

Abstract
Particle size, stiffness and surface functionality are important in determining the injection site, safety and efficacy of injectable soft-tissue fillers. Methods to produce soft injectable biomaterials with controlled particle characteristics are therefore desirable. Here we report a method based on suspension photopolymerization and semi-interpenetrating network (semi-IPN) to synthesize soft, functionalizable, spherical hydrogel microparticles (MP) of independently tunable size and stiffness. MP were prepared using acrylated forms of polyethylene glycol (PEG), gelatin and hyaluronic acid. Semi-IPN MP of PEG-diacrylate and PEG were used to study the effect of process parameters on particle characteristics. The process parameters were systematically varied to produce MP with size ranging from 115 to 515 μm and stiffness ranging from 190 to 1600 Pa. In vitro studies showed that the MP thus prepared were cytocompatible. The ratio and identity of the polymers used to make the semi-IPN MP were varied to control their stiffness and to introduce amine groups for potential functionalization. Slow-release polymeric particles loaded with Rhodamine or dexamethasone were incorporated in the MP as a proof-of-principle of drug incorporation and release from the MP. This work has implications in preparing injectable biomaterials of natural or synthetic polymers for applications as soft-tissue fillers.
Keywords: Soft-tissue filler, Hydrogel, Microparticles, Suspension photopolymerization
1. Introduction
Injectable biomaterials are used in medicine to augment lost or compromised tissue in order to restore form and/or function to the affected region and represent a large market, worth billions of dollars annually. Examples of such biomaterials include dermal fillers, used to treat facial defects; viscosupplements, used to treat osteoarthritis of the knee; and biomaterials, used for augmenting the urethral wall to treat urinary incontinence. Novel injectable biomaterials continue to be developed to address this growing injectable-fillers market. Further, in a small subset of patients, adverse events, such as granulomas, swelling, erythema and nodules, occur even with US Food and Drug Administration (FDA) approved or cleared injectables [1–5]. There is thus significant interest in developing methods to prepare new and putatively better injectable biomaterials.
Most injectable fillers are particulate or break down into particulates upon injection [6]. The size, shape, mechanical properties and surface chemistry of the particulates are known to influence their in vivo performance. For example, silicone particles of less than 70 μm have been shown to migrate away from the periurethral injection site and cause complications [7]. The migration potential of an implant is important for determining its safety as per the FDA’s guidelines for vocal fold applications [8]. Phagocytosis of polymethymethacrylate particles by macrophages is also size dependent [9]. Polymethylmethacrylate particles less than 50 μm in size or irregular in shape induce greater production of tumor necrosis factor alpha, an inflammatory cytokine, than particles that are spherical in shape or are between 50 and 350 μm in size in vivo [10]. Particles with acute angles, such as triangles, also induce a greater foreign body response than spherical particles [11]. Surface chemistry is also known to impact the in vivo response to the injected materials [12–15]. For example, polyurethane functionalized with zwitterionic or anionic functional groups showed lower in vivo inflammatory reaction than those functionalized with cationic chemistries [16]. Similarly, polypropylene particles coated with carboxylic acid groups were less inflammatory than those coated with amine or hydroxyl groups [14].
Particle characteristics also influence the choice of in vivo application of the filler. For example, the stiffness of a filler needs to be different depending upon its injection location, functional requirements and desired residence time. Fillers used to treat urinary incontinence need to be stiff to impart strength, while facial fillers need to be relatively soft. Hyaluronic acid facial fillers with a lower degree of crosslinking, which are therefore softer, have a shorter residence time [17]. Further softer facial fillers are used in superficial injections around the lips and eyes, while stiffer fillers are used to fill deeper wrinkles [18–20]. Also, in the cases of Perlane® and Restylane®, both manufactured by the same company, the former contains larger particulates and is used to fill deeper wrinkles, while Restylane®, with smaller particulates, is suggested for use on superficial wrinkles [19].
The size, morphology, stiffness and surface chemistry of biomaterial particulates contribute to their in vivo application and performance. However, current manufacturing technology platforms offer limited independent control of particle size, shape and surface chemistry. Hyaluronic acid (HA) based soft fillers are made using top-down methods involving bulk polymerization, followed by screening and homogenization to break down the bulk gel into irregularly shaped, polydisperse particles [6,18]. For example, Radiesse VoiceGel® and Restylane® particulates are of irregular shapes and sizes. Other injectable fillers, such as Zyderm® and Cymetra®, result in particulates of irregular shapes and polydisperse sizes upon injection through a needle. Some fillers, such as Radiesse® and Deflux®, contain polydisperse yet controlled-size spherical microparticles (MP) of hydroxylapatite or dextranomers to impart stiffness.
Current methods to synthesize hydrogel MP of controlled size and shape include microfabrication-based techniques [21–24]. While microfabrication techniques provide excellent control over size and shape, the methods are complex and “intrinsically more expensive”, and have low throughput and yield [21]. Furthermore, the use of low molecular weight (MW) polymers at high concentrations to achieve high conversions often result in MP with relatively high stiffness [25,26]. Developing methods that allow preparation of sufficient quantities of injectable fillers, especially with characteristics suitable for use as soft-tissue filler and independent control over its size, mechanical properties and surface chemistry, is desirable.
Here we report a suspension–polymerization-based method to create spherical, controlled-size hydrogel MP with tunable size and stiffness using photopolymerization. While some suspension photopolymerization-based methods have been reported earlier to prepare spherical microparticles for drug delivery [27–29], use of this method to prepare soft MP with tunable properties for use as injectable fillers remains relatively unexplored. Polyethylene glycol (PEG) was chosen to synthesize the MP because of its extensive use in FDA-approved or cleared products and its use in preparing biocompatible materials. HA and gelatin were chosen because of their favorable biocompatibility profiles, history of use in commercial dermal fillers and use in preparing soft, viscoelastic hydrogels [17]. Another motivation for using HA and gelatin to prepare MP was to demonstrate the generality of our method. The semi-interpenetrating technology (semi-IPN), which involves crosslinking of a polymer in the presence of a non-crosslinked polymer, was used to prepare the MP because this technology allows the preparation of materials with tunable properties by varying parameters such as the polymers used, their relative MW and their concentrations. Photocrosslinking was chosen because it can be carried out in ambient conditions, is efficient and affords precise spatiotemporal control over the crosslinking. The size distribution of suspension-polymerized MP can change as the reaction proceeds and, unlike chemical or thermal crosslinking, light-based crosslinking provides better control over the termination of the reaction [30]. Irgacure 2959 (I2959) was chosen as the photoinitiator (PI) because of its better cytocompatibility relative to other commercially available photoinitiators and its extensive use in preparing biomaterials, including those that encapsulate mammalian cells [31–34]. Furthermore, PEG hydrogels prepared using PEG diacrylate (PEG-DA) and I2959 were shown to be safe in humans in a 15-patient clinical trial for articular cartilage repair, thereby making the use of this system attractive [35]. In addition, GelrinC, a CE Mark-approved product in Europe, is also an in situ polymerized PEG-DA and I2959-based hydrogel that was shown to be safe and effective for the treatment of articular cartilage in injured knees over a period of 24 months in a multi-center clinical trial in Europe [36].
