

Background: Calcium-dependent protein kinase (CDPK) regulate key processes in malaria parasite. However, the role of PfCDPK7 has remained unclear.
Results: PfCDPK7 is an effector of PI(4,5)P2 and regulates key parasitic processes.
Conclusion: PfCDPK7 is a target of PI(4,5)P2 and is critical for parasite development.
Significance: The data provides novel insights into the role of PfCDPK7, a key kinase of P. falciparum.
Keywords: Development, Malaria, Phosphoinositide, Plasmodium, Protein Kinase, Kinase, Signal
Abstract
Second messengers such as phosphoinositides and calcium are known to control diverse processes involved in the development of malaria parasites. However, the underlying molecular mechanisms and pathways need to be unraveled, which may be achieved by understanding the regulation of effectors of these second messengers. Calcium-dependent protein kinase (CDPK) family members regulate diverse parasitic processes. Because CDPKs are absent from the host, these kinases are considered as potential drug targets. We have dissected the function of an atypical CDPK from Plasmodium falciparum, PfCDPK7. The domain architecture of PfCDPK7 is very different from that of other CDPKs; it has a pleckstrin homology domain adjacent to the kinase domain and two calcium-binding EF-hands at its N terminus. We demonstrate that PfCDPK7 interacts with PI(4,5)P2 via its pleckstrin homology domain, which may guide its subcellular localization. Disruption of PfCDPK7 caused a marked reduction in the growth of the blood stage parasites, as maturation of rings to trophozoites was markedly stalled. In addition, parasite proliferation was significantly attenuated. These findings shed light on an important role for PfCDPK7 in the erythrocytic asexual cycle of malaria parasites.
Introduction
Malaria continues to be a major global health burden as it inflicts morbidity and mortality on millions every year. Despite recent promises exhibited by some of the vaccine candidates, there are no approved malarial vaccines on the horizon (1). The rapid rise in resistance of malaria parasites against existing antimalarial drugs has emerged as a major problem in the treatment of the disease. The life cycle of Plasmodium falciparum, the most virulent species of malaria parasites, alternates between the human and the mosquito host. The sporozoites injected into the host during the mosquito bite invade hepatocytes and undergo schizogony, leading to the formation of thousands of merozoites. Subsequently, merozoites invade erythrocytes to initiate the asexual erythrocytic cycle, which causes disease pathogenesis. Following invasion, the parasite first displays ring-like morphology, and “starts” to feed on host cell hemoglobin and other nutrients from the extracellular milieu, maturing into a trophozoite. Finally, the parasite divides via the process of schizogony resulting in the formation of up to 30 merozoites. Some of the parasites undergo sexual differentiation into male or female gametocytes, which after ingestion by the Anopheles mosquito, complete the sexual cycle that ultimately leads to the generation of sporozoites that accumulate in the salivary glands of the insect, where they are primed for infection of a new human host.
The involvement of signaling pathways in various stages of malaria parasite development has become increasingly clear, as several protein (2, 3) and lipid kinases (4–6) have been demonstrated to play critical roles in parasite biology. Protein kinases, many of which are regulated by second messengers like cyclic nucleotides and calcium, mediate important parasitic events such as host cell invasion, egress, and sexual differentiation (7–9). The dissection of signaling mechanisms will shed light on novel aspects of parasite biology and may also aid the design of novel intervention strategies.
Second messengers like calcium and phosphoinositides play diverse roles in signaling and trafficking in most eukaryotes (10). Calcium release in Plasmodium is tightly regulated, notably by phospholipase C, and in turn triggers signaling events involved in processes like host cell invasion and sexual development (9, 11).
Recent studies have indicated that phosphoinositides may be generated by various phosphatidylinositol phosphate (PIP)5 kinases expressed by the parasite (4, 5, 12). Although PIPs like PI3P have been implicated in hemoglobin trafficking and export of proteins to the host erythrocyte (4, 13), the role of PIPs in parasite biology remains overall poorly understood. In the present study, we have identified a novel and unexpected effector of PIP signaling, an as yet uncharacterized member of the calcium-dependent protein kinase (CDPK) family. We provide evidence that this enzyme, PfCDPK7, binds to PI(4,5)P2 and controls parasite development in the erythrocyte.
EXPERIMENTAL PROCEDURES
Antibodies
The dilution used for immunofluorescence assays (IFA) is indicated in parentheses: anti-PfCDPK7: rabbit (1:100); anti-BiP: rabbit (1:200) or mouse (1:100); anti-EBA175: rabbit, MR4 (1:100); anti-RAP1 7H8/50 MR4, mouse (1:200 mAb or culture supernatant); anti-RAP2: rabbit (1:100) and anti-MSP1(1–19): mouse and rabbit (1:100), a gift from Dr. Pawan Malhotra; anti-AMA1: rabbit (1:100), a gift from Dr. Chetan Chitnis; anti-GFP: mouse (1:100), Roche Applied Science; and anti-MBP: rabbit (1:1000), Santa Cruz Inc.
Parasite Culture, Transfections, and Generation of Transgenic Parasites
P. falciparum 3D7 strain was cultured in complete RPMI 1640 medium with 0.5% Albumax II (Invitrogen) or human serum at 37 °C as described previously (14). Parasite synchronization was achieved by using 5% sorbitol (15). Typically, 60–100 μg of plasmid DNA was transfected in the parasite by electroporation. Transfected parasites were selected by treatment with blastidicine or WR99210 at ∼2.5 μg/ml and ∼3.5 nm, respectively (16). Parasites transfected with the PfCDPK7-KO plasmid were initially genotyped by PCR to check for integration at the expected locus. These uncloned populations yielded PCR products diagnostic of both disrupted and intact CDPK7 loci. Subsequently, PfCDPK7-KO parasites were cloned by limiting dilution in 96-well plates, and several clones were selected for genotyping. The PfCDPK7-KO parasite clone used in the present study appeared after more than 3 months of drug selection. The information related to various DNA constructs and generation of transgenic parasites is provided below.
