

Abstract
Over the last 50 years, the posttranslational modification (PTM) of proteins has emerged as a central mechanism for cells to regulate metabolism, growth, differentiation, cell-cell interactions, and immune responses. By influencing protein structure and function, PTM leads to a multiplication of proteome diversity. Redox-dependent PTMs, mediated by environmental and endogenously generated reactive species, induce cell signaling responses and can have toxic effects in organisms. PTMs induced by the electrophilic by-products of redox reactions most frequently occur at protein thiols; other nucleophilic amino acids serve as less favorable targets. Advances in mass spectrometry and affinity-chemistry strategies have improved the detection of electrophile-induced protein modifications both in vitro and in vivo and have revealed a high degree of amino acid and protein selectivity of electrophilic PTM. The identification of biological targets of electrophiles has motivated further study of the functional impact of various PTM reactions on specific signaling pathways and how this might affect organisms.
Introduction
Our still-blossoming insight into genetically encoded cellular communication mechanisms stemmed from two seminal advances rooted in the 1950s. When studying steroid hormone function, Mueller and Jensen pioneered the concept that mRNA transcription and translation into protein could be regulated by receptor-ligand interactions (1). The posttranslational modification (PTM) and regulation of proteins by O-phosphorylation, first described by Kennedy, Krebs, and Fischer (2), revealed another exciting aspect of metabolic regulation—that protein function could be modified by secondary chemical reactions that are linked to metabolic and environmental stimuli. Propelled by the discovery of protein O-phosphorylation, additional PTMs of proteins have been discovered that expand the scope of the functional proteome one to two orders of magnitude over that encoded by the genome (3).
Approximately 5% of the genome encodes enzymes that catalyze the PTM of proteins, which dictates proteome diversification and plasticity. In addition to O-phosphorylation, PTMs include acetylation, acylation, adenosine diphosphate–ribosylation, biotinylation, carboxylation, glycosylation, hydroxylation, methylation, ubiquitinylation, SUMOylation, and sulfation, to name a few. These structural modifications of proteins can affect catalytic activity, trafficking, stability, and immunogenicity, thus linking biochemical stimuli with the physiological responses of organisms.
Redox Signaling
Within the ever-expanding universe of PTMs, the regulation of protein structure and function by oxidation-reduction (redox) reactions has emerged as a vibrant area of investigation. Many discoveries in redox biology continue to emerge since McCord and Fridovich’s 1969 discovery of biological superoxide generation and the existence of the oxygen radical–scavenging enzyme superoxide dismutase (4). Soon thereafter, it was appreciated that activated inflammatory cells and various forms of metabolic stress (such as ischemia-reperfusion, hyperoxia, and xenobiotic exposure) increased tissue production rates and steady-state concentrations of reactive oxygen species (5–8). Reactive species, first viewed as toxic by-products of inflammatory responses and xenobiotic exposure (9, 10), are now also appreciated as signaling mediators that are produced and inactivated in a regulated manner. Cellular redox reactions and their regulation rely on an extensive array of proteins and small molecules that evolved (i) to generate reactive species in a regulated manner as host defense and signaling intermediates, (ii) to scavenge catalytically or to inactivate reactive species, and (iii) to sense the cellular redox milieu chemically by undergoing oxidation and reduction reactions.
The discovery of nitric oxide (·NO) as a free radical mediator of cell signaling and inflammation expanded the scope of redox-regulated cell signaling. In addition to activating guanylate cyclase through heme-iron coordination, ·NO reacts directly with superoxide (O2·−) and indirectly with hydrogen peroxide (H2O2)–derived species, to yield some of the most reactive molecules found in biology (9, 11, 12). Through the formation of secondary ·NO-derived species, such as peroxynitrite (ONOO−) and nitrogen dioxide (·NO2), ·NO thus accelerates the chemical reaction rate and increases the breadth of reactions that transduce redox signaling. In cells and tissues, mediators of redox signaling are a diverse population of small molecules and proteins that serve as molecular sensors of cellular metabolic and stress status. When alterations in the redox state of the target molecules act as sensors, organisms respond by modulating many cellular functions, including gene expression, enzyme catalysis, cytoskeletal function, membrane transport, intermediary metabolism, respiratory chain function, and neurotransmission.
The broad reactivities and short half-lives of redox signaling mediators have engendered productive debate regarding the rates of generation, steady-state concentrations, chemical identities, specific molecular targets, and mechanisms of signal transduction of particular reactive species. Notably, nonbiological mediator concentrations are often required to produce detectable responses in model systems, sometimes promoting primary and secondary reactions that would be more limited or absent in vivo. Finally, if labile PTMs are induced by redox mediators, the chemical characterization and quantification of these modifications becomes a challenge. Overall, unappreciated reactions and the analytical challenges inherent in studying short-lived redox intermediates have both churned controversy and motivated investigators to advance knowledge in this area at a rapid pace.
Herein, we focus attention on one class of reactive species that stem from redox reactions, electrophiles. These molecules attract electrons, and by virtue of this property, electrophiles transduce cell signaling by mediating reversible structural and functional changes in susceptible protein targets. We also offer perspective about what constrains the regulated signaling actions of biological electrophiles, as opposed to initiating toxicological outcomes.
Cysteine Thiol—A Primary Target of Redox Reactions
Cysteine is the most nucleophilic amino acid of proteins and readily undergoes Michael addition with electrophiles (13, 14). Simply put, Michael addition is the reaction of a Michael donor (nucleophiles, such as amine or thiol groups) with a Michael acceptor (typically an alkene with an electron-withdrawing substituent, such as a nitro or keto group). Other key biological nucleophiles include the imidazole in the histidine side chain; the amine in the lysine side chain; and the amino-containing substituents of sugars, phospholipids, and DNA. Although less nucleophilic than thiols, these species are also susceptible to electrophile reaction. The thiol of cysteine (Cys-SH) is responsible for its high reactivity with secondary products of oxygen-dependent and NO-dependent reactions and the downstream electrophilic products generated by reaction with biomolecules (15–17). Partially reduced oxygen species (18), NO-derived species (17, 19–21), electrophilic by-products of redox reactions, and glutathione (GSH) can transduce redox signaling by modifying protein cysteine residues to sulfenic (Cys-SO−), sulfinic acid (Cys-SO2−), sulfonic acid (Cys-SO32−), S-nitrosothiol (Cys-S-NO), and disulfide derivatives. Disulfides include cysteinylated proteins (Cys-S-SP), cysteinylated glutathione (Cys-S-SG), and glutathionylated protein (GS-SP), as well as intramolecular disulfides and disulfide cross-links between different proteins (22–24). Because of the diversity of reactive species that are formed during inflammation, metabolic stress, and ·NO signaling, it is not tenable to expect the formation of highly selective thiol-electrophile adduct formation, the generation of a predominant thiol oxidation state, or the formation of predominantly S-nitrosation products. Take, for example, the signaling mediator ·NO, which does not directly react with thiols at an appreciable rate. Rather, ·NO mediates protein S-nitrosation either through transnitrosation from another RS-NO derivative or upon ·NO reaction with a sulfenyl radical (often termed thiyl radical) intermediate after metal-catalyzed thiol oxidation. These reactions are also occurring in a milieu where the generation of O2·−, other partially reduced oxygen species, and by-products of their reactions are ubiquitous under aerobic conditions. This pro-oxidative environment can thus result in an array of biomolecule reactions mediated by ·NO that are independent of NO stimulation of cyclic guanine monophosphate (cGMP) production after activation of soluble guanylate cyclase. Notably, reactions of ·NO in an aerobic environment will also include the concurrent formation of disulfides, the generation of the further-oxidized thiol derivatives noted above, protein and lipid nitration, and the production of various electrophile adducts to be described herein.
Nucleophiles, such as cysteine, react with electrophiles through a mechanism involving donation of a pair of electrons from the nucleophile to the electrophile (electrophilic attack), resulting in the formation of a covalent bond. When this reaction involves an alkyl group as the electrophile, then this reaction is termed S-alkylation (Fig. 1). The nucleophilic reactivity of the cysteine thiol strongly depends on the surrounding chemical milieu and how this in turn affects a thiol’s acid dissociation constant (pKa). The pKa of the thiol of the amino acid cysteine in solution is 8.33, with most protein-associated cysteine thiols also displaying pKa values between 8.2 and 8.5. This translates to relatively low protein thiol reactivity at a typical intracellular pH of 7.2, because only ~10% of thiols are deprotonated to the reactive thiolate anion (Cys-S−) species that is much more readily oxidized and alkylated than neutral protonated thiols. The pKa of the cysteine thiol is decreased when proximal to (i) basic amino acids, such as histidine, lysine, and arginine, that can deprotonate the cysteine and form a hydrogen-bonded ion pair with the cysteine thiolate anion; (ii) the aromatic amino rings of tyrosine or tryptophan that can undergo pi interactions with the SH group; or (iii) metal centers, such as heme-metal complexes, that can interact with the thiol. All of these can facilitate deprotonation to the more reactive thiolate anion, promoting more facile Michael addition or oxidation reactions (25, 26). Therefore, the secondary and tertiary structure of proteins, local pH changes dictated by tissue oxygenation and metabolic state, and the presence of alternative scavenging species will all strongly influence cysteine reaction with electrophiles. Thus, site-specific cysteine deprotonation to the thiolate anion, an event shaped by these anatomic and metabolic factors, can lend target molecule specificity to thiol redox modification. This precept is borne out by a mass spectrometry–based profiling study, where only a limited and reproducible number of highly selective protein sites for electrophilic adduction were identified in a human cell line (897 cysteine residues on 539 proteins). Specific surrounding protein structural motifs influenced the extent of cysteine reactivity with different electrophiles, which indicated that the physical microenvironment of protein cysteine residues undergoing adduction can substantially modulate the reactivity of individual cysteine thiols with electrophiles (27).
Fig. 1.
