

Abstract
Mammalian Hedgehog (Hh) signaling relies on three Gli transcription factors to mediate Hh responses. This process is controlled in part by a major negative regulator, Sufu, through its effects on Gli protein level, distribution and activity. In this report, we showed that Sufu regulates Gli1 protein levels by antagonizing Numb/Itch. Otherwise, Numb/Itch would induce Gli1 protein degradation. This is in contrast to inhibition of Spop-mediated degradation of Gli2/3 by Sufu. Thus, controlling protein levels of all three Gli genes by Sufu is a conserved mechanism to modulate Hh responses albeit via distinct pathways. These findings in cell-based assays were further validated in vivo. In analyzing how Sufu controls Gli proteins in different tissues, we discovered that loss of Sufu in the lung exerts different effects on Hh target genes. Hh targets Ptch1/Hhip are upregulated in Sufu-deficient lungs, consistent with Hh pathway activation. Surprisingly, protein levels of Hh target Gli1 are reduced. We also found that myofibroblasts are absent from many prospective alveoli of Sufu-deficient lungs. Myofibroblast development is dependent on PDGF signaling. Interestingly, analysis of the Pdgfra promoter revealed a canonical Gli-binding site where Gli1 resides. These studies support a model in which loss of Sufu contributes to compromised Pdgfra activation and disrupts myofibroblast development in the lung. Our work illustrates the unappreciated complexity of Hh responses where distinct Hh targets could respond differently depending on the availability of Gli proteins that control their expression.
Keywords: Hedgehog, Sufu, Gli, Lung, Myofibroblast, PDGF
Introduction
Hedgehog (Hh) signaling controls key steps of development in most tissues and organs of invertebrates and vertebrates (Briscoe and Therond, 2013; Ingham et al., 2011; Wilson and Chuang, 2010). The unique cellular composition and morphological movement in individual tissues require distinct modes of Hh signaling. For example, in the mammalian neural tube and limb, Hh expression from a localized source, such as the notochord/floor plate and the zone of polarizing activity (ZPA), is known to exert dose-dependent long-range signaling effects on tissue patterning. By contrast, in several branching organs such as the lung, epithelial Hh signaling to the mesenchyme mediates critical aspects of epithelial-mesenchymal interactions that drive lung branching morphogenesis. Hh signaling thus generates different outputs in diverse tissues, which underlie cellular changes during tissue patterning. Uncovering the whole complement of Hh targets and how they control cellular changes in each tissue is required for understanding the development of a given tissue. This knowledge will also contribute to our mechanistic understanding of tissue regeneration and repair and cancer development, in which Hh signaling is frequently activated (Barakat et al., 2010; Bijlsma and Roelink, 2010; Scales and de Sauvage, 2009).
The Hh pathway has been extensively studied for two decades, culminating in a basic framework of mammalian Hh signal transduction that depends on Gli transcription factors (Gli1-3) to mediate Hh responses (Beachy et al., 2010; Chen and Jiang, 2013; Eggenschwiler and Anderson, 2007; Farzan et al., 2008; Hui and Angers, 2011; Rabinowitz and Vokes, 2012; Robbins et al., 2012; Ryan and Chiang, 2012; Wang et al., 2007). Gli3 (and to some extent Gli2) undergoes limited proteolysis in the absence of the Hh ligand to produce a transcriptional repressor (Pan et al., 2006; Wang et al., 2000). Hh signaling not only inhibits proteolysis of Gli proteins but also promotes the conversion of Gli proteins (primarily Gli2) into transcriptional activators. Gli1, like Ptch1 and Hhip, is a transcriptional target of Hh signaling and Gli1 induction is believed to amplify Hh responses. The combinatorial effects of Gli activators and repressors likely mediate graded Hh responses in diverse tissues. In this regard, a large gap remains in our ability to correlate Hh signaling outputs with phenotypic outcomes since it is difficult to delineate the contributions of individual Gli protein or its processed form. This is further complicated by the differential expression and requirement of Gli proteins (Bai et al., 2004; Bowers et al., 2012; Cao et al., 2013; Ding et al., 1998; Matise et al., 1998) and their complex interactions in diverse tissues (Bowers et al., 2012; Liu et al., 2012).
One of the critical events in mammalian Hh signaling involves regulation of Gli by Suppressor of fused (Sufu), a major negative regulator. Studies of Sufu thus provide a unique opportunity to uncover the molecular mechanisms by which Gli proteins control Hh signaling. Sufu can sequester Gli proteins (Barnfield et al., 2005; Ding et al., 1999; Kogerman et al., 1999; Murone et al., 2000), regulate Gli2/3 protein levels (Chen et al., 2009; Jia et al., 2009; Wang et al., 2010), facilitate the production of Gli repressor and inhibit the generation of Gli activators (Humke et al., 2010; Tukachinsky et al., 2010). Perhaps all of these actions ensure the production of appropriate amounts of Gli activators and repressors as well as a pertinent Gli activator/repressor ratio necessary for tissue development and homeostasis. The relative contribution of multiple effects of Sufu to Gli protein functions has not been clearly delineated.
A key aspect of Hh signaling is to turn on Hh target genes through Gli activators. In this report, we investigate how Sufu controls Gli1 protein levels both in vitro and in vivo (lungs). These studies not only revealed a conserved mechanism by which Sufu controls Gli protein levels but also led to the unexpected finding that Hh targets can exhibit different responses when the Hh pathway is activated. We speculate that this is because different combinations of Gli proteins are present in a particular tissue for activating Hh targets. Thus some Gli targets are upregulated while others are downregulated, depending on the availability of Gli proteins that control their expression in a specific tissue. This result reveals the complexity of Hh responses in diverse tissues and increases our understanding of how the Sufu/Gli circuitry controls Hh pathway activation.
Materials and methods
Animal husbandry
All mice were handled in accordance with the animal care policies of the UCSF Institutional Animal Care and Use Committee. Null and conditional alleles of Sufu, Ptch1LacZ (STOCK Ptch1tm1Mps/J), Dermo1Cre (B6.129X1-Twist2tm1.1(cre)Dor/J), Gli1LacZ (STOCK Gli1tm2Alj/J) and PdgfraH2B-eGFP (B6.129S4-Pdgfratm11(EGFP)Sor/J) mice have been previously described (Bai et al., 2002; Chen et al., 2009; Goodrich et al., 1997; Hamilton et al., 2003; Pospisilik et al., 2010; Yu et al., 2003).
Sufuf mice harboring loxP sites flanking exons 4–8 of Sufu have been previously reported (Chen et al., 2009). Mesenchyme-specific deletion of Sufu was achieved by generating Sufuf/−; Dermo1Cre/+ mice through crosses between Dermo1Cre/+; Sufu+/− and Sufuf/f mice. Crosses were also set up to bring Ptch1-LacZ to Sufuf/−; Dermo1Cre/+ mice. Embryos were obtained from timed pregnancies.