Plasmid Constructs
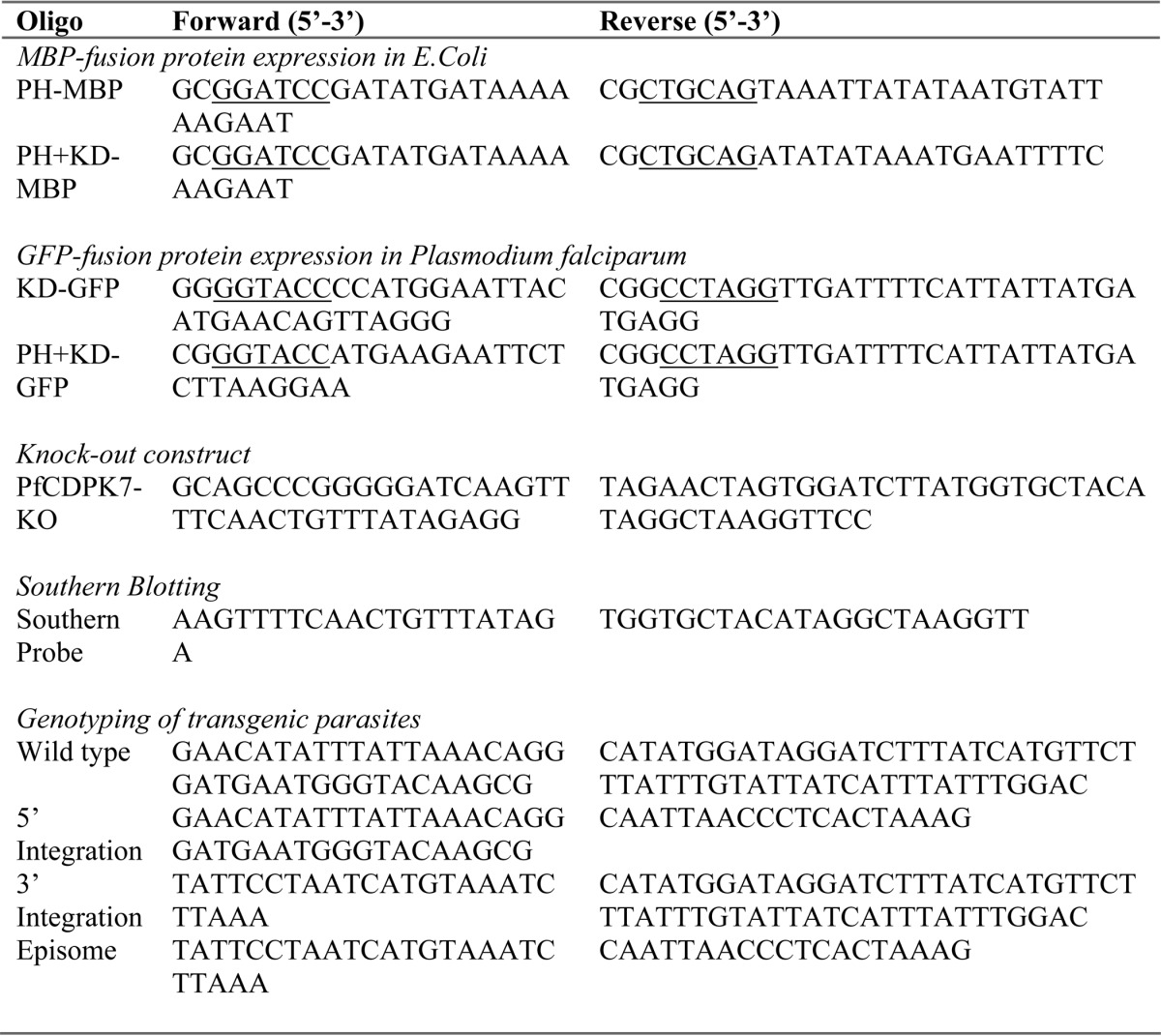
The information related to PCR primers used for all constructs and PCR is provided in Table 1.
TABLE 1.
Description of PCR primers used in the indicated studies
PfCDPK7-KO Construct
The plasmid for PfCDPK7-KO was generated by cloning an amplicon corresponding to the core of the kinase domain of PfCDPK7 in pCAM-BSD vector (2), which has a BSD resistance gene.
Expression Constructs
PH+KD fragment (PH, KD, and a short C-terminal extension, amino acids 1711–2212) and the KD (kinase domain with short C-terminal extension, amino acids 1820–2212), were cloned in KpnI and AvrII sites of pARL vector, which has genes for dihydrofolate reductase and GFP (17) for generating C terminus GFP fusion proteins in the parasite. For expression in Escherichia coli as MBP fusion protein, PH (amino acids 1708–1817) and PH+KD (amino acids 1708–2091) domains were cloned in pMALc2x vector.
