

Abstract
The retinoblastoma (Rb) tumor suppressor gene is frequently inactivated in cancer, resulting in deregulated activation of E2F transcription factors, which promote S-phase entry, p53-dependent and p53-independent apoptosis. Transformed cells evade p53-dependent apoptosis initiated by Rb inactivation by TP53 mutation. However, the mechanisms by which cancer cells circumvent p53-independent apoptosis in this context are poorly understood. Because Rb inactivation primes cells for apoptosis by p53-independent induction of procaspases, we postulated that αB-crystallin, an inhibitor of procaspase-3 activation, would suppress caspase activation in cells with combined Rb and p53 inactivation. Notably, αB-crystallin is commonly expressed in ER/PR/HER2 “triple-negative” breast carcinomas characterized by frequent Rb loss and TP53 mutation. We report that αB-crystallin−/− knock out (KO) MEFs immortalized by dominant negative (DN) p53 are resistant to transformation by the adenovirus E1A oncoprotein, which inactivates Rb, while wild-type (WT) MEFs are readily transformed by DN p53 and E1A. αB-crystallin−/− KO MEFs stably expressing DN p53 and E1A were more sensitive to chemotherapy-induced caspase-3 activation and apoptosis than the corresponding WT MEFs, despite comparable induction of procaspases by E1A. Similarly, silencing Rb in WT and αB-crystallin−/− KO MEFs immortalized by DN p53 increased procaspase levels and sensitized αB-crystallin−/− KO MEFs to chemotherapy. Furthermore, silencing αB-crystallin in triple-negative breast cancer cells, which lack Rb and express mutant p53, enhanced chemotherapy sensitivity compared to non-silencing controls. Our results indicate that αB-crystallin inhibits caspase activation in cells primed for apoptosis by Rb inactivation and plays a novel oncogenic role in the context of combined Rb and p53 inactivation.
Keywords: αB-Crystallin, Apoptosis, Oncogenic transformation, Caspases, Rb
Introduction
The Rb tumor suppressor gene product functions as a transcriptional corepressor, which binds to members of the E2F family of transcription factors and inhibits E2F-dependent transactivation of genes promoting S-phase entry and apoptosis induction [1, 2]. Rb inactivation releases E2F from the Rb–E2F complex and promotes cell proliferation and apoptosis. Inactivation of Rb promotes p53-dependent apoptosis by transcriptional activation of Apaf-1, which is required for procaspase-9 activation in the intrinsic apoptotic pathway, and p53-independent apoptosis by E2F-dependent transcription of procaspases, which “primes” cells for apoptosis induction by lowering the threshold for caspase activation [3, 4]. The apoptosis-sensitizing effect of Rb inactivation in cancer cells is frequently counteracted by mutational inactivation of TP53 [1, 2]. Oncogenic transformation by tumor viruses requires inactivation of both Rb and p53 and is accomplished by the adenovirus E1A and E1B oncoproteins or papillomavirus E7 and E6 oncoproteins, respectively [5–8]. Moreover, Rb gene loss or diminished expression of Rb is frequently accompanied by TP53 mutation in human tumors, including clinically aggressive “triple-negative” breast carcinomas, which lack expression of the hormone receptors ER, PR, and HER2/ErbB2 [9–13]. These findings suggest that TP53 mutation cooperates with Rb loss to transform epithelial cells by suppressing p53-dependent apoptosis.
In contrast to p53-dependent apoptosis, the mechanisms by which transformed cells suppress p53-independent apoptosis initiated by Rb inactivation are poorly understood. Rb loss or inactivation by the adenovirus E1A oncoprotein, which dissociates the Rb-E2F complex, results in E2F-dependent transcriptional activation of pro-caspases [4, 5]. The accumulation of procaspases primes cells for cell death by sensitizing them to apoptotic stimuli. We postulated that the molecular chaperone αB-crystallin, which inhibits apoptosis by suppressing procaspase-3 activation [14–16], might inhibit caspase activation in cells primed for apoptosis by Rb inactivation. Caspase-3 is an effector caspase that executes cell death by cleaving key cellular proteins [17]. Notably, αB-crystallin is commonly expressed in triple-negative breast carcinomas, which frequently harbor Rb gene deletion/reduced Rb expression and TP53 mutations [9–13, 18].
Here, we report that αB-crystallin−/− knock out (KO) MEFs immortalized by dominant negative (DN) p53 are resistant to transformation by E1A, while wild-type (WT) MEFs are transformed by DN p53 and E1A. Deletion of αB-crystallin sensitizes MEFs stably expressing DN p53 and E1A to chemotherapy-induced caspase-3 activation and apoptosis. Similarly, silencing Rb in DN p53-immortalized WT and αB-crystallin−/− KO MEFs increased procaspase levels and enhanced the sensitivity of αB-crystallin−/− KO MEFs to chemotherapy. Moreover, siRNAs targeting αB-crystallin augmented chemotherapy-induced apoptosis in triple-negative breast cancer cells, which lack Rb and express a mutant p53 protein. Our findings indicate that αB-crystallin inhibits caspase activation in cells primed for apoptosis by Rb inactivation and counteracts p53-independent apoptosis in this context.