Histology and in situ hybridization
Embryos were harvested at indicated time points and the embryos or lungs were fixed in 4% paraformaldehyde (PFA) in PBS at 4°C overnight, embedded in paraffin and sectioned at 6 μm. Histological analysis and section in situ hybridization using 33P-labeled riboprobes were performed as reported (Chen et al., 2009).
Standard molecular biology
Standard molecular biology techniques were performed as previously described (Nagy et al., 2003; Sambrook and Russell, 2001).
Isolation of primary lung mesenchymal cells
To derive lung mesenchymal cells, mouse embryonic lungs from wild-type (wt) and Sufuf/−; Dermo1Cre/+ embryos were digested in 0.05% Trypsin/EDTA at 37°C for 10 minutes. Cells were then seeded into culture dishes and incubated at 37°C for 1 hour. Cells in suspension were removed and lung mesenchymal cells that attached to the dishes were harvested for Western blotting, qPCR analysis or immortalization as described (Chen et al., 2009).
Cell culture, transfections, and immunoprecipitation
Wild-type and Sufu−/− mouse embryonic fibroblasts (MEFs) (Chen et al., 2009) and lung mesenchymal cells were maintained as described (Chen et al., 2009).
HEK293T or lung mesenchymal cells were transfected with different combinations of pcDNA3-Gli1-3xFLAG, pcDNA3-Numb-Myc and pcDNA3-Sufu-Myc. 48 hrs post-transfection, cells were lysed with IP buffer (1% Triton X-100, 150mM NaCl, 50mM Tris-Cl, 1mM EDTA) with protease inhibitor cocktail (Roche). The lysates were cleared by centrifugation and the resultant supernatants were bound to 15 μl FLAG M2 agarose in 50% slurry (Sigma) overnight at 4°C. Beads were washed three times with IP buffer and eluted with SDS sample buffer for Western blot analysis. Cycloheximide (CHX) and MG132 were purchased from Sigma.
For Western blotting, samples were run on SDS-glycine gels and transferred to nitrocellulose membrane following standard procedures (Sambrook and Russell, 2001). The membrane was then blocked with Odyssey LI-COR Blocking Buffer (LI-COR), and incubated with primary antibodies overnight. After washes in TBST (0.1% Tween 20), the blots were incubated with IRDye 800CW–conjugated donkey anti-goat IgG, IR Dye 800CW–conjugated donkey anti-rabbit IgG, or IRDye-680LT–conjugated donkey anti-mouse IgG (LI-COR). The signals on the membranes were detected with the Odyssey infrared imaging system (LI-COR). The following primary antibodies were used: rabbit anti-Gli1 (Cell Signaling, 1:1,000), rabbit anti-FLAG (Sigma, 1:2,000), mouse anti-Myc 9E10 monoclonal antibody (1:100), mouse anti-α-tubulin (Sigma, 1:2,000), goat anti-Gli2 (R&D, 1:1,000), goat-anti-Gli3 (R&D, 1:1,000), rabbit anti-Pdgfra (Cell Signaling, 1:2,000).
RNA extraction and qPCR analysis
The following primers for mouse genes were used for qPCR: Gapdh F: 5′ AGGTTGTCTCCTGCGACTTCA 3′; Gapdh R: 5′ CCAGGAAATGAGCTTGACAAAGTT 3′; Ptch1 F: 5′ TGCTGTGCCTGTGGTCATCCTGATT 3′; Ptch1 R: 5′ CAGAGCGAGCATAGCCCTGTGGTTC 3′; Gli1 F: 5′ CCCATAGGGTCTCGGGGTCTCAAAC 3′; Gli1 R: 5′ GGAGGACCTGCGGCTGACTGTGTAA 3′; Numb F: 5′ CGTAGCAATGCCTGTCCGTGAA 3′; Numb R: 5′ AGAGGCAGCACCAGAAGACTGA 3′; Pdgfa F: 5′ CTGGCTCGAAGTCAGATCCACA 3′; Pdgfa R: 5′ GACTTGTCTCCAAGGCATCCTC 3′; Pdgfra F: 5′ GCAGTTGCCTTACGACTCCAGA 3′; Pdgfra R: 5′ GGTTTGAGCATCTTCACAGCCAC 3′; Pdgfrb F: 5′ ACTACATCTCCAAAGGCAGCACCT 3′; Pdgfrb R: 5′ TGTAGAACTGGTCGTTCATGGGCA 3′; Elastin F: 5′ TCCTGGGATTGGAGGCATTGCA 3′; Elastin R: 5′ ACCAGGCACTAAACCTCCAGCA 3′.
shRNA-mediated gene knockdown
shRNAs were designed using the pSicOligomaker program (Reynolds et al., 2004). Oligonucleotides encoding shRNAs were cloned into the pLentiLox3.7 vector. To generate lentiviral supernatant, HEK 293T cells were transfected with the pLentiLox3.7 vector carrying the insert and the packaging vectors pLP1, pLP2, and pLP/VSV-G using Lipofectamine 2000 (Life Technologies). Forty-eight hours post-transfection, supernatants were harvested. Wild-type or Sufu-deficient lung mesenchymal cells at 50% confluency were transduced with lentiviruses supplemented with 8 μg/mL polybrene. Upon reaching confluency, cells were harvested and the lysates were analyzed by Western blotting. The following 19-mer sequences were used for shRNA-mediated gene knockdown: mouse Numb (NM_001136075), 5′ GAAGATGTCACCCTTTAAA 3′ and 5′ GCAGACATTCCCTCAATAT 3′; mouse Itch (NM_001243712), 5′ GAAGCCAAGGTCAGTTAAA 3′ and 5′ GTACTTCTCAGTTGATAAA 3′; GFP, 5′ GCAGACCATTATCAACAAA 3′.
Immunofluorescence and microscopy
Immunohistochemistry was performed following standard procedures (Ausubel et al., 2003). The primary antibodies used were: goat anti-CC10 (Santa Cruz, 1:500), rabbit anti-pro SP-C (Millipore, 1:400), hamster anti-T1α (Developmental Studies Hybridoma Bank, 1:200), mouse anti-Ki67 (BD Biosciences, 1:100), rabbit anti-phospho-Histone H3 (PH3) (Millipore, 1:200), mouse anti-smooth muscle actin (SMA) (Sigma, 1:1,000) and mouse anti-CD31 (PECAM-1) (BD Biosciences, 1:100). PECAM-1 staining was performed using the ABC kit (Vector Laboratories). Antibody against Ki67 required biotin-streptavidin amplification with the TSA kit (PerkinElmer) for optimal signal detection. Secondary antibodies and conjugates used were donkey anti-mouse Alexa Fluor 594 (Life Technologies, 1:2,000), donkey anti-rabbit Alexa Fluor 488 (Life Technologies, 1:2,000), biotinylated horse anti-mouse (Jackson ImmunoResearch Laboratories, 1:1,000) and DAPI (Sigma, 1:10,000).
