syrian Arab Republic (to A. A. A.) and by the Norwegian Cancer Society Grant 3292722-2012 (to M. K.-G.).
- HS
- heparan sulfate
- GlcNS
- glucosamine
- GlcA
- glucuronic acid
- IdoA
- iduronic acid
- HS6ST
- glucosaminyl 6-O-sulfotransferase
- VSV
- vesicular stomatitis virus
- UUO
- unilateral ureteral obstruction
- TRITC
- tetramethylrhodamine isothiocyanate
- FGFR
- FGF receptor
- SULF
- HS-6-O-endosulfatase.
REFERENCES
- 1. Zeisberg M., Neilson E. G. (2010) Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 21, 1819–1834 [DOI] [PubMed] [Google Scholar]
- 2. Lindahl U., Kusche-Gullberg M., Kjellén L. (1998) Regulated diversity of heparan sulfate. J. Biol. Chem. 273, 24979–24982 [DOI] [PubMed] [Google Scholar]
- 3. Esko J. D., Selleck S. B. (2002) Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71, 435–471 [DOI] [PubMed] [Google Scholar]
- 4. Allen B. L., Rapraeger A. C. (2003) Spatial and temporal expression of heparan sulfate in mouse development regulates FGF and FGF receptor assembly. J. Cell Biol. 163, 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feyzi E., Saldeen T., Larsson E., Lindahl U., Salmivirta M. (1998) Age-dependent modulation of heparan sulfate structure and function. J. Biol. Chem. 273, 13395–13398 [DOI] [PubMed] [Google Scholar]
- 6. Jayson G. C., Lyon M., Paraskeva C., Turnbull J. E., Deakin J. A., Gallagher J. T. (1998) Heparan sulfate undergoes specific structural changes during the progression from human colon adenoma to carcinoma in vitro. J. Biol. Chem. 273, 51–57 [DOI] [PubMed] [Google Scholar]
- 7. Ai X., Do A. T., Lozynska O., Kusche-Gullberg M., Lindahl U., Emerson C. P., Jr. (2003) QSulf1 remodels the 6-O-sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J. Cell Biol. 162, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosen S. D., Lemjabbar-Alaoui H. (2010) Sulf-2: an extracellular modulator of cell signaling and a cancer target candidate. Expert Opin. Ther. Targets 14, 935–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sugaya N., Habuchi H., Nagai N., Ashikari-Hada S., Kimata K. (2008) 6-O-sulfation of heparan sulfate differentially regulates various fibroblast growth factor-dependent signalings in culture. J. Biol. Chem. 283, 10366–10376 [DOI] [PubMed] [Google Scholar]
- 10. Schlessinger J., Plotnikov A. N., Ibrahimi O. A., Eliseenkova A. V., Yeh B. K., Yayon A., Linhardt R. J., Mohammadi M. (2000) Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6, 743–750 [DOI] [PubMed] [Google Scholar]
- 11. Ali S., Hardy L. A., Kirby J. A. (2003) Transplant immunobiology: a crucial role for heparan sulfate glycosaminoglycans? Transplantation 75, 1773–1782 [DOI] [PubMed] [Google Scholar]
- 12. Born J., Jann K., Assmann K. J., Lindahl U., Berden J. H. (1996) N-Acetylated domains in heparan sulfates revealed by a monoclonal antibody against the Escherichia coli K5 capsular polysaccharide. Distribution of the cognate epitope in normal human kidney and transplant kidney with chronic vascular rejection. J. Biol. Chem. 271, 22802–22809 [DOI] [PubMed] [Google Scholar]
- 13. Rienstra H., Katta K., Celie J. W., van Goor H., Navis G., van den Born J., Hillebrands J. L. (2010) Differential expression of proteoglycans in tissue remodeling and lymphangiogenesis after experimental renal transplantation in rats. PloS ONE 5, e9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joosten S. A., van Dixhoorn M. G., Borrias M. C., Benediktsson H., van Veelen P. A., van Kooten C., Paul L. C. (2002) Antibody response against perlecan and collagen types IV and VI in chronic renal allograft rejection in the rat. Am. J. Pathol. 160, 1301–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Busse M., Feta A., Presto J., Wilén M., Grønning M., Kjellén L., Kusche-Gullberg M. (2007) Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J. Biol. Chem. 282, 32802–32810 [DOI] [PubMed] [Google Scholar]