Methods
Isolation of MEFs
αB-Crystallin−/− KO mice containing a deletion of αB-crystallin and the adjacent HspB2 gene were described [19]. MEFs were obtained from 13.5-day αB-crystallin+/+ WT and αB-crystallin−/− KO embryos as described [20] and grown DMEM supplemented with 10 % FCS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, and 0.1 mM MEM nonessential amino acids (Life Technologies). Animal experiments were approved by the Animal Care and Use Committee.
Retroviral infection
Retroviruses were produced in Phoenix-Eco and Phoenix-Ampho cells and used to infect MEFs as described [21]. Phoenix cells (2.5 × 106) were transfected with 5 μg pBABE-Puro-DN p53 (GSE56), pLXSN-E1A, pLXSN-H-RasV12 or empty vector using Lipofectamine 2000 (Life Technologies) and grown in DMEM containing 10 % FCS, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Filtered retroviral supernatant was supplemented with 10 μg/ml polybrene (Millipore) and used to infect 3 × 105 MEFs, which were selected for growth in Puromycin (1 μg/ml, Sigma) and/or Geneticin (4 mg/ml, Life Technologies).
Immunoblotting
Immunoblotting was performed as described [22] with antibodies against αB-crystallin (StressGen), α-tubulin, β-actin (Sigma), p53, procaspase-3, procaspase-9, procaspase-6, p780 Rb, p15, cyclin B1, Cdk4, cyclin D1, cyclin D2, cyclin D3 (Cell Signaling), E1A (Neomarkers), and total Rb (Santa Cruz Biotechnology).
Cell growth assays
5 × 104 MEFs were plated and grown as described under “Isolation of wild-type and αB-crystallin−/− knock out MEFs”. Viable cells were scored by trypan blue (Life Technologies) exclusion. Population doubling after each passage was calculated using the formula: log[(cell number after 3 days of growth)/(5 × 104)]/log 2 as described [23, 24]. The total cell number at the end of each passage was determined using the formula: passage (n + 1) = passage (n) × 2(n+1)population doubling.
Senescence-associated β-galactosidase staining
Cells were fixed with 2 % formaldehyde/0.2 % glutaraldehyde and stained with X-Gal. Micrographs of β-galactosidase-stained MEFs were taken using an Axio-plan 2 microscope (Carl Zeiss).
BrdU incorporation
Subconfluent cells were incubated with 10 μM BrdU (Sigma) for 1 h. Cells were then fixed with methanol, incubated with 0.01 % Triton X-100, and subjected to immunofluorescence [24] using a BrdU antibody (Sigma). BrdU positive cells were detected with an AxioVision fluorescence microscope (Carl Zeiss).
Cell cycle analyses
Cell cycle profiles were determined by flow cytometry analysis of DNA content. Cells were fixed in 70 % ethanol for 2 h at 4 °C, incubated with 40 μg/ml propidium iodide and 100 μg/ml RNase A (Sigma) for 1 h at 37 °C, and analyzed using an EPICS Elite ESP Cell Sorter (Beckman Coulter).
Apoptosis assays
Cells were incubated with vehicle, doxorubicin or Taxol (Sigma) for 24 h. Apoptotic cells were scored using an Annexin V-FITC Apoptosis Detection kit (BD Biosciences) using in Epics XL-MCL Flow Cytometer (Beckman Coulter) and FlowJo software.
Matrigel invasion
Serum-starved cells (1 × 105) were seeded onto the upper surface of transwell chamber inserts coated with Matrigel (BD Biosciences) in serum-free DMEM. 10 % FCS was added to the lower compartment as a chemoattractant. After 24 h, the cells on the lower surface of the filter were stained with 1 % crystal violet (Sigma) and scored in 10 randomly selected fields.
Soft agar assay
Cells (1.5 × 105) were plated in 0.7 % agarose (BD Biosciences) on a cell-free 1.4 % agarose bed. Colonies >100 μm in size were scored after 2 weeks in 10 randomly selected fields.
Caspase-3 activity assay
Cells were treated with 250 nM Doxorubicin or 125 nM Taxol for 12 h, and caspase-3 activity was measured with a Caspase 3 Colorimetric Assay Kit (Sigma). Cell lysates were incubated with 20 mM Ac-DEVD-pNA, and the absorbance at 405 nm was measured.
Lentiviral Rb shRNA
Lentiviruses expressing pGIPZ-murine Rb shRNA (Open-BioSystems) were produced by co-transfecting 293 T cells with 5 μg ViraPower mix, and 2 μg non-silencing control vector or murine Rb shRNA using Lipofectamine 2000. Filtered viral supernatant was supplemented with 10 μg/ml polybrene and applied to MEFs.
αB-Crystallin siRNAs
Human MDA-MB-468 triple-negative breast cancer cells (ATCC) were grown in DMEM supplemented with 10 % FCS, 100 IU/ml penicillin, and 100 μg/ml streptomycin. MDA-MB-468 cells were transfected with a pool of siRNA duplexes specific to human αB-crystallin mRNA (100 nM, Dharmacon Research) using Lipofectamine 2000 reagent. Seventy-two hours after transfection, cells were lysed for immunoblotting or treated with chemotherapy.
Statistical analysis
We used the Microsoft Excel program to calculate the SD and statistical significance between samples using the Student’s t test.