Luciferase assays
Analysis of transcription factor binding sites on Pdgfra promoters from different species was performed using ECR Browser (Ovcharenko et al., 2004) and rVISTA 2.0 (Loots and Ovcharenko, 2004). Mouse Pdgfra promoter fragments (−2473 to −2409 and −3027 to −1940; position zero marks the transcriptional start of Pdgfra) were cloned upstream of the firefly luciferase gene in a modified pGL-Basic vector (denoted pGL-TK), which contains a thymidine kinase (TK) minimal promoter. The consensus sequence of Gli binding site (GliBS) (5′GACCACCCA3′) within the Pdgfra promoter was altered to 5′GACTGAAGA3′ or deleted by site-directed mutagenesis. Both mutant constructs gave similar results.
These constructs were co-transfected with pRL-TK (Renilla) and cDNA encoding Gli1 into HEK293T cells. Cells were harvested 48 hours post-transfection for analysis using the Dual Luciferase Reporter Assay System (Promega).
Chromatin immunoprecipitation (ChIP)
MEFs stably expressing Gli1-3xFLAG and Gli2-3xFLAG were used for ChIP analysis following established procedures (Collas, 2011; Weinmann and Farnham, 2002). ChIP was performed using the EZ-ChIP kit (Millipore) according to the manufacturer’s manual. Briefly, cells were cross-linked in 1% formaldehyde and the DNA was sonicated into a range of 100–600 bp in size using a Bioruptor sonicator (Diagenode) for 5 cycles of 30 seconds on/30 seconds off. The extracts were precleared in BSA-blocked protein A/G beads and incubated with antibodies or IgG control overnight. Protein A/G beads were incubated with the lysate-antibody complex for 1 hour. After washes, the DNA was eluted and reverse-crosslinked at 65°C overnight. The DNA was purified and analyzed by qPCR. The antibodies used were mouse anti-FLAG M2 monoclonal antibody (Sigma) and normal mouse IgG (Santa Cruz).
For ChIP analysis, the following primers were used: Pdgfra promoter F: 5′ CTTGGCTAGGCACTGGCACTTGC 3′; Pdgfra promoter R: CCAGCCCAGTTCTTGCCCTGTTC; β-actin promoter F 5′AGAAGGACTCCTATGTGGGTGA 3′, β-actin promoter R 5′ACTGACCTGGGTCATCTTTTC 3′.
Results
Sufu regulates Gli1 protein levels by blocking proteasome-mediated protein degradation
Our prior studies showed that Gli1 protein levels are increased (likely due to transcriptional activation of Gli1) in Sufu-deficient mouse embryonic fibroblasts (MEFs) in contrast to the drastically reduced protein levels of Gli2 and Gli3 (Chen et al., 2009; Jia et al., 2009; Wang et al., 2010). Gli2 and Gli3 are subject to Spop-mediated ubiquitination and degradation, and Sufu antagonizes Spop function to preserve a pool of Gli2 and Gli3 (Chen et al., 2009; Wang et al., 2010). We also demonstrated that Gli1 is immune to Spop-mediated protein degradation (Chen et al., 2009; Zhang et al., 2009). In this study, we revisit the issue of whether Sufu controls Gli1 protein levels by examining Gli1 in Sufu−/− MEFs or Sufu-deficient mouse lung mesenchymal cells (their derivation are described below) treated with proteasome inhibitors (e.g., MG132). To our surprise, Gli1 levels were further increased following MG132 treatment (Fig. 1A), similar to what was observed for Gli2 and Gli3. Since Numb was previously shown to activate the E3 ligase Itch, resulting in Gli1 ubiquitination and degradation (Di Marcotullio et al., 2011), we tested the idea that Sufu stabilizes Gli1 protein by antagonizing Numb/Itch, a mechanism distinct from Sufu control of Gli2/3 protein levels. We found that Gli1 protein levels were reduced upon Numb overexpression in cultured cells (Fig. 1B; supplementary material Fig. S1) and Sufu was capable of blocking Numb activity and restoring Gli1 protein levels (Fig. 1B; supplementary material Fig. S1). Conversely, knockdown of Numb or Itch in Sufu-deficient lung cells led to increased Gli1 protein levels (supplementary material Fig. S2) by eliminating Numb/Itch-mediated Gli1 degradation. Furthermore, Sufu overexpression decreased the binding between Numb and Gli1 (Fig. 1C; supplementary material Fig. S1). This suggests that Sufu controls Gli1 protein levels by antagonizing Numb/Itch (Fig. 1D). Interestingly, Drosophila Su(fu) was also capable of stabilizing Gli1 when expressed in cultured cells (supplementary material Fig. S3). This supports the idea that control of Gli1 protein levels by Sufu is a conserved mechanism.
Figure 1. Control of Gli1 protein levels by Sufu.

(A) Western blot analysis of lysates from Sufu-deficient (Sufuf/−; Dermo1Cre/+) lung cells or Sufu null (Sufu−/−) MEFs treated with MG132 to block proteasome-mediated degradation. Endogenous Gli1 protein levels were elevated when protein degradation was inhibited in Sufu mutants. Similarly, protein levels of transfected Gli1 were increased when Sufu was co-expressed in wild-type (wt) MEFs. These results suggest that Sufu stabilizes Gli1 by preventing proteasome-dependent Gli1 degradation. Note that cycloheximide was added to block new protein synthesis in these studies. (B) Western blot analysis of lysates from HEK293T cells expressing various combinations of Gli1, Numb, Sufu and Acp1 (control). Numb expression resulted in reduction in Gli1 protein levels. This is consistent with previous reports in which Numb was shown to activate the E3 ligase Itch, leading to Gli1 ubiquitination and degradation. Numb-induced Gli1 reduction was reversed when Sufu was co-expressed with Numb. Tubulin was used as the loading control. (C) Western blot analysis of immunoprecipitated Gli1FLAG from HEK293T cell lysates to test the competition between Sufu and Numb in binding to Gli1. Co-immunoprecipitated NumbMyc by Gli1 was significantly reduced when SufuMyc was also pulled down by Gli1. (D) A model in which Sufu stabilizes Gli1 by inhibiting Numb-mediated protein degradation. In, input; IP, immunoprecipitation.