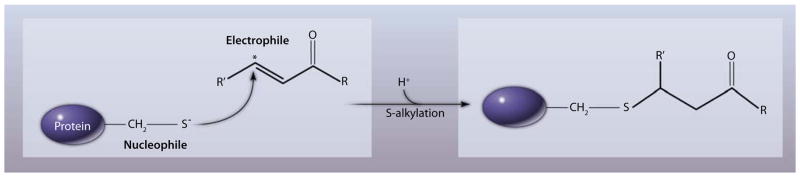
PTM of a protein by an electrophile. The electrophile attacks the nucleophilic thiolate anion of a reactive cysteine residue through Michael addition. Asterisk denotes the electrophilic carbon.
Defining a Biological Electrophile
An electrophile (“electron-lover”) is attracted to electrons and undergoes chemical reactions by accepting an electron pair in order to chemically bond with a nucleophile. Unsaturated compounds with electron-withdrawing substituents are “soft” Michael acceptors and are susceptible to reaction with a “soft” base (nucleophile), such as a thiolate. “Soft” acids and bases tend to interact covalently, as opposed to “hard” acid and bases, which tend to interact ionically. This means that protein thiols interact with the carbonyl carbon (starred in Fig. 1) when undergoing Michael addition reaction.
Arthur Michael first reported in 1887 that olefins (also known as alkenes, organic molecules with at least one carbon-carbon double bond) conjugated with electron-withdrawing groups are susceptible to attack by nucleophiles. This type of reaction was named after Michael, thus leading to the terms Michael acceptor (the electrophile), Michael donor (the nucleophile), and Michael addition reaction (a type of electrophile-nucleophile reaction). In the context of the electrophilic signaling reactions discussed herein, typical electron-withdrawing groups present on biological Michael acceptors can include (but are not limited to) aldehyde (-COH); acyl (-COR, where R typically stands for a carbon if the group is a keto); carboxylic acid (-COOH); ester (-COOR, where R typically stands for a carbon or hydrogen); halide (-Cl, -F, -Br, -I); sulfonyl (-SOn); sulfone (-SO2R, where R typically stands for a hydrogen or carbon); sulfonic acid (-SO3H); and nitro (-NO2) substituents. It can be generalized that alkenes or alkynes (molecules with a triple bond between two carbons, also called acetylenes) that are conjugated to electron-withdrawing groups will display Michael acceptor functionality.
The most prevalent electron-withdrawing group present in both xenobiotic and endogenously produced electrophiles is a carbonyl (or acyl) substituent, with a carbon-oxygen double bond conjugated to an alkene in the electrophiles. Virtually all classes of biomolecules (protein, lipid, carbohydrate, DNA, and RNA) are susceptible to oxidation reactions that can generate the carbonyl (or acyl) functional group. Because of overall abundance and susceptibility to oxidation, the unsaturated fatty acids of membranes and lipoproteins are key cellular sources of electrophilic derivatives after autocatalytic or enzymatically catalyzed peroxidation reactions. Various electrophiles, including fatty acid derivatives, react with the thiol of GSH at second-order reaction rate constants ranging from ~ 1 to 350 M−1s−1 (Table 1). Of note, during oxidative inflammatory conditions, protein carbonylation is primarily attributable to the electrophilic adduction of lipid-derived aldehydes, rather than to the direct oxidation of susceptible amino acids to carbonyl derivatives (28).
Table 1.
Rate constants of electrophile reaction with GSH.
Cells and tissues are continuously exposed to electrophiles that stem from exogenous and endogenous sources. Species having electrophilic reactivity are ingested with foodstuffs and are endogenously produced as metabolic by-products after exposure to compounds that undergo oxidation-reduction reactions (for example, phenolic xenobiotics and anticancer drugs) (29). Electrophiles are also present in and can be generated by environmental sources (for example, combustion products, such as those in cigarette smoke) (30). In addition, insects and plants produce various defensive chemically irritating electrophiles that protect the host from other insects or herbivores (31, 32). Electrophiles are also formed in abundance by the oxidative conditions inherent during aerobic metabolism and inflammation (28, 33). A sampling of the chemical structures of biological electrophiles and their electrophilic centers is provided in Fig. 2. Specific examples of biological electrophiles are described individually below.
Fig. 2.

Chemical structures of selected biological and environmentally induced electrophiles. Asterisk denotes the electrophilic carbon. The target of electrophiles, a protein with the reactive thiol (nucleophile), is shown in the center.
α,β-Unsaturated aldehydes
α,β-Unsaturated aldehydes come from endogenous and exogenous sources and include the lipid peroxidation by-products 4-hydroxynonenal (4-HNE), 4-oxononenal (4-ONE), and acrolein. The last-named is inhaled with cigarette smoke; is formed endogenously during lipid peroxidation, or protein and amino acid oxidation reactions; and is present in cooked oils and other foodstuffs (34, 35). Elevated plasma concentrations of acrolein are detected in patients with chronic renal failure, and the abundance of proteins adducted by acrolein is increased in tissues obtained from patients with Alzheimer’s disease, Parkinson’s disease, atherosclerosis, and chronic obstructive lung disease. The potent electrophilic reactivity of acrolein leads to its S-alkylation of several proteins. For example, animal model studies reveal that acrolein modifies extracellular matrix proteins in the lung, resulting in enhanced macrophage adhesion and activation (36). Proteomic and immunochemical analyses also reveal that acrolein impairs cardiac function by covalently reacting with proteins in the cardiac sarcomeric and cytoskeletal network, as well as with enzymes involved in myocardial energy metabolism (34). Of note, many of these cardiovascular effects of administered acrolein are reversible and can be limited by administering the low-molecular-weight thiol N-acetylcysteine to acrolein-treated animal models (34).
4-HNE and 4-ONE are produced endogenously during peroxidation of membrane and lipoprotein polyunsaturated fatty acids (37, 38). Cell and animal model studies reveal that these electrophiles can traverse intracellularly, escaping reaction with cytosolic GSH and GSH transferase defenses to diffuse to the nucleus and to react with both DNA-associated proteins and DNA bases. Generally considered cytotoxic, 4-HNE and 4-ONE act in part by stimulating cell cycle arrest and c-Jun N-terminal kinase (JNK)–dependent apoptotic signaling (39, 40). In addition to forming Michael adducts with cysteine, histidine, and lysine, and a Schiff’s base with lysine, 4-ONE also can induce intra- and intermolecular cross-links. For example, α- and β-tubulin are differentially modified by 4-HNE and 4-ONE, with 4-HNE leading to adduction of cysteine residues at physiologically relevant concentrations. 4-ONE induces protein cross-linking upon modification of Lys residues by reductive propylation (41). In phagocytes, the PTM of protein kinase C by 4-HNE inhibits the phagocytic function of these cells (42). The Cys120 of human epithelial fatty acid–binding protein (e-FABP) is also a target of 4-HNE reaction. Despite irreversible S-alkylation of this residue, intracellular e-FABP remains functional and continues to bind fatty acids noncovalently; thus, this modification of e-FABP may serve a protective or antioxidant function (43). Finally, the observation that 4-HNE potently can induce multiple stress response–related gene expression pathways suggests that organisms have evolved mechanisms to cope with low endogenous rates of production of 4-HNE and related α,β-unsaturated aldehydes (39, 44).
ω-6 and ω-3 unsaturated fatty acids
A common structural feature of some electrophilic fatty acid derivatives is a cyclopentenone prostaglandin ring containing an α,β-unsaturated carbonyl group [see 15-deoxy-Δ12,14-prostaglandin J2 (15-deoxy-PGJ2) in Fig. 2] or a more linear acyl chain with an α,β-unsaturated carbonyl (4-HNE and acrolein in Fig. 2). Cyclopentenone prostaglandins are formed by enzymatic peroxidation of the ω-6 fatty acid, arachidonic acid (20:4), and include 15-deoxy-PGJ2, as well as E1, E2, G2, F2, and F1α prostaglandin derivatives (45). Arachidonic acid, oxidized by lipoxygenase-dependent catalysis or nonenzymatic oxidative and free radical pathways, also yields α,β-unsaturated carbonyls including 5-, 12-, and 15-oxo-eicosatetraenoic acid and other derivatives (45–47). Longer-chain, more unsaturated ω-3 unsaturated fatty acids [eicosapentaenoic acid (20:5) and docosahexaenoic acid (22:6)] also undergo both enzymatic and nonenzymatic oxidation to yield an array of electrophilic cyclopentenone derivatives and other α,β-unsaturated carbonyl products (48). The signaling actions of oxidized ω-3 unsaturated fatty acids can, in part, account for physiologically beneficial actions ascribed to dietary polyunsaturated fatty acids. This is ascribed to the redox-dependent generation of electrophilic ω-3 unsaturated fatty acid oxidation products that are able to activate the signaling pathways described in the sections below (49–52).
Nitro-fatty acids
Nitro derivatives of unsaturated fatty acids (NO2-FAs) are generated endogenously upon oxidation of unsaturated fatty acids by NO-derived species, a reaction ultimately mediated by ·NO2 (53). These species are formed both during inflammatory conditions, where inducible nitric oxide synthase activity is elevated, and during oxidative stress by both ·NO and nitrite (NO2−)–dependent nitration reactions (54–56). The alkenyl nitro configuration of NO2-FAs is responsible for the electrophilic reactivity of the β-carbon adjacent to the NO2-bonded carbon (57, 58) (see 9-nitro-octadecenoic acid in Fig. 2). In red cells, NO2-FAs react with glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which inhibits its catalytic activity and induces trafficking of this protein to the plasma membrane. This process is facilitated by the hydrophobic interactions that will occur between the GAPDH–NO2-FA adduct and membrane lipids (57). NO2-FAs exhibit broad anti-inflammatory signaling actions through the modulation of transcriptional regulatory mechanisms (53), including serving as agonists of peroxisome proliferator–activated receptors (PPARs) (59–61). NO2-FAs are by-products of oxidative inflammatory reactions that in turn promote signaling reactions that facilitate adaptive gene expression responses to a stressful environment. PTM reactions induced by NO2-FAs are reversible in vivo; the NO2-FAs adducted to protein can transfer between thiols present on both extra- and intracellular proteins, as well as glutathione (55, 57, 62, 63).