Results
Functional inactivation of p53 immortalizes WT and αB-crystallin−/− KO MEFs
αB-Crystallin−/− KO MEFs did not express detectable levels of αB-crystallin protein as determined by immunoblotting (Fig. 1a). In contrast, WT MEFs and human mammary epithelial cells (HMECs) expressed αB-crystallin. To inactivate p53, we retrovirally transduced WT and αB-crystallin−/− KO MEFs with a truncated GSE56 p53 cDNA lacking the p53 transactivation domain, which acts as a dominant negative (DN) inhibitor of WT p53 by binding/stabilizing the WT protein and disrupting its transcriptional activity [25]. Consistent with prior reports [25], stable overexpression of DN p53 in WT and αB-crystallin−/−KO MEFs (abbreviated WT DN p53 MEFs and KO DN p53 MEFs, respectively) resulted in a striking accumulation of endogenous p53 protein (Fig. 1b), which is functionally inactive because these MEFs fail to undergo p53-dependent senescence as determined by senescence-associated β-galactosidase expression (Fig. 1c). These findings indicate that functional inactivation of p53 immortalizes WT and αB-crystallin−/− KO MEFs.
Fig. 1.
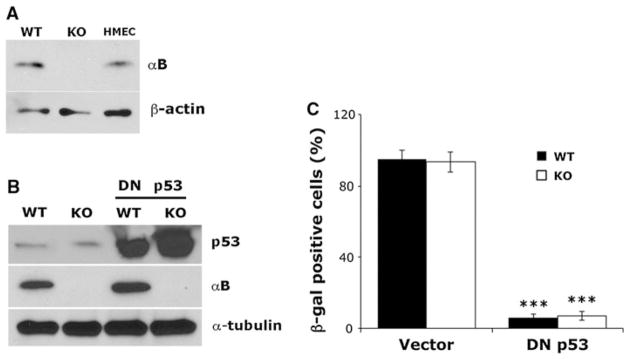
Functional inactivation of p53 immortalizes WT and αB-crystallin−/− KO MEFs. a Immunoblot analysis of WT and αB-crystallin−/− KO MEFs. Human mammary epithelial cells (HMEC) were used as a positive control. b WT and αB-crystallin−/− KO MEFs stably expressing a dominant negative (DN) p53 cDNA or vector control were analyzed by immunoblotting. c WT and αB-crystallin−/−KO MEFs stably expressing empty vector or DN were stained for senescence-associated β-galactosidase activity. The percentage of β-galactosidase positive cells was scored (mean ± SD, n = 3). ***P < 0.001 versus vector controls
αB-Crystallin inhibits cell growth and cell cycle progression in MEFs immortalized by p53 inactivation
To determine the growth rate of immortalized WT and αB-crystallin−/− KO DN p53 MEFs, we measured the doubling rate at each passage by plating 5 × 104 MEFs and counting the number of viable cells after 3 days of growth (Fig. 2a). KO DN p53 MEFs grew more rapidly than WT DN p53 MEFs under normal growth conditions and the differences in growth rates were enhanced with increasing passages. Importantly, WT MEFs immortalized by DN p53 did not express HspB2 (Supplementary Fig. 1), the adjacent gene co-deleted in the αB-crystallin−/− KO MEFs [19], indicating that the observed effects were specifically due to αB-crystallin deletion. These findings demonstrate that αB-crystallin inhibits the growth of MEFs immortalized by p53 inactivation.
Fig. 2.

αB-Crystallin inhibits cell growth and cell cycle progression in MEFs immortalized by p53 inactivation. a Cell number of immortalized WT and αB-crystallin −/− KO MEFs stably expressing DN p53 (abbreviated WT DN p53 and KO DN p53 MEFs, respectively) grown in subconfluent culture with the indicated passage (p) number (p1 at initial plating) reflecting 3-day growth periods (mean ± SD, n = 3). b Immortalized WT and αB-crystallin−/− KO DN p53 MEFs were cultured in media containing 10 or 0.1 % FCS, labeled with BrdU, and the percentage of BrdU positive MEFs was determined by immunohistochemistry (mean ± SD, n = 3). c Flow cytometry analysis of DNA content in immortalized WT and αB-crystallin−/− KO DN p53 MEFs cultured in media containing 0.1 % FCS. d Western blot analysis of paired primary WT and αB-crystallin−/− KO MEFs grown in media containing 10 % FCS for 72 h or immortalized WT and αB-crystallin−/− KO DN p53 MEFs grown in media containing 10 % FCS or 0.1 % FCS for 72 h. In a–c, *P < 0.05, **P < 0.01, or ***P < 0.001 versus control WT DN p53 MEFs
To determine the role of αB-crystallin in DNA replication in MEFs immortalized by p53 inactivation, WT and KO DN p53 MEFs were grown in media containing 10 or 0.1 % FCS for 72 h, and the percentage of BrdU positive cells was determined (Fig. 2b). Although the percentage of BrdU positive cells was similar when MEFs were grown in 10 % FCS, KO DN p53 MEFs exhibited a greater percentage of BrdU positive cells than WT DN p53 MEFs when these MEFs were grown in 0.1 % FCS. These findings indicate that αB-crystallin inhibits DNA replication during S-phase in MEFs immortalized by p53 inactivation.