Loss of Sufu in the mouse lung results in increased Hh signaling but reduced Gli1 protein levels
To further validate regulation of Gli1 protein levels by Sufu in vivo, we examined Gli1 protein in Sufu mutants. Since Sufu mutant embryos die at 9.5 days post coitus (dpc) before major organs develop, conditional inactivation of Sufu is required to investigate how Sufu/Gli interactions affect Hh signaling in various tissues. In this work, we explored tissues not previously examined and focused on the lung. We utilized the Dermo1Cre mouse line (Yu et al., 2003) to convert a conditional allele of Sufu (Sufuf) into a null allele (Sufu−) in the lung mesenchyme as well as the mesenchyme of other tissues. Sufuf/−; Dermo1Cre/+ mice are referred to as Sufu-deficient mice in this study. Sufu-deficient mice died soon after birth likely due to respiratory failure and defects in other tissues.
As a first step toward understanding how the loss of Sufu leads to lung defects, we examined Hh target gene expression in Sufu-deficient lungs by reporter activity, qPCR and Western blotting (Fig. 2A–D). For qPCR analysis of transcript levels or Western blotting, we also isolated lung mesenchyme since Dermo1Cre selectively inactivates Sufu in this compartment (data not shown). We found that Ptch1 reporter activity (Fig. 2A, B) and Ptch1/Hhip mRNA levels (Fig. 2D) were increased in Sufu-deficient lungs, indicative of Hh pathway activation. Similar to Sufu mutant embryos, Gli2 and Gli3 protein levels were reduced in Sufu-deficient lungs (Fig. 2C). To our surprise, protein levels of Gli1, a Hh target, were reduced in Sufu-deficient lungs (Fig. 2C) even though Gli1 mRNA levels were unaltered (Fig. 2D). This result contrasts with the expected outcome of global activation of Hh target genes, implying that our traditional view of Hh pathway activation is oversimplified.
Figure 2. Upregulation of Hh target gene expression with concomitant Gli1 protein reduction in the absence of Sufu.

(A–B) β-galactosidase staining of wild-type (wt) and Sufu-deficient (Sufuf/−; Dermo1Cre/+; Ptch1LacZ/+) lungs to detect Ptch1 expression in lung mesenchyme. Ptch1-LacZ expression was stronger and broader in Sufu mutants compared to wt. (C) Western blot analysis of endogenous Gli1 protein levels in wt and Sufu mutant lungs. Gli1 protein levels were decreased in Sufu mutant lungs collected at various stages of lung development. This resembles reduced protein levels of Gli2/3 in Sufu mutant lungs. Likewise, a reduction in Gli1 protein levels was also detected in Sufu-deficient hearts. (D) qPCR analysis of Ptch1 and Gli1 mRNA in wt and Sufu mutant lungs. Ptch1 and Hhip mRNA levels were elevated in Sufu mutant lungs while Gli1 mRNA levels were unaltered. This suggests that reduced Gli1 protein levels in Sufu-deficient lungs are a result of loss of Sufu and not due to changes in Gli1 transcript levels. All values are means ± standard deviation. * P < 0.05; ** P < 0.01; NS, not significant (unpaired Student’s t-test) (n=6 for 12.5 dpc lungs; n=4 for 16.5 dpc lungs; n=4 for 16.5 dpc heart). Elevation of Hhip mRNA levels at 16.5 dpc was not statistically significant likely due to large variations in transcript levels among different samples. Note that Sufuf/−; Dermo1Cre/+ lungs/heart are abbreviated as Sufu−/− lungs/heart while Sufu−/− embryos represent Sufu null embryos in this figure. FL, full-length; R, repressor. Scale bar = 100 μm for A–B.
We also tested whether reduced Gli protein levels were also present in other Sufu-deficient tissues and found that Sufu mutant hearts had increased Gli1 mRNA but reduced Gli1 protein levels (Fig. 2C, D). This is in contrast to Sufu-deficient neural tubes where Gli1 activity is increased (Cooper et al., 2005; Svard et al., 2006), likely due to transcriptional activation of Gli1. Results from in vitro and in vivo studies on Sufu and Gli1 are consistent with a model in which regulation of Gli1 protein levels by Sufu is a conserved, general mechanism. This is achieved by antagonizing Numb through Sufu; otherwise, Numb would induce Gli1 degradation (supplementary material Fig. S4). Baseline Gli protein levels may differ in various tissues and cell lines depending on Gli1 transcript levels. We suspect that in tissues where Gli1 protein levels are reduced in the absence of Sufu, Hh targets controlled by Gli1 proteins could be affected.
Myofibroblast development is disrupted in Sufu-deficient lungs
To gain insight into how altered Hh target gene expression in Sufu-deficient lungs could impact lung development, we conducted a careful phenotypic analysis of Sufu mutant lungs at various stages of lung development (Fig. 3A–N). The phenotypes in Sufu mutant lungs were completely penetrant and showed little variation from animal to animal. Early patterns of epithelial branching seemed to be established properly in Sufu-deficient lungs, leading to the correct number and positioning of lung lobes (Fig. 3D). However, defective morphogenesis in both the epithelium and mesenchyme became apparent as lung development proceeded (Fig. 3; supplementary material Fig. S5). At birth, Sufu mutant lungs were smaller in size and exhibited reduced branching morphogenesis associated with a compact mesenchyme (Fig. 3N).
Figure 3. Conditional inactivation of Sufu in lung mesenchyme.
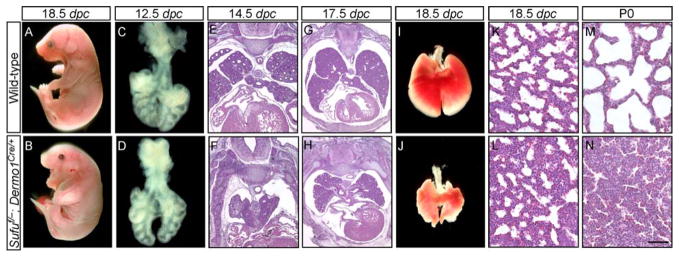
(A–N) External morphology (A, B), dissected lungs (C, D, I, J) and histology (E–H, K–N) of lung sections from wild-type (wt) and Sufuf/−; Dermo1Cre/+ mouse embryos at various embryonic stages and postnatal (p) day 0 as indicated. More than 50 Sufu mutants were examined. The phenotypes in Sufu mutant lungs were completely penetrant and showed little variation from animal to animal. Epithelial and mesenchymal development was defective in Sufu mutants, resulting in a smaller sized lung with a compact mesenchyme. The length of the proximal-distal axis of Sufu mutant lungs is ~80% of that of wt lungs. Sufu mutant mice cannot expand their lungs and died a few hours after birth. Early patterns of epithelial branching appeared to be established properly in Sufu-deficient lungs, leading to the correct number and positioning of lung lobes. Epithelial branching at later stages of development was not as extensive in Sufu-deficient lungs compared to that in wt lungs. dpc, days post coitus. Scale bars: K–N, 50 μm.