Semi-IPN MP of PEG-DA and PEG were prepared using suspension photopolymerization. The effect of process parameters such as stirring speed, surfactant concentration, gelation time, PI concentration and UV intensity on MP size and yield was investigated. In vitro cytocompatibility of the PEG MP was evaluated using the MTT assay. MP were also prepared using acrylated versions of HA and gelatin. The feasibility of preparing functionalizable MP and MP that encapsulate drug-releasing particles was also explored.
2. Materials and methods
2.1. Materials
PEG and PEG-DA (MW of both: 10 kDa) were purchased from Alfa Aesar (Lancaster, UK) and SunBio (Anyang City, South Korea), respectively. Dioctyl sulfosuccinate (AOT, sodium salt, 99%), n-hexane, PEG bis(amine) (MW: 3400 Da), trinitrobenzenesulfonic acid (TNBS), sodium dodecyl sulfate (SDS), sodium bicarbonate, hydrochloric acid, dexamethasone, polyvinyl alcohol (PVA), Rhodamine 6G, gelatin (300 g bloom strength, Type A from porcine skin) and methacrylic anhydride were from Sigma–Aldrich (St. Louis, MO). Sodium hyaluronate (MW: 351–600 kDa) and 2-hydroxy-1-[4-(2-hyroxy-ethoxy)phenyl]-2-methyl-1-propanone (Irgacure 2959, PI) were from Lifecore Biomedical (Chaska, MN) and Ciba (Tarrytown, NY), respectively. Sulfosuccinimidyl acetate (sulfo-NHS-acetate) was from Thermo Scientific (Rockford, IL). Polyoxyethylene (20) Sorbitan Monoeleate (Tween 80) was from VWR (West Chester, PA). Ester-terminated 75:25 poly(DL-lactide-co-glycolide) (PLGA; inherent viscosity: 0.55–0.75 dl g−1) was from Durect Corporation (Cupertino, CA). India ink was from Becton, Dickinson and Company (Cockeysville, MD). The MTT cell proliferation assay kit and NIH/3T3 fibroblast cells were from American Type Culture Collection (Mannassas, VA). Cell culture media, serum, phosphate-buffered saline (PBS), and trypsin were from Invitrogen (Carlsbad, CA). Radiesse Voice Gel®, Restylane® and Zyderm® were from Merz Aesthetics (San Mateo, CA), Medicis Aesthetics (Scottsdale, AZ) and Allergan (Irvine, CA), respectively. All chemicals were used as received.
2.2. Preparation of PEG hydrogel MP
Solutions of PEG and PEG-DA (100 mg ml−1 each) in deionized (DI) water were mixed in a pre-determined volumetric ratio. PI was added to this solution (0.5 mg ml−1 final concentration) to prepare the precursor solution (1 ml), which was then added to an AOT solution in hexane (9 ml) in a 2 inch wide polypropylene beaker. This suspension was then stirred at a predetermined speed using a 1 inch long stir bar on an IKA RET basic ETS-D5 magnetic stir plate. While being stirred, the suspension was photopolymerized at 72 mW cm−2 (measured at 365 nm at the level of the base of the beaker) with an Omnicure Series 2000 UV lamp for 260 s. Unless otherwise noted, the AOT concentration was 2 mM and the stirring speed was 800 rpm. Post-polymerization, the suspension was centrifuged at 150 rcf for 2 min and the organic supernatant was removed. The resulting MP were washed three times, once with 0.1 vol.% Tween-80 and twice with DI water using centrifugation, and finally separated from the solution using a 40 μm nylon cell strainer. Excess water was wicked from the MP using Kimwipes. The volumetric percentage yield was estimated as volumetric yield = (volume of MP judged visually in a calibrated syringe/volume of PEG precursor solution) × 100. The gels or MP prepared using a precursor solution containing 50 vol.% PEG-DA are referred to as PEG50, while those prepared using 75% PEG-DA are referred to as PEG75, and so forth.
2.3. Preparation of PEG hydrogel particulates of uncontrolled size
The PEG precursor solution (1 ml) was photopolymerized at 2 mW cm−2 for 200 s in a 12-well plate. After polymerization, the hydrogel was incubated in PBS at a 1:9 ratio (v/v) at 37 °C overnight. The swollen hydrogels were added to a syringe and broken into smaller particulates by shearing them twice each through sequentially narrower (16, 18, 20 and 22 gauge) needles.
2.4. Preparation of MP from gelatin, HA and PEG-diamine
Gelatin methacrylate (GelMA) was synthesized using gelatin (0.1 g ml−1) and methacrylic anhydride (1 vol.%) as per a method described earlier [37]. To prepare MP, a solution of GelMA (100 mg ml−1) and PI (0.5 mg ml−1) was made in DI water at 50 °C. Photopolymerization was carried out as described earlier for PEG MP at 800 rpm and 72 mW cm−2 for 30 s with an AOT concentration of 1 mM. Post-polymerization, the hexane–AOT was removed and the GelMA MP formed were stirred in water (15 ml) to prevent agglomeration and then washed to remove the residual hexane–AOT, as outlined for PEG MP. HA-methacrylate (HAMA) was synthesized using a method described earlier [38]. HAMA MP were prepared using a method identical to that used for GelMA MP but with HAMA and AOT concentrations of 10 mg ml−1 and 5 mM, respectively. Semi-IPN MP of HAMA and gelatin were prepared using a 75:25 (v/v) ratio of HAMA (10 mg ml−1) and gelatin (100 mg ml−1), with the same polymerization parameters as those for GelMA MP but with an AOT concentration of 2.5 mM. To prepare PEG-diamine MP, PEG was replaced with PEG-diamine (200 mg ml−1) in the procedure described earlier for PEG MP, and a stirring speed of 600 rpm and an AOT concentration of 0.5 mM were used.
2.5. Preparation of NS–MP and MS–MP hybrid particles
PLGA nanospheres (NS) encapsulated with dexamethasone (Dex) were prepared using a method reported earlier, but with dichloromethane and acetone (3:2 volumetric ratio) as the solvent for PLGA and Dex [39]. To prepare Rhodamine-encapsulated PLGA microspheres (MS), PLGA (300 mg) and Rhodamine (30 mg) were co-dissolved in dichloromethane (2.25 ml), added to a PVA solution (10 mg ml−1) and homogenized at 9000 rpm for 5 min with an L4RT-A homogenizer. The organic solvent was removed in a rotary evaporator and by repeated washing with water. The NS and MS were lyophilized overnight to obtain dry powders. To prepare NS–MP or MS–MP conjugates, dry PLGA NS with Dex (40 mg ml−1) or PLGA MS with Rhodamine (10 mg ml−1) were suspended in PEG precursor solution and added to AOT/hexane solution (0.5 mM), photopolymerized and washed as described earlier for PEG MP particles.
2.6. Determination of loading and release rate of dexamethasone in NS–MP hybrid particles
The hybrid NS–MP were thoroughly washed to remove any NS sticking to the surface of the MP. To quantify the amount of Dex encapsulated, the NS–MP hybrid particles (200 mg) were hydrolyzed with NaOH (1 N, 200 μl), neutralized with HCl (1 N), dissolved in acetonitrile (4 ml) and then the Dex concentration in this solution was determined using HPLC as per previous methods [40]. To determine the Dex release rate, the NS–MP particles (~100 mg) were placed inside a cellulose ester dialysis tubing (MWCO 10 kDa, Spectrum Labs, Rancho Dominguez, CA) and then placed in a rotating falcon tubes filled with PBS (50 ml) at 37 °C. The PBS was collected at 6 hours, 1 day and 10 days and replenished with fresh PBS, and the Dex in the collected PBS was determined using HPLC.