To evaluate the role of αB-crystallin in cell cycle progression in MEFs immortalized by p53 inactivation, WT and KO DN p53 MEFs were grown in media containing 0.1 % FCS for 72 h and then analyzed for DNA content by flow cytometry. KO DN p53 MEFs had a greater percentage of cells in S and G2 phase and reduced percentage of cells in G1 compared to WT DN p53 MEFs when grown in 0.1 % FCS (Fig. 2c). Of note, the lack of significant differences in BrdU positive cells between WT and KO DN p53 MEFs grown in 10 % FCS may reflect that fact that these cells were not synchronized, while growth in 0.1 % FCS likely synchronized the cells. Collectively, these results indicate that αB-crystallin inhibits cell growth and S-phase entry in MEFs immortalized by p53 inactivation.
We next examined WT and KO DN p53 MEFs grown in media supplemented with 10 or 0.1 % FCS for expression of genes regulating S-phase entry and cell cycle progression by immunoblotting (Fig. 2d). KO DN p53 MEFs showed higher expression levels of Ser780 phosphorylated Rb protein (p780 Rb) compared to WT DN p53 MEFs, whereas total Rb protein levels were similar. In addition, cyclin B1 levels were increased in KO DN p53 MEFs compared to WT DN p53 MEFs grown in 0.1 % FCS (but not 10 % FCS). No other consistent pattern of differential expression of other cell cycle regulators was observed between WT and KO DN p53 MEFs. Because phosphorylation of Rb is an essential step in the initiation of transcription of many genes required for S-phase entry and cell cycle progression [26], our findings suggest that the growth-inhibiting effects of αB-crystallin in MEFs immortalized by p53 inactivation may be due to diminished phosphorylation of Rb, while reduced cyclin B1 levels may also contribute to the observed growth inhibition in 0.1 % FCS.
αB-Crystallin is required for oncogenic transformation by E1A and inhibits the apoptosis-sensitizing effects of E1A
Primary rodent cells are readily transformed by p53 inactivation in combination with a second oncogenic alteration such as ectopic expression of the E1A or H-RasV12 oncogene [6, 27]. To examine the role of αB-crystallin in oncogenic transformation, we retrovirally transduced immortalized WT and αB-crystallin−/− KO DN p53 MEFs with E1A or H-RasV12 cDNA (or empty vector). In striking contrast to the growth-inhibiting effects of αB-crystallin in MEFs immortalized by p53 inactivation, WT DN p53 MEFs stably expressing E1A (abbreviated WT E1A DN p53 MEFs) grew more rapidly than the corresponding KO E1A DN p53 MEFs (Fig. 3a). Intriguingly, WT E1A DN p53 MEFs formed colonies in soft agar and exhibited anchorage-independent growth, a hallmark of oncogenic transformation, while KO E1A DN p53 MEFs were largely unable to grow in soft agar (Fig. 3b). In contrast, deletion of αB-crystallin did not affect anchorage-independent growth of MEFs transformed by the combination of DN p53 and H-RasV12. Consistent with their transformed phenotype, WT E1A DN p53 MEFs invaded through Matrigel-occluded pores in a transwell invasion chamber, while KO E1A DN p53 MEFs were largely unable to invade in this assay (Fig. 3c). Notably, WT MEFs stably expressing DN p53 and E1A did not express HspB2 (Supplementary Fig. 1) and were therefore unaffected by co-deletion of this adjacent gene. These observations suggest that αB-crystallin plays an essential and previously unrecognized role in oncogenic transformation by E1A overexpression in the setting of p53 inactivation.
Fig. 3.

αB-Crystallin is required for oncogenic transformation by E1A and inhibits the apoptosis-sensitizing effects of E1A. a WT and αB-crystallin−/− KO MEFs stably expressing DN p53 and E1A (abbreviated WT E1A DN p53 and KO E1A DN p53) were grown in subconfluent culture with the indicated passage (p) number reflecting 3-day growth periods (mean ± SD, n = 3). b WT E1A DN p53 MEFs and αB-crystallin−/− KO E1A DN p53 MEFs were grown in soft agar for 2 weeks and the number of colonies was scored (mean ± SD, n = 3). WT and αB-crystallin−/− KO DN p53 MEFs stably expressing H-RasV12 oncogene were also analyzed. c WT and αB-crystallin−/− KO E1A DN p53 MEFs were examined for transwell invasion through Matrigel-occluded pores using 10 % FCS as a chemoattractant. Invading cells were visualized by crystal violet staining and scored (mean ± SD, n = 3). d Immortalized WT and αB-crystallin−/− KO DN p53 MEFs were acutely infected with a retrovirus expressing E1A and the percentage of Annexin V-positive MEFs was determined by flow cytometry (mean ± SD, n = 3). e and f WT E1A DN p53 MEFs and αB-crystallin−/− KO E1A DN p53 MEFs were treated with 250 nM Taxol (e) or 1 μM Doxorubicin (Dox) (f) for 24 h and the percentage of Annexin V-positive cells was determined (mean ± SD, n = 3). *P < 0.05, **P < 0.01, or ***P < 0.001 versus control WT E1A DN p53 MEFs
Given the well-established role of αB-crystallin in suppressing apoptosis by inhibiting caspase-3 activation [14–16], we examined whether αB-crystallin deletion sensitized MEFs to apoptosis. Acute infection of immortalized WT and KO DN p53 MEFs with E1A expressing retroviruses did not result in apoptosis induction compared to MEFs infected with control empty vector retroviruses (Fig. 3d), indicating that E1A overexpression is not suffi-cient to induce apoptosis. Because E1A has been shown to sensitize cancer cells to a variety of chemotherapy drugs, including paclitaxel (Taxol) and Doxorubicin [28, 29], we treated WT and KO E1A DN p53 MEFs with vehicle, Taxol (250 nM) or Doxorubicin (1 μM) for 24 h and measured the percentage of Annexin V-positive cells by flow cytometry (Fig. 3e, f). KO E1A DN p53 MEFs were more sensitive to apoptosis induction by Taxol and Doxorubicin than WT E1A DN p53 MEFs. These findings indicate that αB-crystallin inhibits the apoptosis-sensitizing effects of E1A. Moreover, these data suggest that αB-crystallin plays a key role in oncogenic transformation by E1A by counteracting its apoptosis-sensitizing effects.