Somewhat surprisingly, all the major epithelial cell types including Clara cells (CC10+), ciliated cells (Ac-tubulin+), pulmonary neuroendocrine cells (CGRP+), alveolar type II (SPC+) and type I (T1α+) cells (Morrisey and Hogan, 2010) were properly specified (Fig. 4C–H) in the absence of Sufu. Most mesenchymal cell types including the bronchial smooth muscle (SMA+) (K, L) and blood vessels (PECAM+) (M, N) were also properly produced. In addition, no apparent difference in the rate of cell proliferation (Fig. 4A, B, A′; supplementary material Fig. S6) or cell death (supplementary material Fig. S6) was detected in either the epithelium or mesenchyme between wild-type and Sufu-deficient lungs during lung development.
Figure 4. Disruption of myofibroblast development in Sufu-deficient lungs.

(A–Z) Immunostaining (A–N, Y, Z) and in situ hybridization (O–X) of wild-type (wt) and Sufu-deficient (Sufuf/−; Dermo1Cre/+) lungs. No apparent difference in cell proliferation rate (judged by Ki67 and PH3 staining) or cell death was found between wt (A) and Sufu mutant (B) lungs. Quantification of cell proliferation in the epithelial and mesenchymal compartments at 12.5 dpc was shown in A′. Major epithelial cell types, including Clara cells (CC10+) (C, D), alveolar type II (SPC+), type I (T1α+) cells (E, F) and pulmonary neuroendocrine cells (CGRP+) (G, H) were properly specified in the absence of Sufu. Most mesenchymal cell types such as the bronchial smooth muscle (SMA+) (K, L) and blood vessels (PECAM+) (M, N) were also properly generated. By contrast, myofibroblasts (white arrow; marked by smooth muscle actin [SMA] staining) were greatly reduced in Sufu mutant lungs (I). This was associated with decreased Pdgfra expression (pink signal) (compare V to U) while Pdgf ligand expression was unaltered (compare X to W). In addition, the expression patterns and levels of Shh (O, P), Fgf10 (Q, R) and Bmp4 (S, T) or other components in Hh, Fgf and Bmp signaling were similar between wt and Sufu mutant lungs. Both Gli1 and Pdgfra were detected in the secondary septa of alveoli (arrows in Y, Z). (B′) qPCR analysis of Pdgfa, Pdgfra, Pdgfrb and Elastin mRNA in wt and Sufu mutant lungs. Pdgfra mRNA levels were reduced in Sufu mutant lungs while Pdgfa and Pdgfrb mRNA levels were unaltered. This is consistent with results from in situ hybridization. Elastin transcript levels were also reduced in Sufu mutant lungs, consistent with defective myofibroblast development. All values are means ± standard deviation. * P < 0.05; ** P < 0.01; NS, not significant (unpaired Student’s t-test) (n=3). (C′) Western blot of endogenous Pdgfra protein levels in wt and Sufu mutant lungs. Pdgfra protein levels were decreased in Sufu mutant lungs collected at various stages of lung development. This was likely due to reduced Pdgfra transcript levels in Sufu mutant lungs. dpc, days post coitus. Scale bars: A–F and K–N, 50 μm; G–J and Y–Z, 50 μm; O–X, 100 μm.
We noticed that Sufu-deficient lungs contained disorganized saccules, raising the possibility that alveolar development could be affected (Fig. 3N compared to 3M). In wild-type lungs, by 18.5 dpc (less than 24 hr before birth), myofibroblasts localized to the prospective alveoli (Fig. 4I) and participated in subsequent secondary septa formation during alveolar formation. By contrast, myofibroblasts were absent from many prospective alveoli of Sufu-deficient lungs (Fig. 4J) while smooth muscle cells surrounding the large airways or blood vessels could be detected (Fig. 4K, L). Moreover, reduced myofibroblasts in Sufu-deficient lungs were associated with reduced expression of Elastin (Fig. 4B′). This suggests that Hh pathway perturbation in the absence of Sufu leads to defective myofibroblast maturation and migration.
Defective myofibroblast development in Sufu-deficient lungs is associated with reduced PDGF signaling
We first investigated potential perturbations of major signaling pathways in the absence of Sufu. The expression patterns and levels of components in the Fgf, Bmp and Wnt signaling pathways were similar between wt and Sufu mutant lungs (Fig. 4O–T and data not shown). We then focused on PDGF signaling since it has been shown to play a central role in myofibroblast development. Pdgf ligand or Pdgf receptor α (Pdgfra) knockout mice die without myofibroblasts or secondary septa (Bostrom et al., 1996; Sun et al., 2000). We surmised that defective myofibroblast development in Sufu-deficient lungs is caused by reduced PDGF signaling. To test this idea, we examined the expression of Pdgf ligand and receptor in wild-type and Sufu-deficient lungs by in situ hybridization, qPCR and Western blotting. We found that while Pdgfa expression was indistinguishable between wild-type and Sufu-deficient lungs (Fig. 4W, X and B′), Pdgfra expression was drastically reduced in Sufu mutant lungs (Fig. 4U, V and B′). By contrast, Pdgfrb expression was unaltered in Sufu-deficient lungs (Fig. 4B′). This suggests that disrupted Hh signaling in Sufu mutant lungs leads to reduced Pdgfra expression and consequently defective myofibroblast development.
Pdgfra is a direct transcriptional target of Gli1
We investigated the molecular mechanisms by which loss of Sufu leads to reduced Pdgfra expression. Since a reduction in Pdgfra expression in Sufu-deficient lungs is associated with decreased Gli1 protein levels (despite global Hh pathway activation) and the expression of Pdgfra and Gli1 overlaps along the alveolar wall (Fig. 4Y, Z) and the secondary septum (arrows in Fig. 4Y, Z), we speculate that Pdgfra could be a direct target of Gli1. Indeed, we identified a canonical Gli-binding site (GliBS) in the mouse Pdgfra (but not Pdgfrb) promoter (Fig. 5A). A GliBS was also found in a similar location of the Pdgfra promoter in several vertebrate species (Fig. 5A). This is consistent with our model in which Gli1 controls Pdgfra expression. Reduced Gli1 protein levels in Sufu-deficient lungs could contribute to compromised Pdgfra promoter activation. By contrast, other Hh targets controlled by Gli2 and Gli3 are activated.
Figure 5. Gli1 and regulation of Pdgfra promoter activity.