Detecting Protein Adduction by Electrophiles
The in vivo detection and quantification of protein adduction by electrophiles initially relied on spectrophotometric and radiolabel-based analyses (64, 65). Although useful as indices of the net conversion of endogenous or exogenous molecules to protein-reactive electrophilic species, these approaches often lacked the sensitivity and precision required to shed light on specific protein or amino acid targets. Even when coupled with electrophoretic separation and immunoaffinity-based protein identification and purification strategies, molecular insight was confounded by specificity and sensitivity limitations (66). These studies of the nature and extent of electrophilic PTM reactions laid a foundation of knowledge and provided inspiration for mass spectrometry (MS)–based advances that can be supplemented by immunoaffinity purification of protein adducts, genetic manipulation of susceptible nucleophilic amino acids, and chemistries that more selectively profile electrophilic species and their adduction sites. With these tools, investigators have identified both critical amino acid targets of electrophiles within a particular protein and specific protein targets within different organelles, cell types, and tissues.
Enzymatic protein hydrolyzates obtained from biological sources are expected to have a relatively low abundance of electrophilic PTM in the absence of acute dietary or toxicological exposure; thus, any adducted species that are endogenously present may be below the limits of detection of tandem MS precursor ion or neutral loss scans. The use of scoring algorithms for spectral analysis (SALSA) of MS/MS spectra has enhanced the sensitivity and specificity of detection of peptide PTM by electrophiles (67). Also useful are activity-based protein-profiling strategies that permit fractionation of protein mixtures into electrophile-reactive subproteomes. Below, we describe some different “baits” that can be utilized to “fish” for electrophile targets and to define the factors that discriminate electrophile reactivity (Fig. 3).
Fig. 3.

Three different chemical approaches for detecting specific protein-electrophile reactions. (A) To identify the amino acid target of an electrophile (E) of interest, the azide-coupled electrophile is reacted with a protein and an alkyne-coupled tag (for example, an alkyne adduct of biotin or rhodamine) is added. The azide and the alkyne react irreversibly to form a stable triazole product. The protein-electrophile-triazole tag complex is isolated by affinity purification (for example, with streptavidin beads). After proteolysis with trypsin, labeled peptides are detected by MS or fluorescence-based strategies. (B) After adding a biotinylated electrophile to a protein, the complex is either trypsinized and reacted with avidin or treated with avidin and then trypsinized (the second approach is less sensitive). The covalently modified site on the protein is determined by mass spectrometry. (C) After electrophile reaction with a target protein or tissue, followed by sample solubilization, an excess of β-ME is added to detect and quantify the protein-adducted electrophile. Reversibly bound electrophiles undergo transfer from the protein to β-ME. The E–β-ME complex is detected by mass spectrometry, and the concentration is determined by comparison with an internal standard. The protein to which the electrophile is attached is glyceraldehyde-3-phosphate dehydrogenase (PDB accession number 1rm5).
Click chemistry
This robust reaction strategy capitalizes on the introduction of small and selectively reactive components into a larger structure, so that an irreversible chemical bond is formed with an added target molecule that facilitates target identification and affinity capture. Of great utility in identifying electrophile targets is the process of introducing an azido group into the electrophile and then using a copper (Cu) catalyst to induce covalent linkage with a reporter molecule or an affinity-capture reagent having an alkyne functional group. Overall, this “click” reaction is termed an azide alkyne Huisgen cycloaddition and has the advantage of reducing purification times for electrophile-biomolecule adducts and increasing the sensitivity of adduct detection (68). Modified azido-reactive difluoro cyclooctyne derivatives have been incorporated into living cells, allowing Cu-independent cycloaddition with azido derivatives of biomolecules (for example, lipids and carbohydrates). These difluoro cyclooctyne reagents are useful because they avoid the promotion of secondary and possibly artifactual oxidative reactions that might be mediated by the Cu catalyst that they replace, and they also are amenable to use in live-cell studies (69).
Using a click-chemistry strategy, biomimetic azido and alkynyl derivatives of the fatty acid oxidation product 4-HNE have been prepared and utilized to define protein targets of this electrophilic α,β-unsaturated aldehyde (70). Although both derivatization approaches were informative, the most sensitive and discriminating results were obtained upon affinity capture of azido-HNE–adducted protein. This is accomplished by (i) addition of alkyne-labeled biotin, (ii) enrichment of the “click product” between HNE and biotin with streptavidin beads, (iii) digestion of affinity-purified proteins with trypsin, and (iv) “shotgun proteomic” identification of protein-electrophile adducts by liquid chromatography separation of peptides in concert with electrospray ionization (ESI) liquid chromatography and tandem MS (LC-MS/MS) detection. If adducted peptides are abundant enough, peptide sequencing can also reveal specific amino acids responsible for electrophile adduction. This azido-alkyne click chemistry approach also facilitated comparison of the protein targets of distinctly different carbon electrophiles (a phenylsulfonate ester, linear epoxide, spiral epoxide, and the α,β-unsaturated ketone α-chloroacetamide) (71). Rhodamine azide was used for in-gel fluorescence scanning of protein targets in the soluble fraction of murine organ homogenates or COS-7 cells after resolution by SDS–polyacrylamide gel electrophoresis. More detailed analysis, including the study of engineered cells expressing proteins with specific nucleophilic amino acid mutations, revealed that different electrophile classes displayed unique amino acid specificities after reaction with different proteins. In addition to noncomplexed cysteine residues of proteins, other targets included cysteine thiols participating in metal coordination and the nucleophilic amino acids tyrosine, glutamate, and aspartate (71).
Biotin-electrophile probes
The linkage of biotin to iodoacetamide- and maleimide-based electrophiles has provided a chemical handle for the subsequent harvesting of target proteins from cells after capture by avidin. Alternatively, a more sensitive approach can be used where, after biotin-electrophile reaction, fractionated organelles or whole-cell lysates are subjected to proteolysis and then avidin is used to capture peptides that are conjugated to biotin-electrophile complexes. Once fractionated, peptides and corresponding proteins are identified and sequenced by high-performance liquid chromatography (HPLC)–MS/MS. Addition of biotinylated electrophiles to either intact cells or subcellular fractions (mitochondria, microsomes, nuclei, or cytosol) has provided important new insight into electrophile-sensitive subproteomes (27, 30, 72–74). This strategy has also been extended to the biotin labeling of complex lipids that are then subjected to per-oxyl radical-induced oxidation (75, 76). The parent biotinylated 1-palmitoyl-2-linoleoyl-phosphatidylcholine, upon oxidation, yields a spectrum of electrophilic products that preferentially targeted apolipoprotein A1 when reactions were conducted in human plasma ex vivo (76). Electrophile-reactive amino acids identified by this lipid affinity–tagging approach were located in a functionally important region of the lipoprotein. Notably, product profiles revealed that modified peptides predominantly reflected reactions of carboxylated products of lipid oxidation [such as phospholipid-esterified 9,12-dioxo-10(E)-dodecenoic acid, 9-hydroxy-12-oxo-10(E)-dodecenoic acid, 7-oxoheptanoic acid, 8-oxooctanoic acid, and 9-oxononanoic acid], as opposed to species derived from the ω-terminus of linoleate (such as 4-HNE).
β-Mercaptoethanol exchange
An exchange reaction between an excess of added β-mercaptoethanol (β-ME) and electrophile-adducted biomolecules, when coupled with HPLC-MS/MS, permits sensitive detection and quantification (with standards) of reversibly adducted electrophiles in plasma, organelles, cells, and tissue homogenates (62). Because β-ME is the major ion undergoing neutral loss, the parent electrophile can be further structurally characterized by additional fragmentation. With this strategy, electrophilic NO2-FAs and their metabolites, present in both the free form and after adduction to protein and low-molecular-weight targets, have been profiled (55, 63, 77). This β-ME exchange reaction is applicable to fishing for other reversibly adducted electrophiles and has revealed the presence of electrophilic NO2-FAs in individual proteins that were resolved by affinity capture and electrophoretic separation. Finally, this approach provided evidence that electrophilic fatty acid derivatives could transfer between individual proteins and from protein to GSH and that electrophilic NO2-FAs are protected from secondary decay reactions when adducted to GSH or protein (63, 78).
Electrophiles as Signaling Mediators
Early appreciation of the occurrence and importance of biomolecule reactions with electrophiles came from studies of chemical carcinogenesis and mutations induced by electrophile adduction to DNA bases (79). From this work, an initial viewpoint evolved, namely, that electrophiles mediate noxious reactions by forming adducts with biomolecules. Evolution of a more contemporary view, that electrophile generation and reaction comprises an important aspect of cell adaptation to environmental and metabolic stimuli, was catalyzed by two related discoveries in redox biology. First, the superfamily of GSH S-transferases was described as enzymes that catalytically conjugate GSH with electrophiles (80). This revealed that organisms evolved mechanisms that accelerate the conjugation of xenobiotic-derived and endogenously generated electrophiles and then export these adducts from cells through specific transport mechanisms (81). Second, the landmark discovery was made that a critical denominator of many inhibitors of carcinogenesis was their degree of electrophilic reactivity, which led to the term “chemopreventive agent” (79). Linked with the relative electrophilic reactivity of different compounds was a corresponding relative ability to increase the gene expression of antioxidant response element (ARE)–regulated enzymes, such as GSH S-transferases and quinone reductases (82). This insight propelled the subsequent description of multiple oxidant– and electrophile-regulated transcription factors controlling the transcription of genes encoding proteins that defend against the chemical stress caused by reactive species.
The discovery of integrated systems that control, sense, and respond to the abundance of electrophiles reinforces the concept that organisms have developed regulated mechanisms whereby biochemical detente can be established with electrophiles stemming from the diet, xenobiotics, or oxidative inflammatory conditions. This new understanding of endogenous signaling mechanisms that capitalize on electrophilic reactivity also presents new vistas for drug discovery and the treatment of cell growth disorders, metabolic diseases, and inflammatory disorders. The following are key biological targets of electrophiles that result in downstream signaling events, with additional information detailed in Table 2.