2.7. Determination and blocking of free amine groups
Free amine groups in PEG-diamine MP were estimated using a colorimetric TNBS assay described elsewhere [41,42]. A calibration curve made using PEG-diamine standards ranging from 0 to 10 mg ml−1 was used to estimate the amount of free amines. The amine groups of the PEG-diamine MP were blocked by reacting the MP with an aqueous sulfo-NHS-acetate solution (25× molar excess, 1 ml) for 1 h at room temperature. Excess sulfo-NHS-acetate solution was removed by washing twice with DI water and the remaining free amine groups were determined using the TNBS assay as described above.
2.8. Determination of particle size distribution
Particle size distributions of the MP and MS were determined using a Coulter Counter Multisizer III (Beckman Coulter, Inc., Brea, CA) as per the manufacturer’s protocol. The particle size of the NS was determined using a Zetasizer Nano (Malvern Instruments, Malvern, UK). The particle size was reported as the D90 (weighted by number percentage), where D90 represents the diameter that is equal to or greater than that of 90% of the particles in the sample. The relative span that describes the polydispersity of the particles was calculated as the relative span = (D90 − D10)/D50, where the definitions of D10 and D50 are analogous to that of D90.
2.9. Measurement of viscoelastic properties of MP
The viscoelastic properties of MP were measured using an AR-2000 rheometer (TA Instruments, Inc., New Castle, DE) with a parallel plate geometry at 37 °C. Care was taken to ensure that no excess liquid was present in the MP. Based on previous reports, a gap height of five times the D90 was used [43,44]. For example, for MP prepared by stirring at 800 and 400 rpm, the gap heights were 750 and 4000 μm, respectively. To eliminate wall slip, a 320 grit sand paper (36 μm) was attached to the upper and lower plates. The parallel plate diameter was at least 10 times the gap height. The shear storage modulus and loss modulus of MP were measured as a function of frequency from 1 to 10 Hz at a strain of 0.6% at 37 °C and reported at 10 Hz.
2.10. In vitro cytocompatibility
The cytocompatibility of MP was measured using the MTT cell proliferation assay. PEG50 MP (20 mM AOT, 400 rpm) were synthesized in a laminar flow hood using a precursor solution filtered through a 0.2 μm filter. Fibroblast (NIH/3T3) cells were plated in 24-well plates (40,000 cells ml−1) and allowed to attach in Dulbecco’s modified Eagle’s medium with 10 vol.% calf serum and 1 vol.% penicillin–streptomycin. Sheared hydrogel particulates and PEG50 MP were added separately to the cells. To determine if leachates from the MP and the sheared gel are cytotoxic, both materials were incubated in the medium for 3 days at concentrations varying from 0 to 50 mg ml−1. Samples were centrifuged and the supernatant medium was collected after 3 days. This medium was then used to culture the cells. SDS was used as a positive control. After 3 days of culture, cell proliferation in each well was determined as per the manufacturer’s instructions. Since the purple precipitate produced by viable cells was absorbed by the MP and sheared PEG particulates, the solutions were solubilized in detergent overnight and then read at 570 nm, with the reference read at 670 nm. Normalized viability is reported as ((absorbance at 570 nm − absorbance at 670 nm) − (absorbance of blank))/(absorbance of negative control − absorbance of blank).
2.11. Fluorescent confocal microscopy
Rhodamine–PLGA MS encapsulated within PEG MP were excited at 526 nm and imaged with an Olympus Fluoview 1000 microscope. Images were compiled using ImageJ software.
2.12. Statistics
All data are presented as mean ± standard deviation of at least three separate replicates. Student’s t-test was used with a 95% confidence interval to determine statistical significance unless otherwise noted.
3. Results
3.1. Preparation of MP
PEG hydrogel MP were prepared using suspension photopolymerization of PEG-DA in presence of varying quantities of PEG in a hexane–water–AOT system [45]. The MP thus prepared were spherical in shape, as seen in the optical picture of PEG100 MP (Fig. 1A). A dilute solution of India ink was used as the contrast agent to better visualize the MP. While the MP could be injected through a 22 gauge needle without distortion (Fig. S1), when passed through a 25 gauge needle, a few particles (possibly larger ones) became distorted, though the majority remained unchanged (Fig. 1B). However, no clogging of the 25 gauge needle was observed. The regular shape of the PEG100 MP is in contrast to the irregular shape of commercially available injectable fillers, such as Radiesse® Voice Gel, Restylane® and Zyderm® (Fig. 1D, E and F, respectively). The PEG100 MP were polydisperse in size, with an average diameter of 96 μm, a median diameter of 87 μm, a typical D90 value of 162 μm and a relative span value of 1.47 (Fig. 2). Similar size, shape and injectability data were obtained for MP made using other PEG-DA:PEG ratios (not shown). MP were also prepared using acrylated forms of HA, a biopolymer that has been used extensively to prepare injectables, and gelatin with D90 (span) values of 136 ± 22 μm (1.5 ± 0.2) and 157 ± 4 μm (1.9 ± 0.6), respectively (Figs. 1C and S2). Further semi-IPN MP of HA–methacrylate and gelatin with D90 (span) value of 150 ± 38 μm (2.2 ± 0.2) were also prepared (Fig. S3).
Fig. 1.

Optical micrographs of (A) PEG100 MP, (B) PEG100 MP passed through a 25 gauge needle, (C) hyaluronic acid MP, (D) Radiesse® Voice Gel, (E) Restylane® and (F) Zyderm®, with India ink as the contrast agent.
Fig. 2.

Particle size distribution of PEG100 MP.
3.2. Effect of process parameters on particle characteristics
The effect of a process parameter on particle size distribution and yield was investigated while keeping other parameters constant. The particle size of PEG50 MP decreased with increasing stirring speed. For example, increasing the stirring speed from 400 to 600 rpm decreased the D90 from 515 ± 50 to 140 ± 33 μm (Fig. 3A). However, increasing the stirring speed beyond 600 rpm did not cause a statistically significant change in size. Similarly, increasing the stirring speed from 400 to 1000 rpm decreased the volumetric yield from 110 ± 15 to 43 ± 11%. (The volumetric yield can be greater than 100% because the spherical MP have void spaces when packed together.) In addition, MP swell due to absorption of water during the washing step. Consequently, the volume of the MP obtained may be higher than the precursor solution volume. The relative span of the MP decreased from 2.9 ± 0.2 at 400 rpm to 2.1 ± 0.7 at 500 rpm, though this difference between the two speeds was not statistically significant. Between 600 and 1000 rpm the relative span ranged from 1.4 to 1.7, with no statistically significant difference within this stirring speed range. However, this decrease in the relative span at higher speeds was statistically significant relative to that at 400 or 500 rpm. Doubling the PI concentration to 1 mg ml−1 increased the D90 of PEG50 MP from 142 ± 25 to 225 ± 22 μm, with no statistically significant change in the relative span. Similarly, doubling the UV intensity to 144 mW cm−2 increased the D90 to 231 ± 35 μm, with no change in the relative span. A PI concentration beyond 1 mg ml−1 was not tested because of concerns about potential toxicity [33]. Also, the effect of UV intensity on MP characteristics beyond 144 mW cm−2 was not tested because higher intensities led to heating and evaporation-related changes in the suspension during polymerization. Using other UV exposure times of 30, 65, 130 and 520 s did not change the PEG MP size or the relative span under the conditions tested (data not shown).