(A) Sequence analysis of the Pdgfra promoter from different species. A conserved canonical Gli-binding site (GliBS) is boxed and colored. The numbers represent distances from the transcriptional start site of Pdgfra, which is marked as position zero. (B) Schematic diagram depicting Pdgfra-luc reporter constructs in which mouse Pdgfra promoter fragments are placed upstream of firefly luciferase (luc). A canonical GliBS is present in the Pdgfra promoter and is mutated in the control construct PdgfraΔGliBS-luc (abbreviated as ΔGliBS-luc in the figure). Addition of Gli1 activated Pdgfra-luc and not PdgfraΔGliBS-luc in cell-based assays. Control nuclear protein Smurf did not induce Pdgfra-luc expression. (C) Gli1 but not Gli2 occupied the Pdgfra promoter by ChIP analysis using MEFs expressing FLAG-tagged Gli1 and Gli2. Gli-binding on the Pdgfra promoter was normalized to the β-actin control promoter. All values are means ± standard deviation. * P < 0.05; NS, not significant (unpaired Student’s t-test) (n=3). (D) A model of differential regulation of Gli proteins by Sufu. Loss of Sufu results in reduced protein levels of all three Gli proteins. Reduction in Gli2/3 protein levels is associated with increased Gli activators and reduced Gli repressors. This would lead to overall Hh pathway activation and Hh target gene expression. By contrast, reduced protein levels of the constitutive activator Gli1 result in downregulation of Hh targets that primarily rely on Gli1 for their expression such as Pdgfra in the lung.
We further tested this idea by performing reporter assays using Pdgfra-luc in which a Pdgfra promoter fragment is placed upstream of the luciferase (luc) reporter (Fig. 5B). Gli1 was co-transfected with Pdgfra-luc in MEFs and tested for its ability to activate Pdgfra-luc. A Pdgfra fragment in which the canonical Gli-binding site is mutated (denoted as PdgfraΔGliBS) was used as a control (Fig. 5B). We showed that the addition of Gli1 increased Pdgfra-luc activity but failed to activate PdgfraΔGliBS-luc (Fig. 5B). By contrast, Gli2 has low activity in activating the Pdgfra promoter in vitro (supplementary material Fig. S7). Finally, Hh stimulation led to increased expression levels of Pdgfra but not Pdgfrb in lung mesenchymal cells (supplementary material Fig. S8), suggesting a connection between Hh and PDGF signaling.
To determine whether the endogenous Pdgfra promoter is occupied by Gli1, we performed chromatin immunoprecipitation (ChIP) analysis (Weinmann and Farnham, 2002) on MEFs expressing FLAG-tagged Gli1 and Gli2. Cell lysate from lung mesenchymal cells was immunoprecipitated using FLAG antibodies. We amplified Pdgfra genomic regions using FLAG immunoprecipitates and found significant enrichment of Gli1 (but not Gli2) on the Pdgfra genomic fragment (Fig. 5C). These results suggest that Pdgfra is a direct target of Gli1.
Discussion
In this study, we discover that Sufu stabilizes Gli1 protein levels by antagonizing Numb/Itch-mediated protein degradation. Together with previous work that demonstrates control of Gli2/3 protein levels by Sufu (Chen et al., 2009; Jia et al., 2009; Wang et al., 2010), our new finding indicates that regulation of Gli protein levels by Sufu is a major general mechanism in modulating Hh responses. Interestingly, our analysis of Sufu-deficient lungs led to the unexpected observation that while canonical Hh targets such as Ptch1 are upregulated, Pdgfra, a direct target of Gli1, is downregulated. We propose that Hh targets do not respond uniformly to Hh signaling under different genetic perturbations and this could also vary from tissue to tissue. We speculate that this may be due to differential levels of individual Gli proteins that control common and distinct sets of Hh targets in a given tissue (Fig. 5D). As a result, some Hh targets are activated while others are concomitantly downregulated. Perhaps this enables Hh signaling to generate complex outputs by regulating the availability of different Gli proteins. Testing and generalizing this hypothesis in diverse tissues will significantly increase our understanding of how Hh signaling leads to diverse phenotypic consequences in homeostasis and disease.
Control of Gli protein levels by Sufu
It was previously reported that Sufu controls protein levels of Gli2 and Gli3 by antagonizing Spop-mediated ubiquitination and degradation (Chen et al., 2009; Wang et al., 2010). In addition, Drosophila Su(fu) also controls protein levels of Ci (Gli homolog) by antagonizing HIB/roadkill (Spop homolog) (Zhang et al., 2006). However, Gli1 is immune to Spop-mediated regulation (Chen et al., 2009; Zhang et al., 2009). In this study, we showed that Sufu counters the effects of Numb/Itch-mediated Gli1 protein degradation. Thus, control of Gli protein levels by Sufu is a conserved mechanism employed to modulate Hh responses.
It is unclear how two regulatory circuitries were evolved to differentially control Gli1 and Gli2/3 protein levels mediated by Sufu respectively. In this regard, it is interesting to note that Drosophila Su(fu) not only can substitute for mammalian Sufu and partially restore Gli2/3 protein levels in cell-based assays (Chen et al., 2009), it is also able to stabilize Gli1 protein levels in a similar assay. This suggests that a similar interface of molecular interactions may exist between Sufu-Spop and Sufu-Numb. Alternatively, fly Su(fu) may not have exploited the Numb-Itch pathway for controlling Ci protein levels.
Gli1 is part of a positive feedback loop of Hh responses since Gli1 is an early transcriptional target of Hh signaling. Control of Gli1 protein levels by Sufu may provide a mechanism to dampen Hh signaling when Sufu becomes inactive either by Hh signaling or in disease states. Reduced Gli1 protein levels due to loss of Sufu would limit transcriptional activity mediated by Gli1. This resembles Sufu’s effects on Gli2/3 protein levels, part of which are reflected by the failure to maximally activate Hh signaling in the absence of Sufu (Chen et al., 2009; Liu et al., 2012). In considering the consequence of loss of Sufu on Gli1 protein levels in various tissues, it is important to take into consideration Gli1 transcription. For instance, Gli1 is transcriptionally activated in the neural tube when the Hh pathway is activated (e.g., loss of Sufu). We surmise that increased Gli1 transcript levels in the neural tube would offset the effect of Gli1 protein degradation due to loss of Sufu; as a result, Gli1 protein levels are elevated. By contrast, transcriptional activation of Gli1 in the lungs is not as prominent and thus Sufu-deficient lungs have reduced Gli1 protein levels. However, we cannot rule out the possibility that different tissues may have distinct rates of Gli1 degradation and this could also contribute to variations in Gli1 protein levels in diverse tissues.