Southern Blotting and Genotyping
Southern Blotting
5 μg of genomic DNA prepared from PfCDPK7-KO or 3D7 trophozoites was digested with HindIII followed by agarose gel electrophoresis and transfer on HyBond N+ (Amersham Biosciences) membrane using 6× SSC buffer. Subsequently, the DNA was UV cross-linked. A probe was prepared by PCR using a dNTP mixture supplemented with radioactive [α-32P]dCTP. The radiolabeled PCR product was denatured at 95 °C for 5 min followed by cooling at 4 °C for 5 min and was used for hybridization for 12 h. After washing the membrane with 20× SSC buffer, the membrane was exposed to phosphoimager film, which was read using a Fuji phosphorimager.
Genotyping
For confirmation of 5′ and 3′ integration and detection of the episome, standard PCR was performed using various primer sets (Table 1) and genomic DNA isolated from various parasite lines.
Parasitemia Growth Rate
3D7 and PfCDPK7-KO parasites were synchronized 2–3 times as described above and ring stage parasites were cultured in the presence or absence of blastidicine. For microscopic evaluation, Giemsa-stained thin blood smears were prepared after every 24 h and the number of rings, trophozoites, and schizonts were counted at each time point. In some cases, the number of merozoites per schizont was calculated. For quantitation by FACS, a previously described method was used with minor modifications (18).
Immunofluorescence, Live Cell Imaging, and Immunoelectron Microscopy
Immunofluorescence assays (IFA) were performed on thin blood smears or on parasite suspensions as described previously (19). Typically, cold methanol was used for fixation for smears and permeabilization was performed using 0.05% saponin in PBS followed by blocking with 3% BSA at room temperature. Subsequently, incubation with relevant primary antibodies/antisera and Alexa Fluor 488/594-labeled secondary antibodies (Invitrogen) was done. Finally, Vectashield mounting medium (Vector Laboratories inc.) containing DAPI was used for mounting. For IFAs on parasite suspensions, 4% paraformaldehyde and 0.0075% glutaraldehyde in PBS, pH 7.4, was used as a fixative and the parasites were typically permeabilized with 0.1% Triton X-100 prepared in PBS and fixed parasites were treated with 0.1 mg/ml of solution of NaBH4/PBS. Parasites were visualized using a AxioObserver microscope (Carl Zeiss) with a HRm camera or a Zeiss LSM confocal microsope. Typically, 20–30 Z stacks were collected. The processing of images was done using either Axiovision 4.8.2 or Zen software. For deconvulated images in Fig. 2, A and B, the constrained iterative or inverse algorithms in the deconvolution module of Axiovision Software were used.
FIGURE 2.

PfCDPK7 may be present in the ER and vesicles. A, IFA was performed to localize PfCDPK7 in various asexual stages of the parasite using antisera against PfCDPK7. R, rings; T, trophozoites; S, schizonts; M, merozoites. B, co-staining was done using antisera against PfCDPK7 and BiP on ring and trophozoites (a) or anti-RAP1 mAb on free merozoites (b). C, immuno-EM performed on a trophozoite-stage parasite indicated the presence of PfCDPK7 in ER and vesicular compartments (arrows). Various parasite organelles are indicated: FV, food vacuole; PM/PVM, parasite membrane/parasite vacuolar membrane; ER, endoplasmic reticulum; N, nucleus.
For immunolocalization by electron microscopy, infected RBCs were fixed in 4% paraformaldehyde, 0.05% glutaraldehyde for 1 h at 4 °C. Samples were subsequently embedded in 10% gelatin and infiltrated overnight with 2.3 m sucrose, 20% polyvinyl pyrrolidone in PIPES/MgCl2 at 4 °C. Samples were frozen in liquid nitrogen before sectioning with a Leica Ultracut UCT cryo-ultramicrotome (Leica Microsystems Inc., Bannockburn, IL). Sections of 50 nm thickness were blocked with 5% FBS, 5% normal goat serum for 30 min, and subsequently incubated with CDPK7 antisera followed by secondary anti-rabbit antibody conjugated to 18-nm colloidal gold (Jackson ImmunoResearch Laboratories, Inc., West Grove PA). Sections were washed in PIPES buffer followed by a water rinse, and stained with 0.3% uranyl acetate, 2% methyl cellulose. A JEOL 1200EX transmission electron microscope was used for analyzing the sections. All labeling experiments were performed in parallel with controls by excluding the primary antibody, which were generally negative at the concentration of colloidal gold-conjugated secondary antibodies used in these studies.
Immunoblotting and Antisera Generation
Unless indicated otherwise, cell-free protein extracts from specific parasite stages were prepared in a buffer containing (10 mm Tris, pH 7.5, 100 mm NaCl, 5 mm EDTA, 1% Triton X-100, 20 mm sodium fluoride, 20 mm β-glycerophosphate, 100 μm sodium orthovanadate, and Complete Protease inhibitor mixture (Roche Applied Science)). After separation of lysate proteins on SDS-PAGE gels, proteins were transferred to a nitrocellulose membrane. Immunoblotting was performed using various primary antibodies and antisera and HRP-labeled anti-rabbit IgG. WestPico enhanced chemiluminescence (ECL) substrate (Pierce) was used to develop blots following the manufacturer's instructions. Anti-PfCDPK7 antisera was raised by immunizing rabbits with a keyhole limpet hemocyanin-conjugated peptide (QNVRDQNDTTPHHNNENQN) corresponding to the C terminus of PfCDPK7 using standard protocols.