Table 2.
Biological targets of electrophiles and the reactive amino acid residue(s). BMCC, 1-biotinamido-4-butane; IAB, N-iodoacetyl-N-biotinylhexylenediamine; NEPP, neurite electrophilic outgrowth–promoting prostaglandin.
| Target | Electrophile | Reactive amino acid residue | Selected references |
|---|---|---|---|
| NF-κB (human) | 15-Deoxy-PGJ2 Nitroalkene Acrolein Isothiocyanate |
Cys61 Unknown Cys61, Arg307 Unknown |
(84, 122, 134) (85) (135, 136) (137) |
| IκB (human) | 4-HNE | Unknown | (137–141) |
| Keap1 (human) | Isothiocyanate 4-HNE 15-Deoxy-PGJ2 Nitroalkene IAB, BMCC NEPP |
Unknown Unknown Cys273, Cys288 Unknown Cys288 Cys151 |
(142) (143) (92, 123, 124, 144) (93) (145) (146) |
| PPAR (human) | 15-Deoxy-PGJ2 Nitroalkenes 4-HNE |
Cys285 Cys285 His413 |
(45) (60) (97) |
| TRPV1 TRPA1 (murine) |
Onion or garlic extract Aldehydes or acrolein Isothiocyanate |
Cys157 Cys415, Cys422, Cys622 Cys619, Cys639, Cys663 |
(101) (99) (100) |
| HSF (human) | 15-Deoxy-PGJ2 4-HNE Acrolein |
Unknown Unknown Unknown |
(104, 147) (39) (148) |
| PTP (human) | Acrolein | Cys215 | (108) |
| MAPK (human) | Isothiocyanate 4-HNE |
Unknown Unknown |
(110) (111) |
Nuclear factor κB (NF-κB)
NF-κB is a cytosolic protein heterodimer composed of combinations of the p50, p52, cRel, RebB, and p65 subunits that acts as a rapid-response transcription factor. In unstimulated cells, NF-κB is present in the cytoplasm in an inactive form complexed with inhibitor of κBα (IκB). After exposure to inflammatory-derived reactive species (or alternatively, different viral and bacterial antigen stimuli), NF-κB is released from IκB, translocates to the nucleus, binds to DNA, and induces transcription of various genes involved in inflammatory responses, cell proliferation, and cell survival. Consequently, NF-κB has been closely linked to inflammatory responses and carcinogenesis (83). The p50 and p65 subunits of NF-κB are posttranslationally modified by both oxidants and electrophiles at multiple thiols, which results in either an inhibition of nuclear translocation or impaired DNA binding of NF-κB. For example 15-deoxy-PGJ2 covalently modifies both subunits, which in turn inhibits DNA binding of NF-κB (84). NO2-FAs also form adducts with p65, which inhibits the DNA binding activity of p65 and represses the expression of lipopolysaccharide (LPS)–induced target genes, such as those encoding proinflammatory cytokines (85).
Keap1 and Nrf2
Similar to NF-κB, Nrf2 (nuclear factor erythroid 2–related factor 2) is a transcription factor that is basally regulated by rapid degradation by the proteasome. Nrf2 proteasomal degradation, in turn, is regulated by interactions with the cytosolic Nrf2-specific Cul3 ubiquitin ligase complex adaptor protein Keap1 (Kelch-like ECH-associated protein 1). Keap 1 has multiple electrophile-reactive cysteine residues that, when covalently adducted, result in inhibition of Keap1-dependent Nrf2 ubiquitination, leading to enhanced Nrf2 stability, nuclear translocation, and expression of Nrf2 target genes (86). Both this Nrf2 liberation and the synthesis of Nrf2 in the absence of Keap1 capture facilitate Nrf2 translocation to the nucleus. Nrf2 target genes encode antioxidant enzymes, enzymes of GSH biosynthesis, detoxification enzymes, scavenger receptors, chaperone proteins, and others that are regulated by the Nrf2 regulatory element termed the antioxidant response element or electrophile response element (87). Because the Keap1-Nrf2 pathway and particularly the thiols of Keap1 are sensitive to oxidative modification and adduction by electrophiles, this pathway represents an important detoxification and cell protection mechanism. Electrophiles, such as 15-deoxy-PGJ2, NO2-FAs, isothiocyanates, and 4-HNE, covalently modify Keap1 by Michael addition and thereby promote Nrf2-regulated gene expression (88–93).
Peroxisome proliferator–activated receptors (PPARs)
PPARs are members of the nuclear lipid receptor superfamily and are ligand-activated transcription factors. PPAR agonists can activate transcription by binding PPARs, altering receptor conformation, and thus promoting the association of coactivator proteins with PPAR either before or after binding to target gene promoters. Alternatively, ligand binding can also induce dissociation of corepressor proteins. In aggregate, PPAR-coregulatory protein interactions define the organization of the transcriptional apparatus and promote an open chromatin structure by facilitating PTMs, such as histone acetylation. PPARs also heterodimerize with the retinoid X receptor, and all PPAR-containing transcriptional complexes bind to PPAR response elements in the 5′ untranslated region of target genes (94). Endogenous ligands of PPARγ, a PPAR isoform that plays a pivotal role in cell differentiation and metabolism, include electrophilic keto–, nitro–, and cyclopentenone–fatty acid derivatives (95, 96). These electrophilic fatty acid derivatives can undergo Michael addition with the Cys285 residue in the human PPARγ ligand-binding pocket and thus display nanomolar affinities (60, 97, 98). The covalent adduction of electrophilic ligands with lipid sensors, such as the PPARγ receptor, eliminates traditional equilibrium kinetics as a predictor of PPARγ activation at different ligand concentrations. Also, the patterns of coregulator protein association with PPARγ and the genes that are expressed, compared with those stimulated by synthetic and noncovalently binding ligands (such as thiazolidinediones), are different for electrophilic ligands (60).
Transient receptor potential (TRP) channels
TRP channels are a family of multimeric cation channels. Some TRP channels are located in nociceptive neurons, where they serve to sense peripheral stimuli mediated by temperature extremes and diverse chemical factors, many of which share electrophilic reactivity. For example, TRPA1 and TRPV1 channel activation results in acute pain sensation and neurogenic inflammation. Click chemistry revealed that the electrophiles cinnamaldehyde and mustard oil activated TRPA1 through irreversible S-alkylation of reactive cysteines. In addition, electrophiles, such as acrolein, 4-HNE, and iodoacetamide, activated TRPA1, all with micromolar median effective concentrations (99). Allyl isothiocyanate—responsible for the pungent taste of mustard, wasabi, and horseradish—also activates the TRPA1 channel through covalent PTM of reactive cysteines (100). Of note, the covalent modification of TRPA1 by allyl isothiocyanate is reversible, and the three cysteines susceptible for electrophile adduction and responsible for changes in receptor function by this compound have been identified (100). Although only displaying 13% sequence identity and 22% conservation overall, TRPA1 and TRPV1 are both activated by plant-derived electrophiles, including capsaicin, allyl derivative, and other onion or garlic extracts, through covalent modification of cysteine residues. Only one cysteine seems to be responsible for the activation of TRPV1 by these electrophiles (101). Although NO has also been reported to activate TRPC and TRPV channels through thiol S-nitrosation, high concentrations of NO sources were utilized, creating the distinct possibility that alternative oxidation products and secondary electrophile reactions could be mediating changes in channel function (102). The overall observation that an ion channel activity can be regulated by PTM of functionally important reactive cysteines, in addition to “traditional” noncovalent agonist binding to the active site (ligand-gated ion channels) or activation or inactivation by changes in voltage (voltage-gated ion channels), has broad-reaching implications. For example, this insight can reveal new mechanisms and drug strategies whereby pain and inflammatory signaling may be therapeutically modified.
Heat shock factor (HSF)
HSFs are redox and electrophile-activated transcription factors that regulate the expression of the family of “heat shock response” proteins (HSPs). The HSPs are highly conserved from bacteria to humans and serve as protein chaperones by regulating protein folding, trafficking, and degradation. The expression of genes encoding HSPs is stimulated in response to changes in temperature, the formation of reactive species, and other inflammatory conditions (103). Transcriptional activation is induced by DNA binding of homotrimeric HSFs that interact with promoter sequences termed heat shock elements (HSEs). Through covalent HSF thiol modification, electrophiles, such as 15-deoxy-PGJ2 (104) and 4-HNE (39), activate HSF and stimulate expression of genes encoding HSPs. Enhanced cellular protein thiol oxidation, as well as thiol adduction by electrophilic species, also stimulates HSF DNA binding and transcriptional activation (105, 106). The discovery of small-molecule HSF activators that may include electrophilic species is a promising drug discovery area for treating inflammatory and degenerative diseases.
Protein tyrosine phosphatases (PTPs)
PTPs, responsible for the dephosphorylation of phosphotyrosine residues, are key regulators of receptor tyrosine kinase– and nonreceptor tyrosine kinase–mediated signaling pathways. PTPs are highly conserved and have, in their signature motif, a catalytic cysteine thiolate and several other reactive cysteine residues that, when modified, substantially affect enzyme activity (107). Acrolein and naphthoquinones, representative electrophiles, irreversibly inhibit PTP1B by covalently binding the active-site reactive cysteine. Most electrophiles display one to two orders of magnitude greater second-order rate constants for the inhibition of PTPs than does H2O2. This insight provides useful perspective, as H2O2 is a redox mediator that is typically viewed as a key biological inhibitor of PTPs (108) (109).
Mitogen-activated protein kinases (MAPKs)
When activated by growth factors and stress mediators (including cytokines, irradiation, heat shock, reactive species, and electrophilic lipid derivatives), MAPKs transduce cell signals through the phosphorylation of serine and threonine residues on various target proteins. MAPKs principally regulate cell differentiation, proliferation, and survival. Electrophiles, such as isothiocyanate derivatives and 4-HNE, posttranslationally modify MAPKs to influence their downstream signaling actions (110, 111).