Epithelial-mesenchymal Hh signaling and lung development
Sufu-deficient lungs display overall upregulation of Hh signaling, resulting in a smaller lung with a compact mesenchyme. Somewhat surprisingly, the rate of cell proliferation during early lung development does not seem to be altered. Moreover, despite defective lung development in the absence of Sufu, all major lung cell types are properly specified. We postulate that abnormal mesenchymal development in the absence of Sufu disrupts epithelial-mesenchymal interactions, leading to defective epithelial branching. Consequently, lung size is reduced. Uncovering the cellular and molecular basis of lung defects in Sufu mutants would require future studies that employ tools such as live imaging and whole-genome analysis. This would allow the identification of potential defects in cellular behaviors and genes and pathways involved in this process.
Control of PDGF signaling and myofibroblast development
Removal of Sufu in the lung mesenchyme results in a reduced number of myofibroblasts. This could contribute to defective lung development and even perinatal lethality. Alveolar myofibroblasts play a key role in alveolus formation. They are contractile cells found in the alveolar interstitium during lung development and possess morphological and biochemical features intermediate between fibroblast and smooth muscle. Alveolar myofibroblasts appear to be derived from a population of mesenchymal cells that express the PDGF receptor. These cells subsequently spread to the walls of prospective terminal saccules to become future alveolar myofibroblasts. Alveolar myofibroblasts produce Elastin and participate in septal formation during alveologenesis. Indeed, reduced myofibroblasts in Sufu-deficient lungs are associated with reduced expression of Elastin.
We showed that Sufu-deficient lungs have reduced Pdgfra expression. Since PDGF signaling plays an essential role in myofibroblast development, our findings are consistent with the idea that downregulation of Pdgfra expression in Sufu mutant lungs likely results in a reduced myofibroblast number. Importantly, we demonstrated that Pdgfra is a direct target of Gli1. This allows us to propose a model in which reduced Gli1 protein levels in the absence of Sufu contribute to impaired Pdgfra expression and consequently myofibroblast development. Consistent with these observations, it was previously reported that Pdgfra is regulated by Gli1 in Hh-responsive cell lines (Xie et al., 2001). We also showed that Gli1 and Pdgfra co-localize extensively in the alveolar structure in postnatal lungs, further supporting the notion that Gli1 regulates Pdgfra expression. Future genetic studies that employ Sufu and Pdgfra mutants will further support a functional connection between Hh and PDGF signaling.
Possible distinct and overlapping targets of Gli proteins
Whole genome ChIP-on-chip studies using FLAG-tagged Gli1 and Gli3 in neural tissues and the limb have identified a significant number of genes with bona fide Gli-binding sites (Vokes et al., 2007; Vokes et al., 2008). These studies will serve as the guide for similar efforts in other organs such as the lungs. We anticipate that different sets of Gli targets will be identified in various tissues. A key issue is to determine whether certain Hh targets are controlled by distinct Gli proteins. Certain Hh targets may possess unique binding sequences that can only be recognized by a particular Gli protein. Alternatively, all Gli-binding sites are degenerate and can recruit any Gli proteins. In this case, differential binding would be an outcome of different Gli protein levels. Isolation of distinct cell types from a given tissue for whole-genome ChIP analysis may provide an opportunity to further test these hypotheses.
Gli2 is active in Sufu mutants despite its low levels. However, Gli2 activator fails to elevate Pdgfra expression levels in Sufu-deficient lungs. Gli1 mutants are viable (Park et al., 2000) and do not seem to display apparent defects in myofibroblast development (data not shown). Reduced Gli1 protein levels thus are unlikely the sole cause of defective myofibroblast development in Sufu–deficient lungs. Instead, loss of Sufu likely perturbs other processes, which in conjunction with reduced Gli1 protein levels, lead to the disruption of myofibroblast development. However, it remains possible that reduced Gli2/3 activator levels in the absence of Sufu also contributes to reduced Pdgfra expression in the developing lungs although Gli2 is not detected on the Pdgfra promoter by ChIP analysis using MEFs (Fig. 5C). In this scenario, myofibroblast defects would be observed in mice deficient in multiple Gli proteins. Genetic studies that produce mice deficient in Sufu and one or multiple Gli proteins will be informative in revealing regulation of Gli proteins by Sufu in the lung.
Taken together, our studies highlight the complexity of regulating Gli protein functions in diverse tissues. They also form the basis of further studies to investigate how multiple Gli proteins are regulated at multiple levels to control the expression of a unique set of Hh targets in a given tissue.
Supplementary Material
Sufu regulates Gli1 protein levels by antagonizing Gli1 degraders Numb/Itch
Hh pathway is activated in Sufu mutant lungs but Gli1 protein levels are reduced
Sufu-deficient lungs exhibit defective epithelial and mesenchymal development
Disrupted myofibroblast development is associated with reduced Pdgfra expression
Pdgfra is a direct target of Gli1, linking Hh and PDGF signaling
Acknowledgments
We thank David Ornitz for providing Dermo1Cre mice, Brian Black for the modified pGL-Basic vector, Vivian Chen and Chen-Che Huang for technical assistance, members of the Chuang laboratory for discussion and Ross Metzger for critical reading of the manuscript. This work was supported by grants from the Canadian Cancer Society Research Institute to C.C. H. and the National Institutes of Health (R01 HL091915) to P.T. C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology (Wiley) 2003 [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103–115. doi: 10.1016/s1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Barakat MT, Humke EW, Scott MP. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol Med. 2010;16:337–348. doi: 10.1016/j.molmed.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnfield PC, Zhang X, Thanabalasingham V, Yoshida M, Hui CC. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation; research in biological diversity. 2005;73:397–405. doi: 10.1111/j.1432-0436.2005.00042.x. [DOI] [PubMed] [Google Scholar]
- Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010;24:2001–2012. doi: 10.1101/gad.1951710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlsma MF, Roelink H. Non-cell-autonomous signaling by Shh in tumors: challenges and opportunities for therapeutic targets. Expert opinion on therapeutic targets. 2010;14:693–702. doi: 10.1517/14728222.2010.497488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom H, Willetts K, Pekny M, Leveen P, Lindahl P, Hedstrand H, Pekna M, Hellstrom M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Tornell J, Heath JK, Betsholtz C. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell. 1996;85:863–873. doi: 10.1016/s0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- Bowers M, Eng L, Lao Z, Turnbull RK, Bao X, Riedel E, Mackem S, Joyner AL. Limb anterior-posterior polarity integrates activator and repressor functions of GLI2 as well as GLI3. Dev Biol. 2012;370:110–124. doi: 10.1016/j.ydbio.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nature reviews Molecular cell biology. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- Cao T, Wang C, Yang M, Wu C, Wang B. Mouse limbs expressing only the Gli3 repressor resemble those of Sonic hedgehog mutants. Dev Biol. 2013;379:221–228. doi: 10.1016/j.ydbio.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MH, Wilson CW, Li YJ, Law KK, Lu CS, Gacayan R, Zhang X, Hui CC, Chuang PT. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009;23:1910–1928. doi: 10.1101/gad.1794109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Jiang J. Decoding the phosphorylation code in Hedgehog signal transduction. Cell research. 2013;23:186–200. doi: 10.1038/cr.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collas P. A chromatin immunoprecipitation protocol for small cell numbers. Methods Mol Biol. 2011;791:179–193. doi: 10.1007/978-1-61779-316-5_14. [DOI] [PubMed] [Google Scholar]