αB-Crystallin inhibits E1A-induced caspase-3 activation in response to chemotherapy drugs
Rb inactivation by a variety of mechanisms, including E1A overexpression, leads to an accumulation of free E2F1 and transactivation of E2F-responsive apoptotic genes, including procaspases, resulting in enhanced sensitivity to apoptotic stimuli or “apoptotic priming” [4, 30]. We postulated that αB-crystallin would inhibit caspase activation in MEFs primed for apoptosis by E1A. Consistent with these reports, E1A overexpression in WT and KO DN p53 MEFs resulted in increased Ser780 phosphorylated Rb (inactivated) and elevated levels of procaspases-3, -6, and -9 (Fig. 4a). KO E1A DN p53 MEFs that were primed for apoptosis by elevated procaspase levels were more sensitive to Taxol- and Doxorubicin-induced caspase-3 activation than WT E1A DN p53 MEFs as determined by a colorimetric DEVDase activity assay (Fig. 4b, c). Collectively, these data indicate that αB-crystallin inhibits caspase activation in cells primed for apoptosis by E1A.
Fig. 4.

αB-Crystallin inhibits E1A-induced caspase-3 activation in response to chemotherapy drugs. a Immunoblot analysis of whole cell lysates from immortalized WT and αB-crystallin−/− KO DN p53 MEFs (Control) or immortalized WT and αB-crystallin−/− KO DN p53 MEFs stably expressing E1A. b and c WT E1A DN p53 MEFs and αB-crystallin−/− KO E1A DN p53 MEFs were treated with 125 nM Taxol (b) or 250 nM Doxorubicin (c) for 12 h, and caspase-3 activity was measured using a colorimetric assay and normalized to levels present in WT E1A DN p53 MEFs (mean ± SD, n = 3).
*P < 0.05 versus control WT E1A DN p53 MEFs
αB-Crystallin inhibits the apopotosis-sensitizing effects of Rb silencing
To determine whether the observed apoptosis-sensitizing effects of E1A were indeed due to inactivation of Rb, we stably silenced Rb in immortalized WT and KO DN p53 MEFs using a retroviral shRNA targeting murine Rb (Fig. 5a). Both WT and KO DN p53 MEFs stably expressing shRb had diminished levels of Rb protein by immunoblotting compared with WT and KO DN p53 MEFs stably expressing a non-silencing control construct. Moreover, Rb silencing augmented procaspase-3 levels. Treatment of WT and KO DN p53 shRb MEFs with Taxol or Doxorubicin resulted in an increased cell death in KO DN p53 shRb MEFs compared to their WT counterparts as determined by Annexin V labeling (Fig. 5b). These data indicate that Rb RNAi phenocopies the apoptosis-sensitizing effects of E1A in MEFs harboring p53 inactivation, thereby confirming that Rb is the relevant molecular target responsible for the observed effects.
Fig. 5.

αB-Crystallin inhibits the apoptosis-sensitizing effects of Rb loss. a Immunoblot analysis of immortalized WT and αB-crystallin−/− KO DN p53 MEFs retrovirally transduced with a shRNA targeting murine Rb (shRb) or a non-silencing shRNA (CTRL). b WT and αB-crystallin−/− KO DN p53 MEFs transduced with shRb or non-silencing control were treated with vehicle, 250 nm Taxol or 1 μM Doxorubicin for 24 h. The percentage of Annexin V-positive cells was scored by flow cytometry (mean ± SD, n = 3). c Immunoblot analysis of Rb in MDA-MB-231, GILM2, and MDA-MB-468 triple-negative breast cancer cells and MCF-10A breast epithelial cells. d MDA-MB-468 triple-negative breast cancer cells were transfected with non-silencing siRNA (CTRL) or siRNA pool targeting human αB-crystallin. αB-crystallin levels were determined by immunoblot-ting. e MDA-MB-468 triple-negative breast cancer cells transfected with αB-crystallin siRNA pool or non-silencing control were treated with vehicle, 500 nm Taxol, or 3.4 μM Doxorubicin for 24 h. The percentage of Annexin V-positive cells was scored by flow cytometry (mean ± SD, n = 3). In b and e *P < 0.05 versus control WT E1A DN p53 MEFs
To validate our observations in cancer cells, we chose human MDA-MB-468 triple-negative breast cancer cells, which lack Rb expression and have a TP53 mutation [31, 32]. Immunoblot analysis confirmed that MDA-MB-468 cells lack detectable Rb protein (Fig. 5c). To evaluate the role of αB-crystallin in apoptosis induction in this model, we transfected MDA-MB-468 cells with either non-silencing control siRNAs or a pool of αB-crystallin siRNAs. The αB-crystallin siRNA pool robustly reduced the levels of αB-crystallin in MDA-MB-468 cells compared to that observed in cells transfected with non-silencing control siRNAs (Fig. 5d). Treatment of αB-crystallin siRNA-transfected MDA-MB-468 cells with Taxol or Doxorubicin resulted in greater cell death than in non-silencing control siRNA transfected cells as determined by Annexin V labeling (Fig. 5e). These data indicate αB-crystallin inhibits chemotherapy-induced apoptosis in an established human triple-negative breast cancer cell line with TP53 mutation and Rb loss.