Recombinant Protein Expression and PIP-binding Assay and Kinase Assay
The kinase, PH+KD, and PH domains of PfCDPK7 were expressed as MBP fusion proteins and purified using amylose resin (New England Biolabs) following the manufacturer's instructions. For dot blot assays, various phosphoinsositides (100 μm, Avanti Polar or Calbiochem) were spotted onto nitrocellulose membrane and air dried for 2 h. Subsequently, the membrane was blocked with 3% BSA in buffer A (50 mm Tris-HCl, pH 7.4, 150 mm sodium chloride, 0.1% Tween 20) for 3 h followed by incubation with recombinant MBP-PH or MBP (0.5 μg/ml) for 12 h at 4 °C. The membrane was washed 5 times with buffer A prior to 3 h incubation with anti-MBP antibody (Santa Cruz Biotechnology). Subsequently, the membrane was incubated with HRP-labeled anti-rabbit IgG and PIP-bound protein was detected by chemiluminescence.
Kinase assays using recombinant MBP-PH+KD were performed using Kinase-Glo kit from Promega following the manufacturer's instructions. In this assay, the amount of ATP consumption is measured, which is facilitated by the luciferase enzyme provided with the kit. The assays were performed in a buffer containing 50 mm Tris, pH 7.5, 10 mm magnesium chloride, 20 μm ATP and myelin basic protein. In some cases, lipid vesicles (20) containing PI(4,5)P2 were included in the assay.
RESULTS
PfCDPK7, an Atypical CDPK with a PH Domain
Phosphoinositides interact with specific protein domains like the pleckstrin homology (PH) and FYVE zinc finger domains (named after the four cysteine-rich proteins Fab 1, YOTB, Vac 1, and EEA1 in which they were first described) (21). Our in silico studies lead to the identification of proteins containing PIP-binding FYVE (22) or PH domains6. One of the PH domain-containing proteins possessed a protein kinase domain and an N-terminal EF-hand calcium-binding motif (Fig. 1A). This protein kinase (PLASMODB ID PF11_0242; new GenDB ID PF3D7_1123100) was subsequently annotated as calcium-dependent protein kinase 7 (CDPK7). PfCDPK7 diverges from the canonical architecture of CDPKs (Fig. 1A). Typically, CDPKs have 4 EF-hand motif bearing a C-terminal calmodulin-like domain connected to their kinase domain via a short regulatory junction domain, and have a short and variable N-terminal region. In contrast, PfCDPK7 has two N-terminal calcium-binding EF-hand domains separated by several hundred amino acids from a PH domain, which is immediately N-terminal to the kinase domain present at the C-terminal end.
FIGURE 1.

PfCDPK7 is an atypical CDPK expressed in blood stages of malaria parasites. A, schematic representation of PfCDPK7. Unlike typical PfCDPKs, CDPK7 has a PH domain adjacent to the kinase domain (KD) at the C terminus and two EF-hand motifs near its N-terminal end. Please note that schematics are not drawn to scale, PfCDPK7 is a much larger protein in comparison to other CDPKs. B and C, antisera raised against a C-terminal region of PfCDPK7 and not the preimmune sera recognized a band corresponding to ∼250 kDa, the expected size of the protein (B). Western blot on protein lysates of various asexual blood stages of P. falciarum was performed using anti-PfCDPK7 antisera. Actin was used as a loading control (C).
An antiserum was raised against a PfCDPK7-derived C-terminal peptide, which recognized a ∼250 kDa band, close to the expected size of PfCDPK7 (Fig. 1B). PfCDPK7 protein is expressed throughout the blood stage life cycle of the parasite as indicated by Western blot performed on protein lysates from various parasitic stages (Fig. 1C).
Localization of PfCDPK7 during Parasite Development
IFA were performed to determine the localization of PfCDPK7 during parasite development. The signal was perinuclear at early stages of development (Fig. 2A, R). As the parasite matured, PfCDPK7 exhibited punctate staining indicative of its presence in vesicular structures (Fig. 2A, T and S). Co-staining with the ER marker BiP suggested that these vesicles may be in close proximity of ER (Fig. 2B, a). Immunoelectron microscopy (immuno-EM) of trophozoites (Fig. 2C) suggested that it may be present in the vesicles and possibly ER exit sites. Although co-staining with additional markers for cis- and trans-Golgi did not reveal significant co-localization, PfCDPK7 appeared to be in close proximity of these secretory organelles (data not shown). Various markers for apical organelles whose ontogeny begins in schizonts were used in IFA to evaluate their proximity to PfCDPK7. Co-staining with rhoptry bulb protein RAP1/2 (Fig. 2B, b), neck, and microneme proteins (data not shown) suggested that it is present in vesicles but may not be present in these organelles.
Interaction of PfCDPK7 with PI(4,5)P2 via its PH Domain May Be Critical for Its Subcellular Targeting
The recombinant PH domain of PfCDPK7 was expressed as recombinant maltose-binding protein (MBP) fusion protein to evaluate its interaction with PIPs. A nitrocellulose membrane on which various phosphoinositides were spotted was incubated with the recombinant proteins. Both PH (Fig. 3A) and PH+KD (data not shown) proteins interacted specifically with PI(4,5)P2, without exhibiting detectable binding to any other PIP. These data suggested that PfCDPK7 may be a target of PI(4,5)P2. Next, we tested if this domain could target the kinase to subcellular compartments. To this end, transgenic parasites expressing GFP fusion of PH+KD or KD were generated. The GFP-KD exhibited diffused expression in the parasite cytoplasm (Fig. 3B). Because the GFP fluorescence for PH+KD lines was not strong enough for live imaging, IFA were performed using an anti-GFP antibody (Fig. 3C). The results suggested that the PH+KD construct was in close proximity of the nucleus and ER (Fig. 3C) as observed for PfCDPK7 (Fig. 2, A and B). Based on these data, we conclude that the interaction between PI(4,5)P2 and its PH domain may contribute to subcellular localization of PfCDPK7. Attempts to express and purify the full-length PfCDPK7 protein were not successful, precluding investigations on the role of calcium on its activity. Recombinant PH+KD was expressed and its activity was assayed. The activity of PH+KD was almost unchanged in the presence of PI(4,5)P2 (Fig. 3D). It appears from these results that PI(4,5)P2 may not influence PH+KD activity.