- Cooper AF, Yu KP, Brueckner M, Brailey LL, Johnson L, McGrath JM, Bale AE. Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development. 2005;132:4407–4417. doi: 10.1242/dev.02021. [DOI] [PubMed] [Google Scholar]
- Di Marcotullio L, Greco A, Mazza D, Canettieri G, Pietrosanti L, Infante P, Coni S, Moretti M, De Smaele E, Ferretti E, Screpanti I, Gulino A. Numb activates the E3 ligase Itch to control Gli1 function through a novel degradation signal. Oncogene. 2011;30:65–76. doi: 10.1038/onc.2010.394. [DOI] [PubMed] [Google Scholar]
- Ding Q, Fukami S, Meng X, Nishizaki Y, Zhang X, Sasaki H, Dlugosz A, Nakafuku M, Hui C. Mouse suppressor of fused is a negative regulator of sonic hedgehog signaling and alters the subcellular distribution of Gli1. Current biology: CB. 1999;9:1119–1122. doi: 10.1016/s0960-9822(99)80482-5. [DOI] [PubMed] [Google Scholar]
- Ding Q, Motoyama J, Gasca S, Mo R, Sasaki H, Rossant J, Hui CC. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development. 1998;125:2533–2543. doi: 10.1242/dev.125.14.2533. [DOI] [PubMed] [Google Scholar]
- Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzan SF, Singh S, Schilling NS, Robbins DJ. The adventures of sonic hedgehog in development and repair. III. Hedgehog processing and biological activity. Am J Physiol Gastrointest Liver Physiol. 2008;294:G844–849. doi: 10.1152/ajpgi.00564.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Molecular and cellular biology. 2003;23:4013–4025. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–537. doi: 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010;24:670–682. doi: 10.1101/gad.1902910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nature reviews Genetics. 2011;12:393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- Jia J, Kolterud A, Zeng H, Hoover A, Teglund S, Toftgard R, Liu A. Suppressor of Fused inhibits mammalian Hedgehog signaling in the absence of cilia. Dev Biol. 2009;330:452–460. doi: 10.1016/j.ydbio.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, Sandstedt B, Toftgard R, Zaphiropoulos PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nature cell biology. 1999;1:312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- Liu J, Heydeck W, Zeng H, Liu A. Dual function of suppressor of fused in Hh pathway activation and mouse spinal cord patterning. Dev Biol. 2012;362:141–153. doi: 10.1016/j.ydbio.2011.11.022. [DOI] [PubMed] [Google Scholar]
- Loots GG, Ovcharenko I. rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucleic acids research. 2004;32:W217–221. doi: 10.1093/nar/gkh383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development. 1998;125:2759–2770. doi: 10.1242/dev.125.15.2759. [DOI] [PubMed] [Google Scholar]
- Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18:8–23. doi: 10.1016/j.devcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murone M, Luoh SM, Stone D, Li W, Gurney A, Armanini M, Grey C, Rosenthal A, de Sauvage FJ. Gli regulation by the opposing activities of fused and suppressor of fused. Nature cell biology. 2000;2:310–312. doi: 10.1038/35010610. [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo: A Laboratory Manual. 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2003. [Google Scholar]
- Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic acids research. 2004;32:W280–286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Molecular and cellular biology. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, Joyner AL. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Schramek D, Schnidar H, Cronin SJ, Nehme NT, Zhang X, Knauf C, Cani PD, Aumayr K, Todoric J, Bayer M, Haschemi A, Puviindran V, Tar K, Orthofer M, Neely GG, Dietzl G, Manoukian A, Funovics M, Prager G, Wagner O, Ferrandon D, Aberger F, Hui CC, Esterbauer H, Penninger JM. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140:148–160. doi: 10.1016/j.cell.2009.12.027. [DOI] [PubMed] [Google Scholar]
- Rabinowitz AH, Vokes SA. Integration of the transcriptional networks regulating limb morphogenesis. Dev Biol. 2012;368:165–180. doi: 10.1016/j.ydbio.2012.05.035. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nature biotechnology. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Science signaling. 2012;5:re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KE, Chiang C. Hedgehog secretion and signal transduction in vertebrates. The Journal of biological chemistry. 2012;287:17905–17913. doi: 10.1074/jbc.R112.356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends in pharmacological sciences. 2009;30:303–312. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Sun T, Jayatilake D, Afink GB, Ataliotis P, Nister M, Richardson WD, Smith HK. A human YAC transgene rescues craniofacial and neural tube development in PDGFRalpha knockout mice and uncovers a role for PDGFRalpha in prenatal lung growth. Development. 2000;127:4519–4529. doi: 10.1242/dev.127.21.4519. [DOI] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191:415–428. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vokes SA, Ji H, McCuine S, Tenzen T, Giles S, Zhong S, Longabaugh WJ, Davidson EH, Wong WH, McMahon AP. Genomic characterization of Gli-activator targets in sonic hedgehog-mediated neural patterning. Development. 2007;134:1977–1989. doi: 10.1242/dev.001966. [DOI] [PubMed] [Google Scholar]
- Vokes SA, Ji H, Wong WH, McMahon AP. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 2008;22:2651–2663. doi: 10.1101/gad.1693008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Wang C, Pan Y, Wang B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development. 2010;137:2001–2009. doi: 10.1242/dev.052126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, McMahon AP, Allen BL. Shifting paradigms in Hedgehog signaling. Current opinion in cell biology. 2007;19:159–165. doi: 10.1016/j.ceb.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Weinmann AS, Farnham PJ. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods. 2002;26:37–47. doi: 10.1016/S1046-2023(02)00006-3. [DOI] [PubMed] [Google Scholar]
- Wilson CW, Chuang PT. Mechanism and evolution of cytosolic Hedgehog signal transduction. Development. 2010;137:2079–2094. doi: 10.1242/dev.045021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Aszterbaum M, Zhang X, Bonifas JM, Zachary C, Epstein E, McCormick F. A role of PDGFRalpha in basal cell carcinoma proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9255–9259. doi: 10.1073/pnas.151173398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, Jiang J. Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:21191–21196. doi: 10.1073/pnas.0912008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang L, Wang B, Ou CY, Chien CT, Jiang J. A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell. 2006;10:719–729. doi: 10.1016/j.devcel.2006.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.