Fig. 3.

Particle size (90th percentile particle diameter, D90) and volumetric yield of PEG50 MP as a function of (A) Stirring speed and (B) AOT concentration. *Statistically significant (p < 0.05). Values shown are mean ± standard deviation (SD).
Increasing the AOT concentration from 0.5 to 5 mM decreased the D90 from 254 ± 16 to 103 ± 21 μm and decreased the volumetric yield from 88 ± 9 to 21 ± 4%, while the relative span remained unchanged at between 1.5 and 1.7 (Fig. 3B). Based on the above general relations, MP with D90 as small as 48 and 39 μm were prepared by using the parameters [AOT] = 50 mM, speed = 750 rpm, UV intensity = 365 mW cm−2, UV time = 480 s, [PI] = 1 mg ml−1 and [AOT] = 50 mM, speed = 400 rpm, UV = 72 mW cm−2, UV time = 260 s, [PI] = 0.5 mg ml−1, respectively. However, the yield of particles thus obtained was only 3%, hence these conditions were not explored further. Overall, by varying the process parameters, spherical MP with average D90 in the range of 39–515 μm were prepared.
3.3. MP mechanical characterization
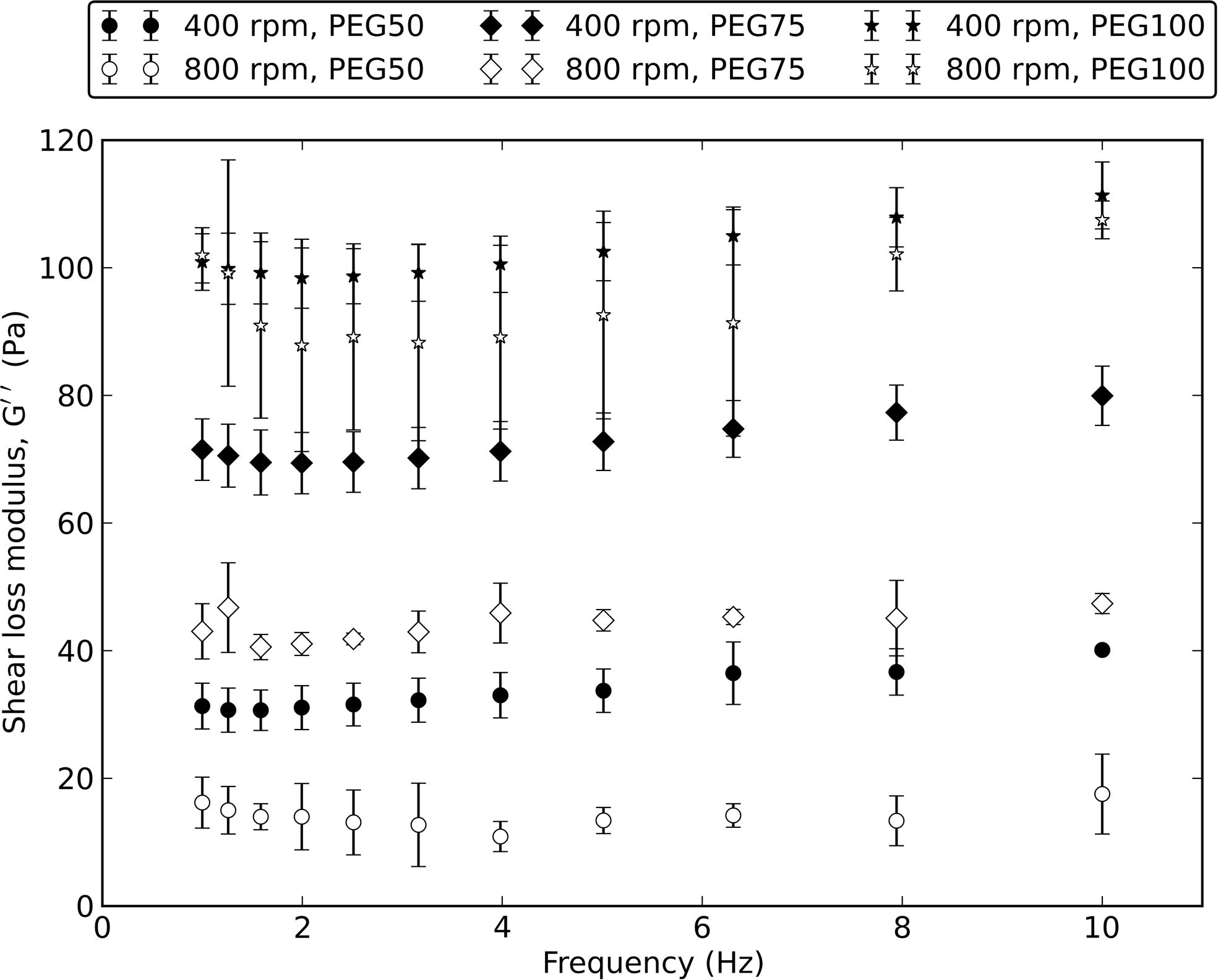
The viscoelastic properties of MP can be controlled by varying the PEG-DA:PEG ratio and by varying the particle size. Regardless of the particle size, increasing the PEG-DA:PEG ratio increased the shear storage modulus (G′) and shear loss modulus (G″) of the MP (Fig. 4A). For MP prepared at 400 rpm, the G′ increased from 523 ± 31 to 1599 ± 110 Pa and the G″ increased from 38 ± 3 to 111 ± 5 Pa when PEG-DA:PEG ratio (v/v) was increased from 50 to 100% while the size stayed the same. Similarly, for MP prepared using a stirring speed of 800 rpm, the G′ increased from 188 ± 63 to 1257 ± 105 Pa and the G″ increased from 15 ± 6 to 100 ± 13 Pa with increasing PEG-DA:PEG ratio without a change in MP size. For a given PEG-DA:PEG ratio, the G′ and G″ increased with decreasing particle size. At all PEG-DA:PEG ratios and particle sizes, G″ is at least one order of magnitude smaller than G′. The G″ was lower than G′ at all frequencies of measurement from 1 to 10 Hz (Fig. S4). The complex viscosity of the MP decreased as the frequency of oscillation was increased, suggesting a shear-thinning behavior for the MP (Fig. 4B). Decreasing the PEG-DA:PEG ratio or decreasing the particle size decreased the complex viscosity of the MP. All of the changes in G′ and G″ described here were statistically significant.
Fig. 4.

Viscoelastic properties of PEG MP. (A) Shear storage modulus (G′) and shear loss modulus (G″) ofMP as a function of PEG-DA:PEG ratio and stirring speeds. (B) Complex viscosity (η*) of PEG MP prepared in this study. *,**Statistically significant (p < 0.05) for G′ and G″, respectively. Values shown are mean ± SD.