FIGURE 3.

PfCDPK7 interacts with PI(4,5)P2 via its PH domain, which may be important for its subcellular localization. A, recombinant MBP-PH fragment of PfCDPK7 exhibited interaction with PI(4,5)P2 in a dot blot assay, MBP was used as a control. B and C, parasites overexpressing GFP fusion proteins of the KD or PH+KD were generated. Panel B, live imaging of the KD-GFP overexpressing parasites suggested that the KD is present in the parasite cytoplasm. Panel C, IFA was performed and PH+KD-GFP was detected using anti-GFP antibody (red) and antisera against ER marker PfBiP. PH+KD-GFP were localized to vesicular compartments in proximity to the ER. D, the PH+KD domain of PfCDPK7 was expressed as a MBP fusion protein and in vitro kinase assays were performed in the presence or absence of lipid vesicles containing PI(4,5)P2 as described under “Experimental Procedures.” Data (S.E. of duplicates from the indicated experiment with S.E.), which is representative of 3 independent experiments is shown.
Disruption of PfCDPK7 in P. falciparum
Gene disruption studies were attempted to elucidate the function of PfCDPK7. Attempts to knock out the homologue of this kinase in Plasmodium berghei were unsuccessful, suggesting that PbCDPK7 may be essential for blood-stage growth of the rodent parasite and therefore may also be important for P. falciparum growth (3). In the context of a kinome-wide reverse genetics study, a single crossover homologous recombination strategy was employed to disrupt PfCDPK7 (2). A fragment corresponding to the core region of the kinase domain was cloned in pCAM-BSD vector for this purpose (Fig. 4A). The blastidicine-resistant parasites obtained after transfection were genotyped by PCR. The drug-resistant parasites at this stage contained a subpopulation in which integration had occurred at the expected locus along with parasites with an intact PfCDPK7 locus (2). The knock-out parasites were cloned by limiting dilution. PCR and Southern blotting (Fig. 4, B and C) confirmed a clone (PfCDPK7-KO) in which the PfCDPK7 locus was disrupted. Western blotting performed using PfCDPK7 antisera confirmed the loss of PfCDPK7 protein in PfCDPK7-KO parasites (Fig. 4D).
FIGURE 4.

Generation of PfCDPK7 knockout parasite line. A, strategy to knock out PfCDPK7. pCAM-BSD-PfCDPK7KO construct contains a fragment of PfCDPK7 kinase domain, which spans the ATP-interacting glycine triad (subdomain I) and the PE di-peptide in subdomain VIII in which the Glu residue is important for conformational stability of the kinase, blastidicine resistance gene (BSD) (2). The location of primers (“Experimental Procedures,” Table 1) used for the detection of integration events is indicated by arrowheads. B, PCR analysis of the PfCDPK7 locus. Parasites were transfected with the indicated construct and selected with blastdicine as described under “Experimental Procedures.” Subsequently, drug-resistant parasites were genotyped for 5′ and 3′ integration followed by limiting dilution cloning. Genomic DNA from wild-type 3D7, PfCDPK7-KO clone was isolated and PCR amplification was performed using the indicated primers. Primer pairs 1/4 and 3/2 were used for integration at the 5′ and 3′ end, respectively, primers 1/2 and 3/4 detect the wild-type PfCDPK7 locus and the episome, respectively. The expected size of PCR products is indicated. C, Southern blot analysis of PfCDPK7 locus in 3D7 and PfCDPK7-KO parasites. After digestion with HindIII, the genomic DNA was transferred to nitrocellulose membrane followed by incubation with α-32P-labeled probe. The sizes of the bands are indicated, which correspond to the expected sizes, are indicated in panel A. In the case of PfCDPK7-KO, two bands of expected size (7.2 and 1.3 kb) indicating the integration at expected locus were observed, an additional 5-kb band corresponding to the episome was also observed. A single band of an expected size of 3.5 kb, which indicated intact PfCDPK7 locus was observed in the case of 3D7 parasites. D, Western blotting using PfCDPK7 antisera was performed on lysates prepared from 3D7 (lane 1), another unrelated modified 3D7 (lane 2), or PfCDPK7-KO (lane 3) line, which confirmed the loss of PfCDPK7 in PfCDPK7-KO parasites. Actin was used as a loading control.
PfCDPK7 Regulates Parasite Development
To examine if the disruption had an impact on parasite growth, the parasitemia of synchronized PfCDPK7-KO or 3D7 parasites was assessed for more than two cycles. The parasitemia of 3D7 parasites increased consistently after each life cycle as expected. PfCDPK7-KO parasites propagated at a significantly slower rate (Fig. 5A), the difference between the two parasite lines being enhanced after each cycle.
FIGURE 5.