- 16. Dennissen M. A., Jenniskens G. J., Pieffers M., Versteeg E. M., Petitou M., Veerkamp J. H., van Kuppevelt T. H. (2002) Large, tissue-regulated domain diversity of heparan sulfates demonstrated by phage display antibodies. J. Biol. Chem. 277, 10982–10986 [DOI] [PubMed] [Google Scholar]
- 17. Robertson H., Ali S., McDonnell B. J., Burt A. D., Kirby J. A. (2004) Chronic renal allograft dysfunction: the role of T cell-mediated tubular epithelial to mesenchymal cell transition. J. Am. Soc. Nephrol. 15, 390–397 [DOI] [PubMed] [Google Scholar]
- 18. Wang S., Ai X., Freeman S. D., Pownall M. E., Lu Q., Kessler D. S., Emerson C. P., Jr. (2004) QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 4833–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lai J., Chien J., Staub J., Avula R., Greene E. L., Matthews T. A., Smith D. I., Kaufmann S. H., Roberts L. R., Shridhar V. (2003) Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J. Biol. Chem. 278, 23107–23117 [DOI] [PubMed] [Google Scholar]
- 20. Celie J. W., Katta K. K., Adepu S., Melenhorst W. B., Reijmers R. M., Slot E. M., Beelen R. H., Spaargaren M., Ploeg R. J., Navis G., van der Heide J. J., van Dijk M. C., van Goor H., van den Born J. (2012) Tubular epithelial syndecan-1 maintains renal function in murine ischemia/reperfusion and human transplantation. Kidney Int. 81, 651–661 [DOI] [PubMed] [Google Scholar]
- 21. Yue X., Li X., Nguyen H. T., Chin D. R., Sullivan D. E., Lasky J. A. (2008) Transforming growth factor-β1 induces heparan sulfate 6-O-endosulfatase 1 expression in vitro and in vivo. J. Biol. Chem. 283, 20397–20407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feyzi E., Lustig F., Fager G., Spillmann D., Lindahl U., Salmivirta M. (1997) Characterization of heparin and heparan sulfate domains binding to the long splice variant of platelet-derived growth factor A chain. J. Biol. Chem. 272, 5518–5524 [DOI] [PubMed] [Google Scholar]
- 23. Lyon M., Deakin J. A., Mizuno K., Nakamura T., Gallagher J. T. (1994) Interaction of hepatocyte growth factor with heparan sulfate. Elucidation of the major heparan sulfate structural determinants. J. Biol. Chem. 269, 11216–11223 [PubMed] [Google Scholar]
- 24. Smeds E., Habuchi H., Do A.-T., Hjertson E., Grundberg H., Kimata K., Lindahl U., Kusche-Gullberg M. (2003) Substrate specificities of mouse heparan sulphate glucosaminyl 6-O-sulphotransferases. Biochem. J. 372, 371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Habuchi H., Miyake G., Nogami K., Kuroiwa A., Matsuda Y., Kusche-Gullberg M., Habuchi O., Tanaka M., Kimata K. (2003) Biosynthesis of heparan sulphate with diverse structures and functions: two alternatively spliced forms of human heparan sulphate 6-O-sulphotransferase-2 having different expression patterns and properties. Biochem. J. 371, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Kuppevelt T. H., Dennissen M. A., van Venrooij W. J., Hoet R. M., Veerkamp J. H. (1998) Generation and application of type-specific anti-heparan sulfate antibodies using phage display technology. Further evidence for heparan sulfate heterogeneity in the kidney. J. Biol. Chem. 273, 12960–12966 [DOI] [PubMed] [Google Scholar]
- 27. Lamanna W. C., Baldwin R. J., Padva M., Kalus I., Ten Dam G., van Kuppevelt T. H., Gallagher J. T., von Figura K., Dierks T., Merry C. L. (2006) Heparan sulfate 6-O-endosulfatases: discrete in vivo activities and functional co-operativity. Biochem. J. 400, 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tátrai P., Egedi K., Somorácz A., van Kuppevelt T. H., Ten Dam G., Lyon M., Deakin J. A., Kiss A., Schaff Z., Kovalszky I. (2010) Quantitative and qualitative alterations of heparan sulfate in fibrogenic liver diseases and hepatocellular cancer. J. Histochem. Cytochem. 58, 429–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rops A. L., van den Hoven M. J., Baselmans M. M., Lensen J. F., Wijnhoven T. J., van den Heuvel L. P., van Kuppevelt T. H., Berden J. H., van der Vlag J. (2008) Heparan sulfate domains on cultured activated glomerular endothelial cells mediate leukocyte trafficking. Kidney Int. 73, 52–62 [DOI] [PubMed] [Google Scholar]