Electrophile modulation of signaling mediator activity—pitfalls
Substantial gaps of knowledge still exist regarding the impact of electrophilic species on key signaling pathways, including those noted above. In many instances, specific target residues in modified proteins are not known, a reaction that is likely to vary depending on the physical characteristics of the electrophile. Also, observations collected from in vitro studies may not accurately reflect in vivo responses, because target molecules subjected to lower concentrations and more diverse populations of electrophilic mediators for longer periods of time may respond differently. Better understanding of the pharmacophore for electrophiles—the aggregate of steric and electronic features that facilitate optimal supramolecular interactions of electrophiles with their biological targets—can propel future drug discovery efforts by elucidating how greater specificity can be obtained for better directing target molecule reactions mediated by a particular electrophile structure. For example, an electrophilic PPARγ ligand having both Cys285 reactivity and strong electrostatic and hydrophobic interactions with other residues in the ligand-binding pocket may show greater promise in treating metabolic and inflammatory disorders than the currently prescribed class of thiazolidinediones. Finally, the nature and extent of many electrophile-signaling mediator interactions in vivo, under more clinically or toxicologically relevant electrophile exposure conditions, frequently has not been evaluated.
Clinical Implications of Electrophile-Stimulated Cell Signaling
Chemopreventive and anti-inflammatory actions induced by administration of (i) synthetic homologs of endogenously produced electrophilic metabolites or (ii) purified electrophilic natural products may be of possible clinical benefit. One might predict therapeutic value upon modulation of the above pathways. In the context of central nervous system disorders, administration of the plant-derived electrophile curcumin is neuroprotective in murine models of ischemia-reperfusion injury and Alzheimer’s disease (112, 113). Similarly, administration of the plant-derived electrophile carnosic acid to mice stimulated Nrf2-dependent gene expression, increased the abundance of GSH in the brain, and protected the brain from middle cerebral artery occlusion and reperfusion injury (114, 115). Through a mechanism dependent on Nrf2, topical administration of a broccoli sprout extract containing the electrophile sulforaphane prevented ultraviolet (UV)–induced carcinogenesis in mice (116). Isothiocyanate- and sulforaphane-based chemopreventive strategies have also shown therapeutic promise—for example, in an experiment in which these were administered to a population at risk for aflatoxin-induced hepatocellular carcinoma (117). Individuals treated with a broccoli sprout tea, which contains both the sulforaphane precursor glucosinolate and various isothiocyanates, increased the abundance of Nrf2-regulated cytoprotective enzymes and excretion of aflatoxin-mercapturic acid conjugates (117–121). Thus, electrophile-based strategies are yielding promising preclinical and early phase clinical study results in the treatment of inflammatory and cancer-related problems.
Regulation and Reversibility of Protein Modification by Electrophiles
Although electrophiles can exert physiologically beneficial effects through PTM and the activation or inactivation of key signaling mediators, the reversibility of most electrophile-protein reactions has not been adequately addressed in the context of protein turnover, transfer of protein-electrophile adducts to different nucleophilic centers on the same or different macromolecule, and cellular export. The potential for unique signaling actions emanating from different classes of electrophiles—sometimes previously viewed as harmful chemical irritants and damaging inflammatory by-products—also warrants more extensive evaluation. It appears that the specific sites and extents of protein adduction, the reversibility of adduct formation after exposure to particular electrophiles, and the outcome variables that are selected for measurement can influence interpretation of electrophile actions in cell and animal models or clinical studies.
Signal selectivity and the existence of mechanisms contributing to the regulation of electrophilic adduction are essential prerequisites if one is to consider electrophiles as a class of cell signaling mediators. The accessibility and preferential reactivity of specific cysteine residues with electrophilic species are two factors that appear to confer a degree of specificity to electrophile signaling. This is confirmed by the observation that mutant proteins lacking a specific reactive cysteine [for example, p65 (122), TRPV1 (101), and Keap1 (123, 124)] show modifications in electrophilic PTM and, in concert with this, subsequent changes in protein function. Besides accessibility and selective amino acid reactivity, subcellular compartmentalization and the formation of more complex protein–macromolecule interactions are additional factors that can affect electrophilic protein modification in cells. This concept, in the context of redox signaling, has been addressed with S-nitrosation and tyrosine nitration reactions (26, 125–127). For these PTMs, surrounding amino acid motifs, protein compartmentalization, and organization of proteins in complexes can either inhibit or facilitate the interaction between molecular targets and reactive species that typically have short half-lives and limited diffusion distances.
GSH biosynthesis, its rereduction following oxidation to a disulfide, and its catalyzed transfer to proteins all contribute to the regulation of electrophilic PTM. GSH is the most abundant cellular reductant and, with its oxidized form, serves as an intra-cellular redox buffer system (15). In addition to its role as a redox buffer, GSH can react with oxidized protein thiols to yield mixed disulfides, thus limiting irreversible oxidation reactions (25, 128). GSH can also undergo an exchange reaction with a disulfide. Disulfide exchange reactions are kinetically slow; however, they are accelerated intracellularly by enzymes, such as glutaredoxins, thioredoxin, and sulfiredoxin. Despite its low reactivity, explained by the relatively high thiol pKa of 9.2, GSH is still an important mediator of redox-based PTMs because of its abundance in cells and its rapid enzymatic regeneration (25).
Some electrophiles permanently and irreversibly modify a target protein and others induce more short-lived and reversible adduction of the target protein (30). From this work, it has been appreciated that reversibly reactive electrophiles display less or no cytotoxic effects at low concentrations. In addition to specificity of reaction, the reversibility of electrophilic PTM supports that these reactions may function as a signaling event that is sensitive to cellular metabolic and redox status. The reversible electrophilic adduction of cysteine can include inter- or intramolecular electrophile exchange between different thiols, with ultimate transfer to GSH and export of the electrophile from cells through specific multidrug resistance protein transport mechanisms (Fig. 4). The clinical promise of electrophile-based therapeutic strategies has motivated additional investigation of factors influencing when redox-mediated PTM reactions cross the threshold from physiological signaling to pathologic reactions. The fact that some redox-based modifications are irreversible and lead to a permanent loss of protein function also supports that some redox-dependent modifications do not exclusively play a role in cell physiology (129, 130).
Fig. 4.

Intracellular transfer of electrophiles. After reversibly binding to a reactive cysteine, the electrophile can be either transferred to a reactive cysteine of another protein or transferred to GSH and exported out of the cell. Most reversibly adducted electrophiles are transferred to GSH and ultimately exported. Irreversible electrophile adduction is typically cleared by protein degradation.
Conclusions and Future Directions
In recent years, redox-dependent and, in particular, electrophile-mediated posttranslational protein modifications have emerged as cell signaling mechanisms that link cell function with inflammatory and metabolic status. Multiple transcription factors have prominent amino acids that are both electrophile reactive and functionally important and so enhance a capability for stress-related adaptive signaling reactions. These highly conserved signaling pathways allow organisms to respond to electrophilic species that are present in the diet, generated as a consequence of toxin exposure, and produced by oxidant and free radical mediators of inflammation and metabolic stress. The availability of more incisive chemical and bioanalytical strategies for identifying and quantifying both the mediators and the molecular targets of electrophile signaling is rapidly propelling new discoveries in this area. Recent advances have shown that there exist subproteomes that are targets of reversible adduction by particular electrophiles. Thus, fertile waters exist for spawning important discoveries related to the consequences of proteomic versus genomic reactions of electrophiles and whether these events have therapeutic or toxicological implications.