3.4. In vitro cytocompatibility
We tested whether the PEG MP or materials leached from the MP are cytotoxic using NIH/3T3 cells as model mammalian cells. PEG50 particulates of uncontrolled size that were not exposed to hexane or AOT at any step of their synthesis were made by shearing a PEG50 gel through needles and used as the control. The same concentrations (ranging from 0 to 50 mg ml−1) of PEG50 MP and PEG50 particulates were incubated over NIH/3T3 cells for 3 days and the cell viability was evaluated using the MTT assay. More than 80% of the cells were viable when exposed to PEG50 at concentrations ranging from 0.1 to 50 mg ml−1, with the lowest normalized viability of 80.9 ± 0.8% seen at the highest MP concentration (Fig. 5A). In addition, minimal to no toxicity was seen when cells were grown in medium that was incubated for 3 days with just the MP or particulates (Fig. 5B).
Fig. 5.

Cytocompatibility of PEG MP. (A) Normalized viability of NIH/3T3 cells incubated with PEG50 MP and hydrogel particulates for 3 days as evaluated by MTT assay. (B) Normalized viability of NIH/3T3 cells incubated with medium exposed to PEG50 MP and hydrogel particulates for 3 days as evaluated by MTT assay. The light gray bars are for PEG50 MP and the dark gray bar for PEG hydrogel particulates. *,**Statistically significant (p < 0.05) for PEG50 MP and PEG hydrogel particulates, respectively. Values shown are mean ± SD.
3.5. Introduction of amine groups for potential surface functionalization
Amine groups were introduced in the MP by replacing the PEG in the PEG50 MP with PEG-diamine. The PEG50-amine MP thus prepared had an average of ~33 ± 2 free amine groups per nm2 of the MP surface, as estimated using the TNBS assay. If the amines are assumed to be uniformly distributed throughout the spherical MP and the outer 5% layer of the MP represents its functionalizable surface, about 14% of the amines, i.e. 4.7 ± 0.3 free amine groups, are present per nm2 of the MP surface. After reaction with amine-reactive sulfo-NHS-acetate, the amine groups of the PEG50-amine MP decreased to ~15%, suggesting that 85 ± 10% of the amines are accessible and reactive to small-molecule reactants.
3.6. Encapsulation and release of dexamethasone from MS–MP hybrid particles
Rhodamine was encapsulated in PLGA MS, and the PLGA–Rhodamine MS were then incorporated inside PEG MP. Rhodamine was used as a model of a small-molecule drug and its fluorescence was used to characterize the distribution of PLGA MS inside the PEG MP. Confocal microscopy revealed that Rhodamine–PLGA MS (median size 4 μm) can be encapsulated inside the PEG MP and the MS are distributed throughout the volume of MP (Fig. 6). No MS were seen to be adsorbed to the surface of the MP. Dexamethasone was used as an example of an anti-inflammatory drug molecule to demonstrate the proof-of-context of incorporation and release of a drug molecule from the hybrid PLGA–drug NS–PEG MP particles. PLGA NS (median size 112 nm) loaded with dexamethasone were also encapsulated in the PEG MP using a similar process. About 18% of the PLGA NS in suspension were encapsulated inside the PEG MP to give a loading of 750 μg of dexamethasone per mg of MP. Preliminary experiments showed that ~74% of the encapsulated drug was released from the PEG NS–MP, compared to 86% that was released from NS alone within the first 24 h (data not shown). However, additional experimentation is needed to fully characterize the drug release profile.
Fig. 6.
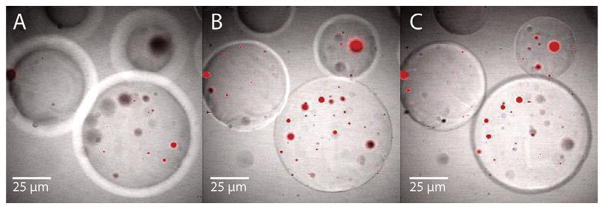
Confocal microscopy pictures of hybrid particles of Rhodamine-loaded PLGA MS incorporated in PEG MP. The micrographs are at optical depths of (A) 100 μm, (B) 350 μm and (C) 450 μm from the surface of the PEG MP.
4. Discussion
The size, stiffness and surface chemistry of injectable materials are important in determining the injection site and in vivo performance. New methods to produce adequate quantities of soft injectable biomaterials which provide independent control over these particle characteristics are therefore desirable. We used suspension photopolymerization and semi-IPN technology to produce spherical hydrogel MP of independently tunable size and stiffness. While the method is generally applicable to acrylated (bio)polymers and was used to prepare MP of acrylated HA, gelatin and their semi-IPNs, PEG MP were used for detailed characterization. The D90 and relative span value were used to report the particle size distribution (PSD) in this study because they are commonly used to report size data of particles with a non-symmetric PSD and have been used for other suspension-polymerized systems [30]. The relative span represents the breadth of size distribution, with a smaller span suggesting a lower polydispersity. Unless otherwise noted the MP prepared in this study had a relative span of ~1.5, which being lower than 2, suggests a low polydispersity [30]. The gelatin MP, the semi-IPN MP of HA methacrylate–gelatin and the PEG MP prepared at twice the normal PI concentration or UV intensity were visibly “sticky”. This stickiness caused some aggregation of the MP that may have contributed to their relatively higher span values.
As with suspension or emulsion polymerization systems used to prepare PLGA MS or polystyrene MP [30,46], increasing stirring speed or surfactant concentration decreased the size of PEG50 MP. The droplet size and eventually the MP size in suspension polymerization systems are decided by the competition between the rate of droplet break-up and coalescence. A higher AOT concentration stabilizes the water-in-oil suspension and reduces the likelihood of PEG-precursor droplets coalescing before polymerization, leading to smaller MP. Similarly, higher energy input in the form of faster stirring helps in overcoming the surface tension forces of smaller droplets and keeps them separate until they have gelled. A larger span at 400 or 500 rpm relative to that at higher speeds suggests that the energy input at lower speeds is probably not high enough to shear the droplets as consistently as at higher speeds, thereby leading to higher polydispersity at lower speeds.
Higher stirring speeds or AOT concentrations also increased the opacity of the suspension, which possibly limited the UV transmittance and hence the yield. Increasing the UV exposure time to ~4 times longer than usual or increasing the UV intensity to the maximum possible using our equipment (365 mW cm−2) to counter the effect of the increase in opacity did not improve the yield, which suggests that either the UV transmittance is not an issue or the increase in UV intensity was not high enough. Further, as the UV intensity was increased to the maximum value, evaporation of hexane due to heating was observed. Although it is conceivable that the yield is lowered by particles smaller than 40 μm passing through the 40 μm cell strainer used to separate the MP, we observed minimal improvement in yield even when the MP were separated solely via centrifugation without the use of a cell strainer. It is possible that the volume of the particles less than 40 μm lost was not significant and/or not many particles were lost during filtration. Indeed, particles smaller than 40 μm were seen in the PSD curves of the MP, suggesting that at least some particles were not lost, possibly due to sticking to the larger particles and/or due to clogging of the cell strainer holes by the larger particles. However, centrifugation was not solely used to separate the MP, as it did not remove enough wash solution to allow reliable measurements of the MP properties. Doubling the PI concentration gave larger and stiffer PEG100 MP, suggesting that the precursor droplets were gelled before they could be broken down further.