PfCDPK7 is important for the asexual development of P. falciparum. A, 3D7 (wild-type, WT) or PfCDPK7-KO parasites were synchronized and ring-infected RBCs were plated to assess the parasite growth. Total parasitemia was determined by counting the parasites either from Giemsa-stained thin blood smears (a) or by FACS analysis (b) at the indicated time points. Data (S.E. of duplicates from the indicated experiment with S.E.), which are representative of more than 5 independent experiments, is shown. B, parasite growth rate assay was setup as described above and parasite smears were prepared periodically at the indicated time (h.p.i) and analyzed microscopically. The individual parasitic stages, rings (red), trophozoites (blue), and schizonts (green), were identified at the indicated time points and represented as % of total number of parasites. Although most 3D7 parasites (column 1) matured to trophozoites (72 and 120 h), a significant number of PfCDPK7-KO (column 2) parasites failed either to mature to trophozoites and/or exhibited abnormal morphology (violet). C, number of nuclei per schizont/segmenter of 3D7 (blue) or PfCDPK7-KO (red) was counted from Giemsa-stained blood smears. The number of schizonts containing more than 20 merozoites was significantly lower in the case of PfCDPK7-KO. Top panel (B and C), light microscopy of Giemsa-stained thin blood smears (left) revealed abnormal morphology of PfCDPK7-KO parasites (right).
The observed reduced growth rate could be due to (i) an extended duration of the asexual cycle; (ii) reduced viability of PfCDPK7-KO parasites; (iii) reduced invasion rate; or (iv) smaller progeny of schizonts. To gain insight into the basis for the attenuated growth of PfCDPK7-KO parasites, we monitored the appearance of developmental stages during asexual proliferation. It was striking that the maturation of ∼50% of PfCDPK7-KO rings into trophozoites was abrogated in subsequent cycles (Fig. 5B). A close examination of parasites revealed abnormal stunted morphology of the PfCDPK7-KO parasites (Fig. 5B, Giemsa-stained parasites), which most likely were rings or early trophozoites with aborted development (Fig. 5B). The number of these abnormal parasites was not significant in the first cycle (Fig. 5B, 24h). Because the experiments were initiated with sorbitol-synchronized parasites, it is possible that due to altered PVM and other defects (discussed below) these abnormal parasites were unable to survive sorbitol treatment. As a result they were excluded from the parasite population, which was used to initiate the assay. Therefore, mainly normal parasites were observed after 24 h. Because abnormal PfCDPK7-KO rings formed in the next cycle continued to be present in the assay, a significant number of parasites with stunted growth were observed in subsequent cycles (72 and 120 h). This was confirmed by using parasites without sorbitol treatment. In this case, ∼40% CDPK7-KO rings that did not successfully mature to trophozoites were observed even in the first cycle.
To ascertain the effect of PfCDPK7 on parasite division, merozoites per schizont were counted. The number of schizonts with 21–25 merozoites was significantly smaller (and with 11–15 merozoites much larger) in PfCDPK7-KO in comparison to the wild-type parasites (Fig. 5C). These data suggested that the PfCDPK7 may regulate the number of progeny merozoites, which would also contribute to the observed slower growth rate. Thus, the slow growth phenotype appears to result from both a smaller progeny for each schizont, and defects in ring to trophozoite development.
PfCDPK7-KO Parasites Exhibit Abnormal Morphology
Erythrocyte invasion assays carried out with PfCDPK7-KO parasites did not suggest a major change in invasion efficiency (data not shown). The growth and morphology of parasites post-invasion was monitored by IFA for RAP1/2 and MSP1, which are located in PV and PVM, respectively (23). The rhoptry bulb proteins such as the low molecular weight rhoptry bulb complex proteins RAP1 and 2 are transferred to the PV post-invasion (23), (24). The staining of parasites with MSP1 and RAP1/2 revealed a significant number of parasites with abnormal morphology. Typically, 3D7 parasites attained “round” or “amoeboid” shape 3–12 h post-invasion (Fig. 6, A and B) with MSP1 and RAP1/2 in close proximity as described previously (23). In contrast, a significant number of PfCDPK7-KO parasites possessed “abnormal” shapes in which fragmented (and discontinuous) MSP1 staining was observed (Fig. 6, A, B, and D). In addition, strikingly, blobs and extensions containing RAP1 were found in the host RBC outside the PV (Fig. 6C) in erythrocytes infected with PfCDPK7-KO parasites. These defects corroborate well with stalled ring to trophozoite maturation observed in growth assays (Fig. 5B). It is possible that the formation of PV and/or PVM may have been impaired, resulting in unusual structures observed in PfCDPK7-KO parasites.
FIGURE 6.

PfCDPK7-KO parasites exhibit abnormal morphology post-invasion. A-C, IFA were performed on parasites 6–12 h post-invasion (A–C) to detect rhoptry bulb proteins RAP1 (A and C) and RAP2 (B), which are transferred to PV post-invasion, and PVM was stained with MSP1. The 3D7 parasites exhibited round or amoeboid shape with MSP1 and RAP1 staining in close proximity. The CDPK7-KO abnormal parasites exhibited significantly smaller vaculoar space and often MSP1 or RAP1 staining was at distinct locations. C, typically, RAP1 is confined to the parasite/PV as seen in the 3D7 parasites. In contrast, several PfCDPK7-KO parasites exhibited extra-vacuolar RAP1 (arrow). D, parasites exhibiting abnormal morphology were counted from IFAs described in panels A and B and are represented as % of total parasites. Values are mean ± S.E. from three experiments.
DISCUSSION
To our knowledge, PfCDPK7 is the only protein kinase in Plasmodium to have been demonstrated to interact with a phosphoinositide. PfCDPK7 may be transiently localized to the ER early in development and as the parasite develops it may be incorporated in the budding vesicles. IFA performed with markers of other secretory organelles like GRASP and Rab6 did not reveal a significant co-localization, even though PfCDPK7 was present in close proximity (data not shown). The interaction between the PH domain and PI(4,5)P2 may guide the localization of the enzyme to the ER and associated vesicles, which is supported by the observed punctate staining of the PH domain constructs. Most PH domain probes that interact with PI(4,5)P2 mainly localize to the plasma membrane (25). Therefore, the vesicular localization of PfCDPK7 and its PH domain constructs was surprising. It is possible that these organelles may be enriched in PI(4,5)P2, and/or additional interactions may recruit the kinase to these locations.