Discussion
The Rb tumor suppressor gene is frequently inactivated in cancer, resulting in dysregulated E2F activity and transcriptional induction of genes, which promote S-phase entry as well as p53-dependent and p53-independent apoptosis [1, 2]. Transformed cells frequently evade p53-dependent apoptosis initiated by Rb inactivation via mutational inactivation of TP53, thereby accounting for the cooperativity of Rb and p53 inactivation in oncogenic transformation, the coordinated inactivation of Rb and p53 by tumor viruses, and the frequent association of Rb gene loss/reduced Rb expression and TP53 mutation in human tumors [1, 2, 10–12]. However, the mechanisms by which tumors suppress p53-independent apoptosis in the setting of Rb inactivation are largely unknown. Rb inactivation promotes p53-independent cell death via E2F-depedendent transcription of procaspases [4]. The accumulation of procaspases primes cells for apoptosis by lowering the threshold for stress-induced caspase activation.
We have demonstrated that the antiapoptotic molecular chaperone αB-crystallin, previously reported to inhibit procaspase-3 activation [14–16], is a novel negative regulator of p53-independent apoptosis initiated by Rb inactivation. Several lines of evidence support this conclusion. First, αB-crystallin−/− KO MEFs stably expressing DN p53 and E1A were more sensitive to caspase-3 activation and apoptosis induction by chemotherapy than WT MEFs stably expressing DN p53 and E1A, even though the induction of procaspase levels by E1A was comparable in both models. Importantly, WT E1A DN p53 MEFs did not express HspB2, the adjacent gene co-deleted in the αB-crystallin−/− KO MEFs [19], indicating that the observed effects were specifically due to αB-crystallin deletion. Second, stably silencing Rb with a shRNA in WT and αB-crystallin−/− KO MEFs immortalized by DN p53 resulted in increased levels of several procaspases and enhanced sensitivity of αB-crystallin−/− KO MEFs to chemotherapy. Third, silencing αB-crystallin in human MDA-MB-468 triple-negative breast cancer cells, which lack Rb and express mutant p53, increased their sensitivity to chemotherapy compared to non-silencing controls. Importantly, this human triple-negative breast cancer cell line recapitulates several molecular abnormalities frequently observed in clinical triple-negative breast tumors: Rb loss, TP53 mutation, and αB-crystallin expression [9–13, 18]. Taken together, our results indicate that αB-crystallin inhibits caspase activation in cells primed for apoptosis by Rb inactivation and plays a key role in promoting cell survival in this context. Moreover, our findings point to αB-crystallin as a potential therapeutic target: αB-crystallin expression would be predicted to confer chemotherapy resistance, while inhibition of αB-crystallin activity or expression would be predicted to sensitize tumors to chemotherapy. Indeed, we have previously demonstrated that αB-crystallin expression in breast cancer was associated with resistance to neoadjuvant (pre-surgery) chemotherapy in women with invasive breast cancer [33].
We also showed that αB-crystallin plays an essential role in oncogenic transformation by the combination of E1A-mediated Rb inactivation and p53 inhibition. Specifically, αB-crystallin−/− KO MEFs immortalized by a DN p53 construct are resistant to transformation by E1A, while wild-type MEFs are readily transformed by the combination of DN p53 and E1A. Consistent with their transformed phenotype, WT E1A DN p53 MEFs grow more rapidly than αB-crystallin−/− KO E1A DN p53 MEFs and are much more invasive. The growth-promoting effects of αB-crystallin in transformed WT E1A DN p53 MEFs were particularly striking given our observation that αB-crystallin inhibited cell growth and S-phase entry in MEFs immortalized by DN p53. These latter results suggest that αB-crystallin promotes dysregulated growth in the setting of E1A-induced Rb inactivation by opposing its apoptosis-sensitizing effects, rather than by augmenting its mitogenic actions. Intriguingly, the oncogenic actions of αB-crystallin appear to be context-dependent: deletion of αB-crystallin did not confer resistance to oncogenic transformation by the combination of H-RasV12 and DN p53 in MEFs. Our results suggest that αB-crystallin promotes oncogenic transformation in the setting of combined Rb and p53 inactivation by inhibiting caspase activation in cells primed for apoptosis by the accumulation of procaspases.