Importantly, the effect of the process parameters on the size and yield of MP was independent of the PEG-DA:PEG ratio used to make the MP. Therefore we were able to vary the stiffness (G′) of the MP from ~190 to 1600 Pa by controlling the PEG-DA:PEG ratio without compromising the control on the MP size. The data in Fig. 4A suggests that it should be possible to vary MP size while keeping the stiffness constant. The range of G′ values is similar to that of the HA-based soft-tissue fillers, which have G′ values of ~90–3000 Pa [47]. In addition, the shear loss modulus is at least an order of magnitude lower than the shear storage modulus. This suggests that the MP behave as a viscoelastic gel system. The range of D90 for the controlled-size MP that also had practically useful yields was from ~100 to 500 μm. This size range is similar to that of HA-based dermal fillers, which have an average size range of 300–700 μm [19]. The complex viscosity (η*) of a filler is a measure of its injectability through a needle and impacts on its utility for different injection sites and purposes [47]. For example, fillers with high η*are generally used for facial sculpting and lifting, which need materials that would not spread easily. The η*of the PEG MP (230–22 Pa s at 1 Hz) is lower than the range of η* reported for HA-based fillers [47], possibly because of the lower MW and the branching of the PEG used here relative to the HA. However, the ranges of stiffness and viscosity can be increased by using other polymers of different MW. For example, we prepared MP with a G′ of 15,760 Pa by using PEG-DA and PEG (1:1, 300 mg ml−1 both) of MW 700 and 545 Da, respectively. Overall, hydrogel MP of tunable size, shape and viscoelastic properties that are in the range of current clinically used injectables can be prepared with natural or synthetic polymers using the method reported here.
The MTT results for cytotoxicity of PEG50 MP and hydrogel particulates were similar irrespective of whether the cells were incubated directly with the material or with medium exposed to the material. These results suggest that the 20% decrease in cell viability seen with higher concentrations of PEG50 MP was not due to toxicity specific to PEG50 MP, but could possibly be due to the decrease in nutrients reaching the cells because of a layer of MP on the cells. In addition, the higher cell viability observed when cells were grown in medium exposed to MP/hydrogel particulates relative to those incubated under MP/hydrogel particulates suggests that the leachates, if any, did not cause any measurable cytotoxicity. However, in vivo studies in animal models are required to fully characterize the biocompatibility of the PEG MP.
It is known that the cellular response to an implanted material can be influenced by surface properties such as electric charge or the presence of peptides [12,13,48–50]. In the case of fillers used to treat urinary incontinence, it has been suggested that surface functionalization of the MP with tissue-binding proteins may limit their migration away from the injection site [51]. The semi-IPN platform used to prepare the PEG MP not only provides opportunities to control the viscoelastic properties but also allows the possibility of altering the surface chemistry of the MP. To enable the functionalization of the MP, we introduced amine groups in the PEG MP because amines have been used to modify surfaces by covalent attachment of (bio)molecules [38,52]. The PEG50-amine MP reported here had ~4.7 amine groups per nm2 on the surface of the MPm and ~85% of the amines were accessible to small molecules. These amines can be potentially conjugated with (bio)molecules such as the RGD peptide. Since only 1 fmol cm−2 or six molecules of RGD peptide per μm2 is sufficient to increase cell adhesion, the surface amine density of the MP is about 106 times this minimum requirement. Further experiments on the conjugation of bioactive molecules to MP, however, are needed to fully explore the possibility of surface functionalization of the MP.
Filler materials may be mixed with therapeutics to alter their in vivo effects on or tolerability to the patient. For example, dermal fillers are often mixed with lidocaine to reduce pain during injection [53]. Using another small hydrophobic drug, such as dexamethasone, and a fluorescent dye, such as Rhodamine, we showed that PLGA NS/MS can be encapsulated inside the PEG MP and that the dexamethasone was released from the PEG MP over time. Incorporation of drugs in fillers via encapsulation in PLGA NS/MS may provide opportunities to prepare fillers with tunable drug loading and controlled release profile (e.g. by encapsulating monodisperse drug-loaded PLGA NS/MS [54]). Such NS/MS–MP conjugates can be used instead of or in conjunction with the physical mixing the drugs with the fillers. These results demonstrate a proof of principle that this method can be used to load drugs in the hydrogel MP. Additional experimentation is needed to further determine and control the drug release through the MP.
The method reported here may be improved by addressing the limitations of scale and the polydisperse nature of the MP. Scale-up of suspension polymerization systems has been well studied, and such reactors are routinely used industrially [55]. While photopolymerization-based systems may pose unique challenges for scaling up, batch reactors ~2 m high with special UV lamps have been reported [56]. While the polydispersity of the MP may not be as important as the particle size threshold in determining the in vivo reaction, lower polydispersity may be desirable for manufacturers that wish to differentiate products based on particle size, as is currently done in the case of Restylane® and Perlane®. Using specially designed reactors or optimizing the viscosity and density of the dispersed and continuous phase may help in lowering the polydispersity of MP [57]. The polydispersity may also be decreased by filtering the MP through sieves. Upon filtering an MP suspension through a metallic sieve 115 μm in size, we found that the D90 decreased from 135 to 61 μm and, importantly, the relative span decreased from 1.54 to 0.74. Experimental variables such as the geometry of the vessel, the stirrer size and the experimental conditions, which are known significantly affect the size, polydispersity and yield of the MP [30,58], may be systematically optimized to obtain the desired MP. Changing the identity, concentration and functional groups of the non-crosslinked (bio)polymers can also be used to prepare MP with desired characteristics, such as pH sensitivity or surface charge.
5. Conclusions
In conclusion, a suspension photopolymerization-based platform to synthesize soft, spherical hydrogel MP of independently tunable size and stiffness for use as injectable biomaterials has been reported. Acrylated PEG, HA and gelatin were used to prepare MP that were injectable through 22 and 25 gauge needles with minimal distortion. Semi-IPN-based PEG MP were used as a model system to evaluate the effect of process parameters on MP characteristics. Increasing the stirring speed or the surfactant concentration decreased the size of the MP. Doubling the UV intensity or the photoinitiator concentration increased the MP size, but changing the UV exposure time had a minimal impact on the MP size. PEG MP with D90 ranging from 115 to 515 μm and stiffness values varying from 190 to 1600 Pa were prepared by systematically varying the process parameters. Importantly, the stiffness of the MP could be varied independent of their size. In vitro studies suggested that the PEG MP were cytocompatible. PEG MP with accessible amine groups at sufficient concentrations for potential surface modification were prepared. In addition, PLGA MS and NS were encapsulated inside the MP and the release of dexamethasone was demonstrated. Overall, this work reports a method to prepare soft, injectable and functionalizable hydrogel microparticles of natural or synthetic polymers with independently tunable size and stiffness for use as biomedical fillers.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Matias Porras Paniagua and Keriann Durgin for assistance with drug release studies and Dr. Jie Zhao for assistance with confocal microscopy. This work was funded by the Voice Health Institute.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.actbio.2014.02.021.