References and Notes
- 1.Fannon SA, Vidaver RM, Marts SA. An abridged history of sex steroid hormone receptor action. J Appl Physiol. 2001;91:1854–1859. doi: 10.1152/jappl.2001.91.4.1854. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 3.Walsh C. Post-Translational Modification of Proteins: Expanding Nature’s Repertoire. Roberts and Company; Greenwood Village, CO: 2006. [Google Scholar]
- 4.McCord JM, Fridovich I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 5.Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Granger DN, Rutili G, McCord JM. Superoxide radicals in feline intestinal ischemia. Gastroenterology. 1981;81:22–29. [PubMed] [Google Scholar]
- 7.Hassan HM, Fridovich I. Intracellular production of superoxide radical and of hydrogen peroxide by redox active compounds. Arch Biochem Biophys. 1979;196:385–395. doi: 10.1016/0003-9861(79)90289-3. [DOI] [PubMed] [Google Scholar]
- 8.Freeman BA, Crapo JD. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J Biol Chem. 1981;256:10986–10992. [PubMed] [Google Scholar]
- 9.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeman BA, Crapo JD. Biology of disease: Free radicals and tissue injury. Lab Invest. 1982;47:412–426. [PubMed] [Google Scholar]
- 11.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, Van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 12.Castro L, Freeman BA. Reactive oxygen species in human health and disease. Nutrition. 2001;17:161–165. doi: 10.1016/s0899-9007(00)00570-0. [DOI] [PubMed] [Google Scholar]
- 13.Isom AL, Barnes S, Wilson L, Kirk M, Coward L, Darley-Usmar V. Modification of cytochrome c by 4-hydroxy-2-nonenal: Evidence for histidine, lysine, and arginine-aldehyde adducts. J Am Soc Mass Spectrom. 2004;15:1136–1147. doi: 10.1016/j.jasms.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 14.Marnett LJ, Riggins JN, West JD. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest. 2003;111:583–593. doi: 10.1172/JCI18022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 16.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 17.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls: The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 18.Ying J, Clavreul N, Sethuraman M, Adachi T, Cohen RA. Thiol oxidation in signaling and response to stress: Detection and quantification of physiological and pathophysiological thiol modifications. Free Radic Biol Med. 2007;43:1099–1108. doi: 10.1016/j.freeradbiomed.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: Purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 20.Lane P, Hao G, Gross SS. S-Nitrosylation is emerging as a specific and fundamental post-translational protein modification: Head-to-head comparison with O-phosphorylation. Sci STKE. 2001:re1. doi: 10.1126/stke.2001.86.re1. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Ruiz A, Lamas S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: Convergences and divergences. Cardiovasc Res. 2007;75:220–228. doi: 10.1016/j.cardiores.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Ghezzi P, Bonetto V. Redox proteomics: Identification of oxidatively modified proteins. Proteomics. 2003;3:1145–1153. doi: 10.1002/pmic.200300435. [DOI] [PubMed] [Google Scholar]
- 23.Ying J, Clavreul N, Sethuraman M, Adachi T, Cohen RA. Thiol oxidation in signaling and response to stress: Detection and quantification of physiological and pathophysiological thiol modifications. Free Radic Biol Med. 2007;43:1099–1108. doi: 10.1016/j.freeradbiomed.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SER-CA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 25.Netto LE, de Oliveira MA, Monteiro G, Demasi AP, Cussiol JR, Discola KF, Demasi M, Silva GM, Alves SV, Faria VG, Horta BB. Reactive cysteine in proteins: Protein folding, antioxidant defense, redox signaling and more. Comp Biochem Physiol C Toxicol Pharmacol. 2007;146:180–193. doi: 10.1016/j.cbpc.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 26.Derakhshan B, Hao G, Gross SS. Balancing reactivity against selectivity: The evolution of protein S-nitrosylation as an effector of cell signaling by nitric oxide. Cardiovasc Res. 2007;75:210–219. doi: 10.1016/j.cardiores.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dennehy MK, Richards KA, Wernke GR, Shyr Y, Liebler DC. Cytosolic and nuclear protein targets of thiol-reactive electrophiles. Chem Res Toxicol. 2006;19:20–29. doi: 10.1021/tx050312l. [DOI] [PubMed] [Google Scholar]
- 28.Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem. 2008;283:21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 30.Liebler DC. Protein damage by reactive electrophiles: Targets and consequences. Chem Res Toxicol. 2008;21:117–128. doi: 10.1021/tx700235t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piskorski R, Hanus R, Vasickova S, Cvacka J, Sobotnik J, Svatos A, Valterova I. Nitroalkenes and sesquiterpene hydrocarbons from the frontal gland of three Prorhinotermes termite species. J Chem Ecol. 2007;33:1787–1794. doi: 10.1007/s10886-007-9341-y. [DOI] [PubMed] [Google Scholar]
- 32.Farmer EE, Davoine C. Reactive electrophile species. Curr Opin Plant Biol. 2007;10:380–386. doi: 10.1016/j.pbi.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 33.Burcham PC. Potentialities and pitfalls accompanying chemico-pharmacological strategies against endogenous electrophiles and carbonyl stress. Chem Res Toxicol. 2008;21:779–786. doi: 10.1021/tx700399q. [DOI] [PubMed] [Google Scholar]
- 34.Luo J, Hill BG, Gu Y, Cai J, Srivastava S, Bhatnagar A, Prabhu SD. Mechanisms of acrolein-induced myocardial dysfunction: Implications for environmental and endogenous aldehyde exposure. Am J Physiol Heart Circ Physiol. 2007;293:H3673–H3684. doi: 10.1152/ajpheart.00284.2007. [DOI] [PubMed] [Google Scholar]
- 35.Dalle-Donne I, Scaloni A, Butterfield DA. Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases. Wiley-Inter-science; Hoboken, NJ: 2006. [DOI] [PubMed] [Google Scholar]
- 36.Kirkham PA, Spooner G, Ffoulkes-Jones C, Calvez R. Cigarette smoke triggers macrophage adhesion and activation: Role of lipid peroxidation products and scavenger receptor. Free Radic Biol Med. 2003;35:697–710. doi: 10.1016/s0891-5849(03)00390-3. [DOI] [PubMed] [Google Scholar]
- 37.Uchida K, Szweda LI, Chae H-Z, Stadtman ER. Immunochemical detection of 4-hydroxynonenal protein adducts in oxidized hepatocytes. Proc Natl Acad Sci USA. 1993;90:8742–8746. doi: 10.1073/pnas.90.18.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase: A possible involvement of intra- and intermolecular cross-linking reaction. J Biol Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 39.Jacobs AT, Marnett LJ. Heat shock factor 1 attenuates 4-hydroxynonenal-mediated apoptosis: Critical role for heat shock protein 70 induction and stabilization of Bcl-XL. J Biol Chem. 2007;282:33412–33420. doi: 10.1074/jbc.M706799200. [DOI] [PubMed] [Google Scholar]
- 40.Biasi F, Vizio B, Mascia C, Gaia E, Zarkovic N, Chiarpotto E, Leonarduzzi G, Poli G. c-Jun N-terminal kinase upregulation as a key event in the proapoptotic interaction between transforming growth factor-β1 and 4-hydroxynonenal in colon mucosa. Free Radic Biol Med. 2006;41:443–454. doi: 10.1016/j.freeradbiomed.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 41.Stewart BJ, Doorn JA, Petersen DR. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem Res Toxicol. 2007;20:1111–1119. doi: 10.1021/tx700106v. [DOI] [PubMed] [Google Scholar]
- 42.Leonarduzzi G, Robbesyn F, Poli G. Signaling kinases modulated by 4-hydroxynonenal. Free Radic Biol Med. 2004;37:1694–1702. doi: 10.1016/j.freeradbiomed.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 43.Bennaars-Eiden A, Higgins L, Hertzel AV, Kapphahn RJ, Ferrington DA, Bernlohr DA. Covalent modification of epithelial fatty acid-binding protein by 4-hydroxynonenal in vitro and in vivo: Evidence for a role in antioxidant biology. J Biol Chem. 2002;277:50693–50702. doi: 10.1074/jbc.M209493200. [DOI] [PubMed] [Google Scholar]
- 44.West JD, Marnett LJ. Alterations in gene expression induced by the lipid peroxidation product, 4-hydroxy-2-nonenal. Chem Res Toxicol. 2005;18:1642–1653. doi: 10.1021/tx050211n. [DOI] [PubMed] [Google Scholar]
- 45.Shiraki T, Kamiya N, Shiki S, Kodama TS, Kakizuka A, Jingami H. α,β-Unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor γ. J Biol Chem. 2005;280:14145–14153. doi: 10.1074/jbc.M500901200. [DOI] [PubMed] [Google Scholar]
- 46.Refsgaard HH, Tsai L, Stadtman ER. Modifications of proteins by polyunsaturated fatty acid peroxidation products. Proc Natl Acad Sci USA. 2000;97:611–616. doi: 10.1073/pnas.97.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erlemann KR, Rokach J, Powell WS. Oxidative stress stimulates the synthesis of the eosinophil chemoattractant 5-oxo-6,8,11,14-eicosatetraenoic acid by inflammatory cells. J Biol Chem. 2004;279:40376–40384. doi: 10.1074/jbc.M401294200. [DOI] [PubMed] [Google Scholar]
- 48.Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, Morrow JD, Freeman ML. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem. 2007;282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- 49.Brooks JD, Milne GL, Yin H, Sanchez SC, Porter NA, Morrow JD. Formation of highly reactive cyclopentenone isoprostane compounds (A3/J3-isoprostanes) in vivo from eicosapentaenoic acid. J Biol Chem. 2008;283:12043–12055. doi: 10.1074/jbc.M800122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milne GL, Morrow JD. Isoprostanes and related compounds: Update 2006. Antioxid Redox Signal. 2006;8:1379–1384. doi: 10.1089/ars.2006.8.1379. [DOI] [PubMed] [Google Scholar]
- 51.Yin H, Musiek ES, Gao L, Porter NA, Morrow JD. Regiochemistry of neuroprostanes generated from the peroxidation of docosahexaenoic acid in vitro and in vivo. J Biol Chem. 2005;280:26600–26611. doi: 10.1074/jbc.M503088200. [DOI] [PubMed] [Google Scholar]
- 52.Marszalek JR, Lodish HF. Docosahexaenoic acid, fatty acid-intreating proteins and neuronal function: Breastmilk and fish are good for you. Annu Rev Cell Dev Biol. 2005;21:633–657. doi: 10.1146/annurev.cellbio.21.122303.120624. [DOI] [PubMed] [Google Scholar]