Our findings have important implications for triple-negative breast cancer, which frequently harbor TP53 mutations, Rb gene loss and/or reduced Rb expression, and αB-crystallin expression [9–12, 18]. RB1 loss of hetero-zygosity was reported in 72 % of human breast cancers with a basal-like gene expression pattern [11]; these basal-like tumors are typically triple-negative by ER/PR/HER2 hormone receptor expression [13]. Moreover, enhanced p53 expression by immunohistochemistry, indicative of stabilization of the WT p53 protein by mutant p53 protein, is frequently observed in basal-like tumors with reduced Rb expression [12]. TP53 mutations also cooperate with Rb deletion to promote triple-negative mammary tumors in mice [34]. We recently reported that αB-crystallin is expressed in about half of basal-like breast tumors and promotes an invasive, apoptosis-resistant phenotype in breast epithelial cells [18]. Others have confirmed the association of αB-crystallin expression with poor outcomes and the basal-like/triple-negative subtype of breast cancer and described a similar oncogenic role for αB-crystallin in the pathogenesis of glioblastoma multiforme [16, 35, 36]. It is tempting to speculate that the observed role of αB-crystallin in suppressing caspase activation in setting of Rb inactivation and p53 inhibition might account for the frequent occurrence of these three molecular events (Rb loss, TP53 mutation, and αB-crystallin expression) in clinically aggressive triple-negative breast cancers, although it remains to be determined how frequently all three of these events occur in individual basal-like/triple-negative tumors. Nevertheless, our results suggest that αB-crystallin may represent a unique molecular target to enhance the vulnerability of these tumors to cytotoxic agents, a hypothesis we are actively exploring in murine models.
Supplementary Material
Acknowledgments
We are indebted to Eric Wawrousek for providing the αB-crystallin−/− knock out mice, Scott Lowe for providing H-RasV12 and E1A cDNAs, and Nissim Hay for providing the DN p53 cDNA. We also thank Paul Lambert and Ming Zhang for their critical reading of the manuscript. This research was supported by a grant from the Breast Cancer Research Foundation and Grants KG091372 and KG101575 from Susan G. Komen for the Cure.
Abbreviations
- Rb
Retinoblastoma
- KO
Knock out
- MEFs
Murine embryonic fibroblasts
- DN
Dominant negative
- WT
Wild-type
- DMEM
Dulbecco’s modified Eagle’s medium
- PBS
Phosphate-buffered saline
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Electronic supplementary material The online version of this article (doi:10.1007/s10549-013-2465-6) contains supplementary material, which is available to authorized users.
References
- 1.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, Wang S, Herschkowitz JI, Egan SE, Perou CM, Zacksenhaus E. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle. 2011;10:1563–1570. doi: 10.4161/cc.10.10.15703. [DOI] [PubMed] [Google Scholar]
- 3.Moroni MC, Hickman ES, Lazzerini Denchi E, Caprara G, Colli E, Cecconi F, Muller H, Helin K. Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol. 2001;3:552–558. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- 4.Nahle Z, Polakoff J, Davuluri RV, McCurrach ME, Jacobson MD, Narita M, Zhang MQ, Lazebnik Y, Bar-Sagi D, Lowe SW. Direct coupling of the cell cycle and cell death machinery by E2F. Nat Cell Biol. 2002;4:859–864. doi: 10.1038/ncb868. [DOI] [PubMed] [Google Scholar]
- 5.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 6.Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- 7.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retino-blastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 8.Scheffner M, Munger K, Byrne JC, Howley PM. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci USA. 1991;88:5523–5527. doi: 10.1073/pnas.88.13.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gauthier ML, Berman HK, Miller C, Kozakeiwicz K, Chew K, Moore D, Rabban J, Chen YY, Kerlikowske K, Tlsty TD. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell. 2007;12:479–491. doi: 10.1016/j.ccr.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008;10:R75. doi: 10.1186/bcr2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subhawong AP, Subhawong T, Nassar H, Kouprina N, Begum S, Vang R, Westra WH, Argani P. Most basal-like breast carcinomas demonstrate the same Rb−/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. Am J Surg Pathol. 2009;33:163–175. doi: 10.1097/PAS.0b013e31817f9790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toft DJ, Cryns VL. Minireview: basal-like breast cancer: from molecular profiles to targeted therapies. Mol Endocrinol. 2011;25:199–211. doi: 10.1210/me.2010-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamradt MC, Chen F, Cryns VL. The small heat shock protein αB-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J Biol Chem. 2001;276:16059–16063. doi: 10.1074/jbc.C100107200. [DOI] [PubMed] [Google Scholar]