- 30. Masola V., Gambaro G., Tibaldi E., Brunati A. M., Gastaldello A., D'Angelo A., Onisto M., Lupo A. (2012) Heparanase and syndecan-1 interplay orchestrates fibroblast growth factor-2-induced epithelial-mesenchymal transition in renal tubular cells. J. Biol. Chem. 287, 1478–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Strutz F., Zeisberg M. (2006) Renal fibroblasts and myofibroblasts in chronic kidney disease. J. Am. Soc. Nephrol. 17, 2992–2998 [DOI] [PubMed] [Google Scholar]
- 32. Jastrebova N., Vanwildemeersch M., Rapraeger A. C., Giménez-Gallego G., Lindahl U., Spillmann D. (2006) Heparan sulfate-related oligosaccharides in ternary complex formation with fibroblast growth factors 1 and 2 and their receptors. J. Biol. Chem. 281, 26884–26892 [DOI] [PubMed] [Google Scholar]
- 33. Mohammadi M., Olsen S. K., Goetz R. (2005) A protein canyon in the FGF-FGF receptor dimer selects from an à la carte menu of heparan sulfate motifs. Curr. Opin. Struct. Biol. 15, 506–516 [DOI] [PubMed] [Google Scholar]
- 34. Mulloy B., Rider C. C. (2006) Cytokines and proteoglycans: an introductory overview. Biochem. Soc. Trans. 34, 409–413 [DOI] [PubMed] [Google Scholar]
- 35. Dai Y., Yang Y., MacLeod V., Yue X., Rapraeger A. C., Shriver Z., Venkataraman G., Sasisekharan R., Sanderson R. D. (2005) HSulf-1 and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo. J. Biol. Chem. 280, 40066–40073 [DOI] [PubMed] [Google Scholar]
- 36. Guimond S., Maccarana M., Olwin B. B., Lindahl U., Rapraeger A. C. (1993) Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J. Biol. Chem. 268, 23906–23914 [PubMed] [Google Scholar]
- 37. Kamimura K., Koyama T., Habuchi H., Ueda R., Masu M., Kimata K., Nakato H. (2006) Specific and flexible roles of heparan sulfate modifications in Drosophila FGF signaling. J. Cell Biol. 174, 773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Do A. T., Smeds E., Spillmann D., Kusche-Gullberg M. (2006) Overexpression of heparan sulfate 6-O-sulfotransferases in human embryonic kidney 293 cells results in increased N-acetylglucosaminyl 6-O-sulfation. J. Biol. Chem. 281, 5348–5356 [DOI] [PubMed] [Google Scholar]
- 39. Ai X., Do A. T., Kusche-Gullberg M., Lindahl U., Lu K., Emerson C. P., Jr. (2006) Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. J. Biol. Chem. 281, 4969–4976 [DOI] [PubMed] [Google Scholar]
- 40. Chevalier R. L., Forbes M. S., Thornhill B. A. (2009) Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 75, 1145–1152 [DOI] [PubMed] [Google Scholar]
- 41. van den Born J., Salmivirta K., Henttinen T., Ostman N., Ishimaru T., Miyaura S., Yoshida K., Salmivirta M. (2005) Novel heparan sulfate structures revealed by monoclonal antibodies. J. Biol. Chem. 280, 20516–20523 [DOI] [PubMed] [Google Scholar]
- 42. Ten Dam G. B., Kurup S., van de Westerlo E. M., Versteeg E. M., Lindahl U., Spillmann D., van Kuppevelt T. H. (2006) 3-O-sulfated oligosaccharide structures are recognized by anti-heparan sulfate antibody HS4C3. J. Biol. Chem. 281, 4654–4662 [DOI] [PubMed] [Google Scholar]
- 43. Lensen J. F., Rops A. L., Wijnhoven T. J., Hafmans T., Feitz W. F., Oosterwijk E., Banas B., Bindels R. J., van den Heuvel L. P., van der Vlag J., Berden J. H., van Kuppevelt T. H. (2005) Localization and functional characterization of glycosaminoglycan domains in the normal human kidney as revealed by phage display-derived single chain antibodies. J. Am. Soc. Nephrol. 16, 1279–1288 [DOI] [PubMed] [Google Scholar]
- 44. Wong W. K., Robertson H., Carroll H. P., Ali S., Kirby J. A. (2003) Tubulitis in renal allograft rejection: role of transforming growth factor-β and interleukin-15 in development and maintenance of CD103+ intraepithelial T cells. Transplantation 75, 505–514 [DOI] [PubMed] [Google Scholar]