- 53.Freeman BA, Baker PR, Schopfer FJ, Woodcock SR, Napolitano A, d’Ischia M. Nitro-fatty acid formation and signaling. J Biol Chem. 2008;283:15515–15519. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferreira AM, Ferrari MI, Trostchansky A, Batthyany C, Souza JM, Alvarez MN, Lopez GV, Baker PR, Schopfer FJ, O’Donnell V, Freeman BA, Rubbo H. Macrophage activation induces formation of the anti-inflammatory lipid cholesteryl-nitrolinoleate. Biochem J. 2009;417:223–234. doi: 10.1042/BJ20080701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nadtochiy SM, Baker PR, Freeman BA, Brookes PS. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: Implications for cardioprotection. Cardiovasc Res. 2008 doi: 10.1093/cvr/cvn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chem Res Toxicol. 1999;12:83–92. doi: 10.1021/tx980207u. [DOI] [PubMed] [Google Scholar]
- 57.Batthyany C, Schopfer FJ, Baker PRS, Duran R, Baker LMS, Huang Y, Cervenansky C, Branchaud BP, Freeman BA. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baker LM, Baker PR, Golin-Bisello F, Schopfer FJ, Fink M, Woodcock SR, Branchaud BP, Radi R, Freeman BA. Nitro-fatty acid reaction with glutathione and cysteine: Kinetic analysis of thiol alkylation by a Michael addition reaction. J Biol Chem. 2007;282:31085–31093. doi: 10.1074/jbc.M704085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: An endogenous peroxisome proliferator-activated receptor γ ligand. Proc Natl Acad Sci USA. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Zhang J, Schopfer FJ, Martynowski D, Garcia-Barrio MT, Kovach A, Suino-Powell K, Baker PRS, Freeman BA, Chen YE, Xu HE. Molecular recognition of nitrated fatty acids by PPARγ. Nat Struct Mol Biol. 2008;15:865–867. doi: 10.1038/nsmb.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. Fatty acid transduction of nitric oxide signaling: Multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schopfer FJ, Batthyany C, Baker PR, Bonacci G, Cole MP, Rudolph V, Groeger A, Rudolph TK, Nadtochiy S, Brookes PS, Freeman BA. Detection and quantification of protein adduction by electrophilic fatty acids: Mitochondrial generation of fatty acid nitroalkene derivatives. Free Radic Biol Med. 2009;46:1250–1259. doi: 10.1016/j.freeradbiomed.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rudolph V, Schopfer FJ, Khoo NK, Rudolph TK, Cole MP, Woodcock SR, Bonacci G, Groeger AL, Golin-Bisello F, Chen CS, Baker PR, Freeman BA. Nitro-fatty acid metabolome: Saturation, desaturation, β-oxidation, and protein adduction. J Biol Chem. 2009;284:1461–1473. doi: 10.1074/jbc.M802298200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller JA, Sapp RW, Miller EC. The absorption spectra of certain carcinogenic aminoazo dyes and the protein-bound derivatives formed from these dyes in vivo. J Am Chem Soc. 1948;70:3458–3463. doi: 10.1021/ja01190a072. [DOI] [PubMed] [Google Scholar]
- 65.Green LC, Skipper PL, Turesky RJ, Bryant MS, Tannenbaum SR. In vivo dosimetry of 4-aminobiphenyl in rats via a cysteine adduct in hemoglobin. Cancer Res. 1984;44:4254–4259. [PubMed] [Google Scholar]
- 66.Codreanu SG, Zhang B, Sobecki SM, Billheimer DD, Liebler DC. Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol Cell Proteomics. 2009;8:670–680. doi: 10.1074/mcp.M800070-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hansen BT, Jones JA, Mason DE, Liebler DC. SALSA: A pattern recognition algorithm to detect electrophile-adducted peptides by automated evaluation of CID spectra in LC-MS-MS analyses. Anal Chem. 2001;73:1676–1683. doi: 10.1021/ac001172h. [DOI] [PubMed] [Google Scholar]
- 68.Kolb HC, Finn MG, Sharpless KB. Click chemistry: Diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 69.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vila A, Tallman KA, Jacobs AT, Liebler DC, Porter NA, Marnett LJ. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem Res Toxicol. 2008;21:432–444. doi: 10.1021/tx700347w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weerapana E, Simon GM, Cravatt BF. Disparate proteome reactivity profiles of carbon electrophiles. Nat Chem Biol. 2008;4:405–407. doi: 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wong HL, Liebler DC. Mitochondrial protein targets of thiol-reactive electrophiles. Chem Res Toxicol. 2008;21:796–804. doi: 10.1021/tx700433m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shin NY, Liu Q, Stamer SL, Liebler DC. Protein targets of reactive electrophiles in human liver microsomes. Chem Res Toxicol. 2007;20:859–867. doi: 10.1021/tx700031r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Higdon AN, Dranka BP, Hill BG, Oh JY, Johnson MS, Landar A, Darley-Usmar VM. Methods for imaging and detecting modification of proteins by reactive lipid species. Free Radic Biol Med. 2009;47:201–212. doi: 10.1016/j.freeradbiomed.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tallman KA, Kim HY, Ji JX, Szapacs ME, Yin H, McIntosh TJ, Liebler DC, Porter NA. Phospholipid-protein adducts of lipid peroxidation: Synthesis and study of new biotinylated phosphatidylcholines. Chem Res Toxicol. 2007;20:227–234. doi: 10.1021/tx600331s. [DOI] [PubMed] [Google Scholar]
- 76.Szapacs ME, Kim HY, Porter NA, Liebler DC. Identification of proteins adducted by lipid peroxidation products in plasma and modifications of apolipoprotein A1 with a novel bi-otinylated phospholipid probe. J Proteome Res. 2008;7:4237–4246. doi: 10.1021/pr8001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rudolph V, Rudolph TK, Schopfer FJ, Bonacci G, Woodcock SR, Cole MP, Baker PR, Ramani R, Freeman BA. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischemia and reperfusion. Cardiovasc Res. doi: 10.1093/cvr/cvp275. Published online 7 August 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schopfer FJ, Baker PR, Giles G, Chumley P, Batthyany C, Crawford J, Patel RP, Hogg N, Branchaud BP, Lancaster JR, Jr, Freeman BA. Fatty acid transduction of nitric oxide signaling: Nitrolinoleic acid is a hydrophobically stabilized nitric oxide donor. J Biol Chem. 2005;280:19289–19297. doi: 10.1074/jbc.M414689200. [DOI] [PubMed] [Google Scholar]
- 79.Lawley PD, Brookes P. The action of alkylating agents on deoxyribonucleic acid in relation to biological effects of the alkylating agents. Exp Cell Res. 1963;9(suppl 1):512–520. doi: 10.1016/0014-4827(63)90291-x. [DOI] [PubMed] [Google Scholar]
- 80.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–520. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 81.Kaplowitz N, Fernandez-Checa JC, Kannan R, Garcia-Ruiz C, Ookhtens M, Yi JR. GSH transporters: Molecular characterization and role in GSH homeostasis. Biol Chem Hoppe Seyler. 1996;377:267–273. doi: 10.1515/bchm3.1996.377.5.267. [DOI] [PubMed] [Google Scholar]
- 82.Talalay P, De Long MJ, Prochaska HJ. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc Natl Acad Sci USA. 1988;85:8261–8265. doi: 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc Natl Acad Sci USA. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, Batthyany C, Chacko BK, Feng X, Patel RP, Agarwal A, Freeman BA, Chen YE. Nitrated fatty acids: Endogenous anti-inflammatory signaling mediators. J Biol Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol. 2008;21:705–710. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 88.Colburn NH, Kensler TW. Targeting transcription factors for cancer prevention—the case of Nrf2. Cancer Prev Res (Philadelphia, Pa) 2008;1:153–155. doi: 10.1158/1940-6207.CAPR-08-0025. [DOI] [PubMed] [Google Scholar]
- 89.Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ceaser EK, Moellering DR, Shiva S, Ramachandran A, Landar A, Venkartraman A, Crawford J, Patel R, Dickinson DA, Ulasova E, Ji S, Darley-Usmar VM. Mechanisms of signal transduction mediated by oxidized lipids: The role of the electrophile-responsive proteome. Biochem Soc Trans. 2004;32:151–155. doi: 10.1042/bst0320151. [DOI] [PubMed] [Google Scholar]
- 91.Oh JY, Giles N, Landar A, Darley-Usmar V. Accumulation of 15-deoxy-Δ12,14-prostaglandin J2 adduct formation with Keap1 over time: Effects on potency for intracellular antioxidant defence induction. Biochem J. 2008;411:297–306. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Villacorta L, Zhang J, Garcia-Barrio M, Chen X, Freeman BA, Chen YE, Cui T. Nitrolinoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. Am J Physiol Heart Circ Physiol. 2007;293:H770–H776. doi: 10.1152/ajpheart.00261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–935. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nettles KW. Insights into PPARγ from structures with endogenous and covalently bound ligands. Nat Struct Mol Biol. 2008;15:893–895. doi: 10.1038/nsmb0908-893. [DOI] [PubMed] [Google Scholar]
- 96.Waku T, Shiraki T, Oyama T, Fujimoto Y, Maebara K, Kamiya N, Jingami H, Morikawa K. Structural insight into PPARγ activation through covalent modification with endogenous fatty acids. J Mol Biol. 2009;385:188–199. doi: 10.1016/j.jmb.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 97.Coleman JD, Prabhu KS, Thompson JT, Reddy PS, Peters JM, Peterson BR, Reddy CC, Vanden Heuvel JP. The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) Free Radic Biol Med. 2007;42:1155–1164. doi: 10.1016/j.freeradbiomed.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shiraki T, Kodama TS, Shiki S, Nakagawa T, Jingami H. Spectroscopic analyses of the binding kinetics of 15d-PGJ2 to the PPARγ ligand-binding domain by multi-wavelength global fitting. Biochem J. 2006;393:749–755. doi: 10.1042/BJ20050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- 100.Hinman A, Chuang HH, Bautista DM, Julius D. TRP channel activation by reversible covalent modification. Proc Natl Acad Sci USA. 2006;103:19564–19568. doi: 10.1073/pnas.0609598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Salazar H, Llorente I, Jara-Oseguera A, Garcia-Villegas R, Munari M, Gordon SE, Islas LD, Rosenbaum T. A single N-terminal cysteine in TRPV1 determines activation by pungent compounds from onion and garlic. Nat Neurosci. 2008;11:255–261. doi: 10.1038/nn2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- 103.Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zingarelli B, Hake PW, Mangeshkar P, O’-Connor M, Burroughs TJ, Piraino G, Denenberg A, Wong HR. Diverse cardioprotective signaling mechanisms of peroxisome proliferator-activated receptor-γ ligands, 15-deoxy-Δ12,14-prostaglandin J2 and ciglitazone, in reperfusion injury: Role of nuclear factor-κB, heat shock factor 1, and Akt. Shock. 2007;28:554–563. doi: 10.1097/shk.0b013e31804f56b9. [DOI] [PubMed] [Google Scholar]