The analysis of PfCDPK7-KO parasites suggested severe defects in the growth of the parasite, indicating the importance of PfCDPK7 for blood stage life cycle. Gene disruption of the CDPK7 homologue in P. berghei was not possible indicating that it is essential for the rodent parasite (3). It took prolonged efforts to clone the PfCDPK7-KO parasites as they propagated extremely slowly. It is likely that CDPK7 has a similar deleterious effect on the blood stage growth of P. berghei. As a result, PbCDPK7-KO parasites could not be obtained, presumably due to rapid clearance by the rodent immune system.
A close examination of PfCDPK7-KO parasites suggested that the development of rings was markedly affected, which was indicated by stunted parasite morphology. In addition, the number of merozoites per PfCDPK7-KO schizont was significantly lower than in the wild-type parasites. It is indeed possible that developmental defects in early life cycle stages, such as inefficient nutrient uptake, affected parasite division and/or PfCDPK7 may have a direct role in cell division. These possibilities need to be examined by performing further studies. A recent study reported gene disruption of the PfCDPK7 orthologue in Toxoplasma gondii, TgCDPK7, suggested that it is essential for parasite growth. Using a conditional knock-out strategy, TgCDPK7-KO parasites were obtained that exhibited significant defects in parasite division, asynchronous development, and impaired centrosome duplication (26). Because Plasmodium and Toxoplasma divide via the rather divergent processes of schizogony and endodyogeny, respectively, CDPK7 may regulate the division in these parasites via different mechanisms. TgCDPK7−KO parasites (26) do not seem to share any other phenotypic trait with PfCDPK7-KO parasites.
A reduced growth rate caused by a decrease in the average number of nuclear bodies per segmenter has been reported for two other plasmodial protein kinases, PfPK7 (16) and Pfcrk-5 (27). It will be interesting to explore if PfCDPK7 functions in a pathway that also includes one of these kinases. The identification of PfCDPK7 substrates will shed light on how this kinase controls parasitic processes. Given the importance of PfCDPK7 demonstrated here, it will also be worth evaluating this kinase as an anti-malarial drug target.
Acknowledgments
We thank all investigators for sharing reagents, like antibodies (see “Experimental Procedures”). The cryo-immuno-EM studies were carried out by Wandy Beatty, Molecular Microbiology Imaging Facility, Washington University, St. Louis, we appreciate her support. We also appreciate Roseleen Ekka for performing kinase assays.
This work was supported in part by a Swarnajayanti Fellowship Grant from Department of Science and Technology (DST), India (to P. S.), the MALSIG project funded by the European Union under FP7, and a Indo-German collaborative grant by Department of Biotechnology (DBT), India, and IR-BMBF (Federal Ministry of Education and Research) Grant IND 09/024, Germany (to P. S. and T. G.).
R. Ranjan and P. Sharma, unpublished data.
- PIP
- phosphatidylinositol phosphate
- BSD
- blastidicine S-deaminase
- CDPK
- calcium-dependent kinase
- CDPK7
- calcium-dependent kinase 7
- IFA
- immunofluorescence assay
- MBP
- maltose-binding protein
- Pf
- Plasmodium falciparum
- PH domain
- pleckstrin homology domain
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PVM
- parasitophorous vacuole membrane
- RBC
- red blood corpuscles
- Tg
- Toxoplasma gondii
- MSP1
- merozoite surface protein 1
- RAP
- rhoptry-associated protein.
REFERENCES
- 1. Schwartz L., Brown G. V., Genton B., Moorthy V. S. (2012) A review of malaria vaccine clinical projects based on the WHO rainbow table. Malar. J. 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Solyakov L., Halbert J., Alam M. M., Semblat J. P., Dorin-Semblat D., Reininger L., Bottrill A. R., Mistry S., Abdi A., Fennell C., Holland Z., Demarta C., Bouza Y., Sicard A., Nivez M. P., Eschenlauer S., Lama T., Thomas D. C., Sharma P., Agarwal S., Kern S., Pradel G., Graciotti M., Tobin A. B., Doerig C. (2011) Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2, 565. [DOI] [PubMed] [Google Scholar]
- 3. Tewari R., Straschil U., Bateman A., Böhme U., Cherevach I., Gong P., Pain A., Billker O. (2010) The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe 8, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vaid A., Ranjan R., Smythe W. A., Hoppe H. C., Sharma P. (2010) PfPI3K, a phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host erythrocyte and is involved in hemoglobin trafficking. Blood 115, 2500–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tawk L., Chicanne G., Dubremetz J. F., Richard V., Payrastre B., Vial H. J., Roy C., Wengelnik K. (2010) Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium, localizes to the food vacuole membrane and the apicoplast. Eukaryot. Cell 9, 1519–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McNamara C. W., Lee M. C., Lim C. S., Lim S. H., Roland J., Nagle A., Simon O., Yeung B. K., Chatterjee A. K., McCormack S. L., Manary M. J., Zeeman A. M., Dechering K. J., Kumar T. R., Henrich P. P., Gagaring K., Ibanez M., Kato N., Kuhen K. L., Fischli C., Rottmann M., Plouffe D. M., Bursulaya B., Meister S., Rameh L., Trappe J., Haasen D., Timmerman M., Sauerwein R. W., Suwanarusk R., Russell B., Renia L., Nosten F., Tully D. C., Kocken C. H., Glynne R. J., Bodenreider C., Fidock D. A., Diagana T. T., Winzeler E. A. (2013) Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504, 248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baker D. A. (2011) Cyclic nucleotide signalling in malaria parasites. Cell Microbiol. 13, 331–339 [DOI] [PubMed] [Google Scholar]