Appendix B. Figures with essential color discrimination
Certain figures in this article, particularly Fig. 6, is difficult to interpret in black and white. The full color images can be found in the on-line version, at http://dx.doi.org/10.1016/j.actbio.2014.02.021.
References
- 1.Lowe NJ, Maxwell CA, Patnaik R. Adverse reactions to dermal fillers: review. Dermatol Surg. 2005;31:1616–25. [PubMed] [Google Scholar]
- 2.Sankar V, McGuff HS. Foreign body reaction to calcium hydroxylapatite after lip augmentation. J Am Dent Assoc. 2007;138:1093–6. doi: 10.14219/jada.archive.2007.0321. [DOI] [PubMed] [Google Scholar]
- 3.Zielke H, Wolber L, Wiest L, Rzany B. Risk profiles of different injectable fillers: results from the Injectable Filler Safety Study (IFS Study) Dermatol Surg. 2008;34:326–35. doi: 10.1111/j.1524-4725.2007.34066.x. [DOI] [PubMed] [Google Scholar]
- 4.Herschorn S. Current use of injectable agents for female stress urinary incontinence. Rev Urol. 2005;7(Suppl 1):S12–21. [PMC free article] [PubMed] [Google Scholar]
- 5.Yang JH, Lee SM, Won CH, Chang SE, Lee MW, Choi JH, et al. Foreign body granuloma caused by hyaluronic acid/dextranomer microsphere filler injection. Int J Dermatol. 2012;51:1517–8. doi: 10.1111/j.1365-4632.2010.04795.x. [DOI] [PubMed] [Google Scholar]
- 6.Tezel A, Fredrickson GH. The science of hyaluronic acid dermal fillers. J Cosmet Laser Ther. 2008;10:35–42. doi: 10.1080/14764170701774901. [DOI] [PubMed] [Google Scholar]
- 7.Henly DR, Barrett DM, Weiland TL, O’Connor MK, Malizia AA, Wein AJ. Particulate silicone for use in periurethral injections: local tissue effects and search for migration. J Urol. 1995;153:2039–43. [PubMed] [Google Scholar]
- 8.FDAU. Guidance for industry and FDA staff vocal fold medialization devices –premarket notification [510 (k)] Submissions. 2004. p. 510. [Google Scholar]
- 9.Morhenn VB, Lemperle G, Gallo RL. Phagocytosis of different particulate dermal filler substances by human macrophages and skin cells. Dermatol Surg. 2002;28:484–90. doi: 10.1046/j.1524-4725.2002.01273.x. [DOI] [PubMed] [Google Scholar]
- 10.Gelb H, Schumacher HR, Cuckler J, Ducheyne P, Baker DG. In vivo inflammatory response to polymethylmethacrylate particulate debris: effect of size, morphology, and surface area. J Orthop Res. 1994;12:83–92. doi: 10.1002/jor.1100120111. [DOI] [PubMed] [Google Scholar]
- 11.Matlaga BF, Yasenchak LP, Salthouse TN. Tissue response to implanted polymers: the significance of sample shape. J Biomed Mater Res. 1976;10:391–7. doi: 10.1002/jbm.820100308. [DOI] [PubMed] [Google Scholar]
- 12.Bellis SL. Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials. 2011;32:4205–10. doi: 10.1016/j.biomaterials.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brodbeck WG, Patel J, Voskerician G, Christenson E, Shive MS, Nakayama Y, et al. Biomaterial adherent macrophage apoptosis is increased by hydrophilic and anionic substrates in vivo. Proc Natl Acad Sci U S A. 2002;99:10287–92. doi: 10.1073/pnas.162124199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamath S, Bhattacharyya D, Padukudru C, Timmons RB, Tang L. Surface chemistry influences implant-mediated host tissue responses. J Biomed Mater Res A. 2008;86:617–26. doi: 10.1002/jbm.a.31649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thevenot P, Hu W, Tang L. Surface chemistry influences implant biocompatibility. Curr Top Med Chem. 2008;8:270–80. doi: 10.2174/156802608783790901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khandwekar AP, Patil DP, Shouche Y, Doble M. Surface engineering of polycaprolactone by biomacromolecules and their blood compatibility. J Biomater Appl. 2011;26:227–52. doi: 10.1177/0885328210367442. [DOI] [PubMed] [Google Scholar]
- 17.Fakhari A, Berkland C. Applications and emerging trends of hyaluronic acid in tissue engineering, as a dermal filler and in osteoarthritis treatment. Acta Biomater. 2013;9:7081–92. doi: 10.1016/j.actbio.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edsman K, Nord LI, Ohrlund A, Larkner H, Kenne AH. Gel properties of hyaluronic acid dermal fillers. Dermatol Surg. 2012;38:1170–9. doi: 10.1111/j.1524-4725.2012.02472.x. [DOI] [PubMed] [Google Scholar]
- 19.Kablik J, Monheit GD, Yu L, Chang G, Gershkovich J. Comparative physical properties of hyaluronic acid dermal fillers. Dermatol Surg. 2009;35(Suppl 1):302–12. doi: 10.1111/j.1524-4725.2008.01046.x. [DOI] [PubMed] [Google Scholar]
- 20.Baumann L, Blyumin M, Saghari S. Dermal fillers. In: Weisberg E, editor. Cosmetic dermatology: principles and practice. Philadelphia, PA: McGraw-Hill Professional; 2007. pp. 191–211. [Google Scholar]
- 21.Helgeson ME, Chapin SC, Doyle PS. Hydrogel microparticles from lithographic processes: novel materials for fundamental and applied colloid science. Curr Opin Colloid Interface Sci. 2011;16:106–17. doi: 10.1016/j.cocis.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merkel TJ, Jones SW, Herlihy KP, Kersey FR, Shields AR, Napier M, et al. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc Natl Acad Sci U S A. 2011;108:586–91. doi: 10.1073/pnas.1010013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dendukuri D, Pregibon DC, Collins J, Hatton TA, Doyle PS. Continuous-flow lithography for high-throughput microparticle synthesis. Nat Mater. 2006;5:365–9. doi: 10.1038/nmat1617. [DOI] [PubMed] [Google Scholar]
- 24.Xu S, Nie Z, Seo M, Lewis P, Kumacheva E, Stone HA, et al. Generation of monodisperse particles by using microfluidics: control over size, shape, and composition. Angew Chem Int Ed Engl. 2005;44:724–8. doi: 10.1002/anie.200462226. [DOI] [PubMed] [Google Scholar]
- 25.Haghgooie R, Toner M, Doyle PS. Squishy non-spherical hydrogel microparticles. Macromol Rapid Commun. 2010;31:128–34. doi: 10.1002/marc.200900302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Acharya G, Shin CS, McDermott M, Mishra H, Park H, Kwon IC, et al. The hydrogel template method for fabrication of homogeneous nano/microparticles. J Control Release. 2010;141:314–9. doi: 10.1016/j.jconrel.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 27.Sahiner N, Jha AK, Nguyen D, Jia XQ. Fabrication and characterization of cross-linkable hydrogel particles based on hyaluronic acid: potential application in vocal fold regeneration. J Biomater Sci-Polym Ed. 2008;19:223–43. doi: 10.1163/156856208783432462. [DOI] [PubMed] [Google Scholar]
- 28.John G, Morita M. Synthesis and characterization of photo-cross-linked networks based on L-lactide/serine copolymers. Macromolecules. 1999;32:1853–8. [Google Scholar]
- 29.Kumar A, Lahiri SS, Singh H. Development of PEGDMA: MAA based hydrogel microparticles for oral insulin delivery. Int J Pharm. 2006;323:117–24. doi: 10.1016/j.ijpharm.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 30.Rodrigo R, Toro CA, Cuellar J. Influence of the geometric factors of the experimental device used in suspension polymerization on the properties of poly(styrene-co-divinylbenzene) microparticles. J Appl Polym Sci. 2012;124:1431–46. [Google Scholar]
- 31.Bryant SJ, Nuttelman CR, Anseth KS. Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J Biomater Sci Polym Ed. 2000;11:439–57. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 32.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials. 2009;30:6702–7. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams CG, Malik AN, Kim TK, Manson PN, Elisseeff JH. Variable cytocompatibility of six cell lines with photoinitiators used for polymerizing hydrogels and cell encapsulation. Biomaterials. 2005;26:1211–8. doi: 10.1016/j.biomaterials.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 34.Panda P, Ali S, Lo E, Chung BG, Hatton TA, Khademhosseini A, et al. Stop-flow lithography to generate cell-laden microgel particles. Lab Chip. 2008;8:1056–61. doi: 10.1039/b804234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma B, Fermanian S, Gibson M, Unterman S, Herzka DA, Cascio B, et al. Human cartilage repair with a photoreactive adhesive-hydrogel composite. Sci Transl Med. 2013;5 doi: 10.1126/scitranslmed.3004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ICRS. Hydrogel Implant GelrinC Demonstrates Impressive Recovery Rates for Patients with Knee Cartilage Damage. 2013 press release. http://www.regentis.co.il/files/files/ICRS%202013%20press%20release.pdf.