- 15.Kamradt MC, Lu M, Werner ME, Kwan T, Chen F, Strohecker A, Oshita S, Wilkinson JC, Yu C, Oliver PG, Duckett CS, Buchsbaum DJ, LoBuglio AF, Jordan VC, Cryns VL. The small heat shock protein αB-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J Biol Chem. 2005;280:11059–11066. doi: 10.1074/jbc.M413382200. [DOI] [PubMed] [Google Scholar]
- 16.Stegh AH, Kesari S, Mahoney JE, Jenq HT, Forloney KL, Protopopov A, Louis DN, Chin L, DePinho RA. Bcl2L12-mediated inhibition of effector caspase-3 and caspase-7 via distinct mechanisms in glioblastoma. Proc Natl Acad Sci USA. 2008;105:10703–10708. doi: 10.1073/pnas.0712034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 18.Moyano JV, Evans JR, Chen F, Lu M, Werner ME, Yehiely F, Diaz LK, Turbin D, Karaca G, Wiley E, Nielsen TO, Perou CM, Cryns VL. αB-Crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J Clin Invest. 2006;116:261–270. doi: 10.1172/JCI25888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brady JP, Garland DL, Green DE, Tamm ER, Giblin FJ, Wawrousek EF. αB-Crystallin in lens development and muscle integrity: a gene knockout approach. Invest Ophthalmol Vis Sci. 2001;42:2924–2934. [PubMed] [Google Scholar]
- 20.Petrovic V, Costa RH, Lau LF, Raychaudhuri P, Tyner AL. FoxM1 regulates growth factor-induced expression of kinase-interacting stathmin (KIS) to promote cell cycle progression. J Biol Chem. 2008;283:453–460. doi: 10.1074/jbc.M705792200. [DOI] [PubMed] [Google Scholar]
- 21.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrovic V, Costa RH, Lau LF, Raychaudhuri P, Tyner AL. Negative regulation of the oncogenic transcription factor FoxM1 by thiazolidinediones and mithramycin. Cancer Biol Ther. 2010;9:1008–1016. doi: 10.4161/cbt.9.12.11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben Azouna N, Jenhani F, Regaya Z, Berraeis L, Ben Othman T, Ducrocq E, Domenech J. Phenotypical and functional characteristics of mesenchymal stem cells from bone marrow: comparison of culture using different media supplemented with human platelet lysate or fetal bovine serum. Stem Cell Res Ther. 2012;3:6. doi: 10.1186/scrt97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ossovskaya VS, Mazo IA, Chernov MV, Chernova OB, Strezoska Z, Kondratov R, Stark GR, Chumakov PM, Gudkov AV. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc Natl Acad Sci USA. 1996;93:10309–10314. doi: 10.1073/pnas.93.19.10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee WH, Chen PL, Riley DJ. Regulatory networks of the retinoblastoma protein. Ann N Y Acad Sci. 1995;752:432–445. doi: 10.1111/j.1749-6632.1995.tb17453.x. [DOI] [PubMed] [Google Scholar]
- 27.Kortum RL, Johnson HJ, Costanzo DL, Volle DJ, Razidlo GL, Fusello AM, Shaw AS, Lewis RE. The molecular scaffold kinase suppressor of Ras 1 is a modifier of RasV12-induced and replicative senescence. Mol Cell Biol. 2006;26:2202–2214. doi: 10.1128/MCB.26.6.2202-2214.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ueno NT, Yu D, Hung MC. Chemosensitization of HER-2/neu-overexpressing human breast cancer cells to paclitaxel (Taxol) by adenovirus type 5 E1A. Oncogene. 1997;15:953–960. doi: 10.1038/sj.onc.1201250. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Z, Jia SF, Hung MC, Kleinerman ES. E1A sensitizes HER2/neuoverexpressing Ewing’s sarcoma cells to topoisomerase II-targeting anticancer drugs. Cancer Res. 2001;61:3394–3398. [PubMed] [Google Scholar]
- 30.Almasan A, Yin Y, Kelly RE, Lee EY, Bradley A, Li W, Bertino JR, Wahl GM. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc Natl Acad Sci USA. 1995;92:5436–5440. doi: 10.1073/pnas.92.12.5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blick T, Widodo E, Hugo H, Waltham M, Lenburg ME, Neve RM, Thompson EW. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis. 2008;25:629–642. doi: 10.1007/s10585-008-9170-6. [DOI] [PubMed] [Google Scholar]
- 32.Carlson CA, Ethier SP. Lack of RB protein correlates with increased sensitivity to UV-radiation-induced apoptosis in human breast cancer cells. Radiat Res. 2000;154:590–599. doi: 10.1667/0033-7587(2000)154[0590:lorpcw]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 33.Ivanov O, Chen F, Wiley EL, Keswani A, Diaz LK, Memmel HC, Rademaker A, Gradishar WJ, Morrow M, Khan SA, Cryns VL. αB-Crystallin is a novel predictor of resistance to neo-adjuvant chemotherapy in breast cancer. Breast Cancer Res Treat. 2008;111:411–417. doi: 10.1007/s10549-007-9796-0. [DOI] [PubMed] [Google Scholar]
- 34.Jiang Z, Deng T, Jones R, Li H, Herschkowitz JI, Liu JC, We-igman VJ, Tsao MS, Lane TF, Perou CM, Zacksenhaus E. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest. 2010;120:3296–3309. doi: 10.1172/JCI41490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsang JY, Lai MW, Wong KH, Chan SK, Lam CC, Tsang AK, Yu AM, Tan PH, Tse GM. αB-Crystallin is a useful marker for triple negative and basal breast cancers. Histopathology. 2012;61:378–386. doi: 10.1111/j.1365-2559.2012.04234.x. [DOI] [PubMed] [Google Scholar]
- 36.Kim HS, Lee Y, Lim YA, Kang HJ, Kim LS. αB-Crystallin is a novel oncoprotein associated with poor prognosis in breast cancer. J Breast Cancer. 2011;14:14–19. doi: 10.4048/jbc.2011.14.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.