- 8. Gaur D., Chitnis C. E. (2011) Molecular interactions and signaling mechanisms during erythrocyte invasion by malaria parasites. Curr. Opin. Microbiol. 14, 422–428 [DOI] [PubMed] [Google Scholar]
- 9. Billker O., Lourido S., Sibley L. D. (2009) Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe 5, 612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vanhaesebroeck B., Leevers S. J., Ahmadi K., Timms J., Katso R., Driscoll P. C., Woscholski R., Parker P. J., Waterfield M. D. (2001) Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 70, 535–602 [DOI] [PubMed] [Google Scholar]
- 11. Sharma P., Chitnis C. E. (2013) Key molecular events during host cell invasion by apicomplexan pathogens. Curr. Opin. Microbiol. 16, 432–437 [DOI] [PubMed] [Google Scholar]
- 12. Leber W., Skippen A., Fivelman Q. L., Bowyer P. W., Cockcroft S., Baker D. A. (2009) A unique phosphatidylinositol 4-phosphate 5-kinase is activated by ADP-ribosylation factor in Plasmodium falciparum. Int. J. Parasitol. 39, 645–653 [DOI] [PubMed] [Google Scholar]
- 13. Bhattacharjee S., Stahelin R. V., Speicher K. D., Speicher D. W., Haldar K. (2012) Endoplasmic reticulum PI(3)P lipid binding targets malaria proteins to the host cell. Cell 148, 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vaid A., Thomas D. C., Sharma P. (2008) Role of Ca2+/calmodulin-PfPKB signaling pathway in erythrocyte invasion by Plasmodium falciparum. J. Biol. Chem. 283, 5589–5597 [DOI] [PubMed] [Google Scholar]
- 15. Lambros C., Vanderberg J. P. (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420 [PubMed] [Google Scholar]
- 16. Dorin-Semblat D., Sicard A., Doerig C., Ranford-Cartwright L., Doerig C. (2008) Disruption of the PfPK7 gene impairs schizogony and sporogony in the human malaria parasite Plasmodium falciparum. Eukaryot. Cell 7, 279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marti M., Good R. T., Rug M., Knuepfer E., Cowman A. F. (2004) Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science 306, 1930–1933 [DOI] [PubMed] [Google Scholar]
- 18. Theron M., Hesketh R. L., Subramanian S., Rayner J. C. (2010) An adaptable two-color flow cytometric assay to quantitate the invasion of erythrocytes by Plasmodium falciparum parasites. Cytometry A 77, 1067–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tonkin C. J., van Dooren G. G., Spurck T. P., Struck N. S., Good R. T., Handman E., Cowman A. F., McFadden G. I. (2004) Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method. Mol. Biochem. Parasitol. 137, 13–21 [DOI] [PubMed] [Google Scholar]
- 20. Andjelković M., Alessi D. R., Meier R., Fernandez A., Lamb N. J., Frech M., Cron P., Cohen P., Lucocq J. M., Hemmings B. A. (1997) Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272, 31515–31524 [DOI] [PubMed] [Google Scholar]
- 21. Lemmon M. A. (2003) Phosphoinositide recognition domains. Traffic 4, 201–213 [DOI] [PubMed] [Google Scholar]
- 22. McIntosh M. T., Vaid A., Hosgood H. D., Vijay J., Bhattacharya A., Sahani M. H., Baevova P., Joiner K. A., Sharma P. (2007) Traffic to the malaria parasite food vacuole: a novel pathway involving a phosphatidylinositol 3-phosphate-binding protein. J. Biol. Chem. 282, 11499–11508 [DOI] [PubMed] [Google Scholar]
- 23. Riglar D. T., Richard D., Wilson D. W., Boyle M. J., Dekiwadia C., Turnbull L., Angrisano F., Marapana D. S., Rogers K. L., Whitchurch C. B., Beeson J. G., Cowman A. F., Ralph S. A., Baum J. (2011) Super-resolution dissection of coordinated events during malaria parasite invasion of the human erythrocyte. Cell Host Microbe 9, 9–20 [DOI] [PubMed] [Google Scholar]
- 24. Richard D., Kats L. M., Langer C., Black C. G., Mitri K., Boddey J. A., Cowman A. F., Coppel R. L. (2009) Identification of rhoptry trafficking determinants and evidence for a novel sorting mechanism in the malaria parasite Plasmodium falciparum. PLoS Pathog. 5, e1000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Watt S. A., Kular G., Fleming I. N., Downes C. P., Lucocq J. M. (2002) Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase Cδ1. Biochem. J. 363, 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morlon-Guyot J., Berry L., Chen C. T., Gubbels M. J., Lebrun M., Daher W. (2014) The Toxoplasma gondii calcium dependent protein kinase 7 is involved in early steps of parasite division and is crucial for parasite survival. Cell Microbiol. 16, 95–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dorin-Semblat D., Gil Carvalho T., Paule Nivez M., Halbert J., Poullet P., Philippe Semblat J., Goldring D., Chakrabarti D., Mehra P., Dhar S., Paing M. M., Goldberg D. E., McMillan P. J., Tilley L., Doerig C. (2013) An atypical cyclin-dependent kinase controls Plasmodium falciparum proliferation rate. Kinome 1, 4–16 [Google Scholar]