- 37.Van Den Bulcke AI, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M, Berghmans H. Structural and rheological properties of methacrylamide modified gelatin hydrogels. Biomacromolecules. 2000;1:31–8. doi: 10.1021/bm990017d. [DOI] [PubMed] [Google Scholar]
- 38.Camci-Unal G, Nichol JW, Bae H, Tekin H, Bischoff J, Khademhosseini A. Hydrogel surfaces to promote attachment and spreading of endothelial progenitor cells. J Tissue Eng Regen Med. 2012;7:337–47. doi: 10.1002/term.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim DH, Martin DC. Sustained release of dexamethasone from hydrophilic matrices using PLGA nanoparticles for neural drug delivery. Biomaterials. 2006;27:3031–7. doi: 10.1016/j.biomaterials.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 40.Ju YM, Yu B, West L, Moussy Y, Moussy F. A dexamethasone-loaded PLGA microspheres/collagen scaffold composite for implantable glucose sensors. J Biomed Mater Res A. 2010;93:200–10. doi: 10.1002/jbm.a.32512. [DOI] [PubMed] [Google Scholar]
- 41.Habeeb AF. Determination of free amino groups in proteins by trinitrobenzenesulfonic acid. Anal Biochem. 1966;14:328–36. doi: 10.1016/0003-2697(66)90275-2. [DOI] [PubMed] [Google Scholar]
- 42.Snyder SL, Sobocinski PZ. An improved 2,4,6-trinitrobenzenesulfonic acid method for the determination of amines. Anal Biochem. 1975;64:284–8. doi: 10.1016/0003-2697(75)90431-5. [DOI] [PubMed] [Google Scholar]
- 43.Brown E, Zhang H, Forman NA, Maynor BW, Betts DE, DeSimone JM, et al. Shear thickening in densely packed suspensions of spheres and rods confined to few layers. J Rheol. 2010;54:1023–46. [Google Scholar]
- 44.Menut P, Seiffert S, Sprakel J, Weitz DA. Does size matter? Elasticity of compressed suspensions of colloidal- and granular-scale microgels. Soft Matter. 2012;8:156–64. [Google Scholar]
- 45.Saralidze K, Koole LH, Knetsch MLW. Polymeric microspheres for medical applications. Materials. 2010;3:3537–64. [Google Scholar]
- 46.Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int J Pharm. 2008;364:298–327. doi: 10.1016/j.ijpharm.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 47.Sundaram H, Voigts B, Beer K, Meland M. Comparison of the rheological properties of viscosity and elasticity in two categories of soft tissue fillers: calcium hydroxylapatite and hyaluronic acid. Dermatol Surg. 2010;36(Suppl 3):1859–65. doi: 10.1111/j.1524-4725.2010.01743.x. [DOI] [PubMed] [Google Scholar]
- 48.Barrera DA, Zylstra E, Lansbury PT, Langer R. Synthesis and RGD peptide modification of a new biodegradable copolymer – poly(lactic acid-co-lysine) J Am Chem Soc. 1993;115:11010–1. [Google Scholar]
- 49.Bhatnagar P, Nixon AJ, Kim I, Kameoka J. Protein functionalized micro hydrogel features for cell–surface interaction. Biomed Microdevices. 2008;10:567–71. doi: 10.1007/s10544-008-9167-6. [DOI] [PubMed] [Google Scholar]
- 50.Rimmer S, Johnson C, Zhao B, Collier J, Gilmore L, Sabnis S, et al. Epithelialization of hydrogels achieved by amine functionalization and co-culture with stromal cells. Biomaterials. 2007;28:5319–31. doi: 10.1016/j.biomaterials.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 51.Saralidze K, Knetsch ML, van Hooy-Corstjens CS, Koole LH. Radio-opaque and surface-functionalized polymer microparticles: potentially safer biomaterials for different injection therapies. Biomacromolecules. 2006;7:2991–6. doi: 10.1021/bm0603903. [DOI] [PubMed] [Google Scholar]
- 52.Rowley JA, Madlambayan G, Mooney DJ. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials. 1999;20:45–53. doi: 10.1016/s0142-9612(98)00107-0. [DOI] [PubMed] [Google Scholar]
- 53.Alam M, Gladstone H, Kramer EM, Murphy JP, Jr, Nouri K, Neuhaus IM, et al. ASDS guidelines of care: injectable fillers. Dermatol Surg. 2008;34:S115–48. doi: 10.1111/j.1524-4725.2008.34253.x. [DOI] [PubMed] [Google Scholar]
- 54.Berkland C, King M, Cox A, Kim K, Pack DW. Precise control of PLG microsphere size provides enhanced control of drug release rate. J Control Release. 2002;82:137–47. doi: 10.1016/s0168-3659(02)00136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brooks BW. Suspension polymerization processes. Chem Eng Technol. 2010;33:1737–44. [Google Scholar]
- 56.Gao ZQ, Grulke EA, Ray AK. Synthesis of monodisperse polymer microspheres by photopolymerization of microdroplets. Colloid Polym Sci. 2007;285:847–54. [Google Scholar]
- 57.Dowding PJ, Goodwin JW, Vincent B. Production of porous suspension polymers using a continuous tubular reactor. Colloid Polym Sci. 2000;278:346–51. [Google Scholar]
- 58.Arshady R. Suspension, emulsion and dispersion polymerization – a methodological survey. Colloid Polym Sci. 1992;270:717–32. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.