- 105.Liu H, Lightfoot R, Stevens JL. Activation of heat shock factor by alkylating agents is triggered by glutathione depletion and oxidation of protein thiols. J Biol Chem. 1996;271:4805–4812. [PubMed] [Google Scholar]
- 106.Paroo Z, Meredith MJ, Locke M, Haist JV, Karmazyn M, Noble EG. Redox signaling of cardiac HSF1 DNA binding. Am J Physiol Cell Physiol. 2002;283:C404–C411. doi: 10.1152/ajpcell.00051.2002. [DOI] [PubMed] [Google Scholar]
- 107.Zhang ZY. Protein tyrosine phosphatases: Structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 108.Seiner DR, LaButti JN, Gates KS. Kinetics and mechanism of protein tyrosine phosphatase 1B inactivation by acrolein. Chem Res Toxicol. 2007;20:1315–1320. doi: 10.1021/tx700213s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Iwamoto N, Sumi D, Ishii T, Uchida K, Cho AK, Froines JR, Kumagai Y. Chemical knockdown of protein-tyrosine phosphatase 1B by 1,2-naphthoquinone through covalent modification causes persistent transactivation of epidermal growth factor receptor. J Biol Chem. 2007;282:33396–33404. doi: 10.1074/jbc.M705224200. [DOI] [PubMed] [Google Scholar]
- 110.Kong AN, Owuor E, Yu R, Hebbar V, Chen C, Hu R, Mandlekar S. Induction of xenobiotic enzymes by the MAP kinase pathway and the antioxidant or electrophile response element (ARE/EpRE) Drug Metab Rev. 2001;33:255–271. doi: 10.1081/dmr-120000652. [DOI] [PubMed] [Google Scholar]
- 111.Zhang H, Liu H, Iles KE, Liu RM, Postlethwait EM, Laperche Y, Forman HJ. 4-Hydroxynonenal induces rat γ-glutamyl transpeptidase through mitogen-activated protein kinase-mediated electrophile response element/nuclear factor erythroid 2-related factor 2 signaling. Am J Respir Cell Mol Biol. 2006;34:174–181. doi: 10.1165/rcmb.2005-0280OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Satoh T, Lipton SA. Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 2007;30:37–45. doi: 10.1016/j.tins.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 113.Balogun E, Foresti R, Green CJ, Motterlini R. Changes in temperature modulate heme oxygenase-1 induction by curcumin in renal epithelial cells. Biochem Biophys Res Commun. 2003;308:950–955. doi: 10.1016/s0006-291x(03)01517-1. [DOI] [PubMed] [Google Scholar]
- 114.Satoh T, Izumi M, Inukai Y, Tsutsumi Y, Nakayama N, Kosaka K, Shimojo Y, Kitajima C, Itoh K, Yokoi T, Shirasawa T. Carnosic acid protects neuronal HT22 cells through activation of the antioxidant-responsive element in free carboxylic acid- and catechol hydroxyl moieties-dependent manners. Neurosci Lett. 2008;434:260–265. doi: 10.1016/j.neulet.2008.01.079. [DOI] [PubMed] [Google Scholar]
- 115.Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton SA. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem. 2008;104:1116–1131. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dinkova-Kostova AT, Jenkins SN, Fahey JW, Ye L, Wehage SL, Liby KT, Stephenson KK, Wade KL, Talalay P. Protection against UV-light-induced skin carcinogenesis in SKH-1 high-risk mice by sulforaphane-containing broccoli sprout extracts. Cancer Lett. 2006;240:243–252. doi: 10.1016/j.canlet.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 117.Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, Ye L, Coady JL, Wang JB, Wu Y, Sun Y, Zhang QN, Zhang BC, Zhu YR, Qian GS, Carmella SG, Hecht SS, Benning L, Gange SJ, Groopman JD, Talalay P. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China. Cancer Epidemiol Biomarkers Prev. 2005;14:2605–2613. doi: 10.1158/1055-9965.EPI-05-0368. [DOI] [PubMed] [Google Scholar]
- 118.Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem Res Toxicol. 2005;18:1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- 119.Shapiro TA, Fahey JW, Dinkova-Kostova AT, Holtzclaw WD, Stephenson KK, Wade KL, Ye L, Talalay P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: A clinical phase I study. Nutr Cancer. 2006;55:53–62. doi: 10.1207/s15327914nc5501_7. [DOI] [PubMed] [Google Scholar]
- 120.Yates MS, Kensler TW. Keap1 eye on the target: Chemoprevention of liver cancer. Acta Pharmacol Sin. 2007;28:1331–1342. doi: 10.1111/j.1745-7254.2007.00688.x. [DOI] [PubMed] [Google Scholar]
- 121.Yates MS, Kensler TW. Chemopreventive promise of targeting the Nrf2 pathway. Drug News Perspect. 2007;20:109–117. doi: 10.1358/dnp.2007.20.2.108343. [DOI] [PubMed] [Google Scholar]
- 122.Cernuda-Morollon E, Pineda-Molina E, Canada FJ, Perez-Sala D. 15-Deoxy-Δ12,14-prostaglandin J2 inhibition of NF-κB-DNA binding through covalent modification of the p50 subunit. J Biol Chem. 2001;276:35530–35536. doi: 10.1074/jbc.M104518200. [DOI] [PubMed] [Google Scholar]
- 123.Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, Warabi E, Noguchi N, Itoh K, Yamamoto M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem. 2005;280:27244–27250. doi: 10.1074/jbc.M502551200. [DOI] [PubMed] [Google Scholar]
- 124.Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, Kelly V, Sekizawa K, Uchida K, Yamamoto M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Δ12,14-prostaglandin J2. Mol Cell Biol. 2004;24:36–45. doi: 10.1128/MCB.24.1.36-45.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 126.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: Biochemistry pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 127.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α 1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 128.Cross JV, Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid β-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 130.Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-Nitrosylation of parkin regulates ubiquiti-nation and compromises parkin’s protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 131.Tacka KA, Dabrowiak JC, Goodisman J, Souid AK. Kinetic analysis of the reactions of 4-hydroperoxycyclophosphamide and acrolein with glutathione, mesna, and WR-1065. Drug Metab Dispos. 2002;30:875–882. doi: 10.1124/dmd.30.8.875. [DOI] [PubMed] [Google Scholar]
- 132.Doorn JA, Petersen DR. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem Biol Interact. 2003;143–144:93–100. doi: 10.1016/s0009-2797(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 133.Podhradsky D, Drobnica L, Kristian P. Reactions of cysteine, its derivatives, glutathione coenzyme A, and dihydrolipoic acid with isothiocyanates. Experientia. 1979;35:154–155. doi: 10.1007/BF01920581. [DOI] [PubMed] [Google Scholar]
- 134.Pande V, Ramos MJ. Molecular recognition of 15-deoxy-Δ12,14-prostaglandin J2 by nuclear factor-κB and other cellular proteins. Bioorg Med Chem Lett. 2005;15:4057–4063. doi: 10.1016/j.bmcl.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 135.Horton ND, Biswal SS, Corrigan LL, Bratta J, Kehrer JP. Acrolein causes inhibitor κB-independent decreases in nuclear factor κB activation in human lung adenocarcinoma (A549) cells. J Biol Chem. 1999;274:9200–9206. doi: 10.1074/jbc.274.14.9200. [DOI] [PubMed] [Google Scholar]
- 136.Lambert C, Li J, Jonscher K, Yang TC, Reigan P, Quintana M, Harvey J, Freed BM. Acrolein inhibits cytokine gene expression by alkylating cysteine and arginine residues in the NF-κB1 DNA binding domain. J Biol Chem. 2007;282:19666–19675. doi: 10.1074/jbc.M611527200. [DOI] [PubMed] [Google Scholar]
- 137.Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhauser C. Nuclear factor κB is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem. 2001;276:32008–32015. doi: 10.1074/jbc.M104794200. [DOI] [PubMed] [Google Scholar]
- 138.Jeong WS, Kim IW, Hu R, Kong AN. Modulatory properties of various natural chemopreventive agents on the activation of NF-κB signaling pathway. Pharm Res. 2004;21:661–670. doi: 10.1023/b:pham.0000022413.43212.cf. [DOI] [PubMed] [Google Scholar]
- 139.Kim SJ, Kang SY, Shin HH, Choi HS. Sulforaphane inhibits osteoclastogenesis by inhibiting nuclear factor-κB. Mol Cells. 2005;20:364–370. [PubMed] [Google Scholar]
- 140.Xu C, Shen G, Chen C, Gelinas C, Kong AN. Suppression of NF-κB and NF-κB-regulated gene expression by sulforaphane and PEITC through IκBα, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24:4486–4495. doi: 10.1038/sj.onc.1208656. [DOI] [PubMed] [Google Scholar]
- 141.Ji C, Kozak KR, Marnett LJ. IκB kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. J Biol Chem. 2001;276:18223–18228. doi: 10.1074/jbc.M101266200. [DOI] [PubMed] [Google Scholar]
- 142.Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, Lin W, Reddy B, Chan JY, Kong AN. Identification of Nrf2-regulated genes induced by chemopreventive isothiocyanate PEITC by oligonucleotide microarray. Life Sci. 2006;79:1944–1955. doi: 10.1016/j.lfs.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 143.Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, Mann GE. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 144.Gong P, Stewart D, Hu B, Li N, Cook J, Nel A, Alam J. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Δ12,14-prostaglandin J2 is mediated by the stress response elements and transcription factor Nrf2. Antioxid Redox Signal. 2002;4:249–257. doi: 10.1089/152308602753666307. [DOI] [PubMed] [Google Scholar]
- 145.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280:31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 146.Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M, Tohyama K, Lipton SA. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic phase II inducers. Proc Natl Acad Sci USA. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang X, Lu L, Dixon C, Wilmer W, Song H, Chen X, Rovin BH. Stress protein activation by the cyclopentenone prostaglandin 15-deoxy-Δ 12,14-prostaglandin J2 in human mesangial cells. Kidney Int. 2004;65:798–810. doi: 10.1111/j.1523-1755.2004.00454.x. [DOI] [PubMed] [Google Scholar]
- 148.Horton ND, Mamiya BM, Kehrer JP. Relationships between cell density, glutathione and proliferation of A549 human lung adenocarcinoma cells treated with acrolein. Toxicology. 1997;122:111–122. doi: 10.1016/s0300-483x(97)00086-3. [DOI] [PubMed] [Google Scholar]
- 149.B. A. F. is the Chief Scientific Officer of Complexa, Inc.