

Abstract
A water-soluble anthracycline antibiotic drug (daunorubicin, DNR) was loaded into oxidized porous silicon (pSiO2) microparticles and then encapsulated with a layer of polymer (poly lactide-co-glycolide, PLGA) to investigate their synergistic effects in control of DNR release. Similarly fabricated PLGA-DNR microspheres without pSiO2, and pSiO2 microparticles without PLGA were used as control particles. The composite microparticles synthesized by a solid-in-oil-in-water (S/O/W) emulsion method have mean diameters of 52.33±16.37 μm for PLGA-pSiO2_21/40-DNR and the mean diameter of 49.31±8.87 μm for PLGA-pSiO2_6/20-DNR. The mean size, 26.00±8 μm, of PLGA-DNR was significantly smaller, compared with the other two (p<0.0001). Optical microscopy revealed that PLGA-pSiO2-DNR microsphere contained multiple pSiO2 particles. In vitro release experiments determined that control PLGA-DNR microspheres completely released DNR within 38 days and control pSiO2-DNR microparticles (with no PLGA coating) released DNR within 14 days, while the PLGA-pSiO2-DNR microspheres released DNR for 74 days. Temporal release profiles of DNR from PLGA-pSiO2 composite particles indicated that both PLGA and pSiO2 contribute to the sustained release of the payload. The PLGA-pSiO2 composite displayed a more constant rate of DNR release than the pSiO2 control formulation, and it displayed a significantly slower release of DNR than either the PLGA or pSiO2 formulations. We conclude that this system may be useful in managing unwanted ocular proliferation when formulated with anti-proliferation compounds such as DNR.
Keywords: Porous silicon oxide, Poly (dl-lactide-co-glycolide), Daunorubicin, Ocular drug delivery
Introduction
Proliferative vitreoretinopathy (PVR) is the most frequent cause of failure for retinal detachment surgery [1]. Previous studies have shown that daunorubicin (DNR) is effective in inhibiting PVR formation [2], and it also has been shown to be effective for the treatment of experimental PVR [3–5]. However, DNR has a short half-life in the vitreous and also a narrow therapeutic concentration range, which would require too frequent injections to allow intravitreal DNR to be a practical therapeutic [6, 7]. A drug appropriate for the control of PVR needs to inhibit cell proliferation effectively and maintain a therapeutic level in the targeting tissue for a minimum 2 months, which is the median time for PVR development [8]. Porous silicon (pSi) is a nanostructured material with a surface area of 400–800 m2 /g that is commonly produced from bulk single crystal silicon by electrochemical anodization in hydrofluoric acid [9]. An oxidized form of pSi that retains the porous nanostructure and displays a lower reactivity with redox-active drugs [10] can be prepared by thermal oxidation of pSi. From a biological and biomedical perspective, pSi and pSiO2 are attractive materials as they are both biocompatible and biodegradable, meaning that they are able to undergo complete degradation in the body to produce silicic acid (Si(OH)4) that is a nontoxic soluble form of silicon [11]. It has been established that Si(OH)4 is readily cleared from intraocular fluid [12]. Furthermore, surface chemistries such as silanol condensation and hydrosilylation are available for this material that allows adjustment of degradation rate in biological systems [13–15]. It has been shown that therapeutic payloads can be loaded into the pores of pSi or pSiO2 by adsorption or surface grafting [10, 14, 16, 17]. These properties, in addition to the very large internal surface area [18] renders pSi a versatile drug delivery platform [19]. In previous works, we reported the possibility of using pSi and pSiO2 microparticles as an intraocular drug delivery system. Whereas pSi was found to react with and degrade redox-active DNR, pSiO2 formulations were inert with respect to chemical reaction with the drug [20]. In a study with the pSiO2 formulation, DNR was loaded into pSiO2 microparticles using two methods, covalent attachment and physical adsorption [10]. The study revealed an obvious difference in the release profiles for the two drug-loading strategies. Covalently loaded particles released less than 1% of the loaded DNR within 8 days in excised rabbit vitreous while particles loaded by physical adsorption released more than 75% of loaded DNR within the same time period. A subsequent in vivo study demonstrated localized retinal toxicity from adsorption loaded particles due to rapid release of drug [10]. Particles prepared by covalent loading of DNR did not show retinal toxicity during a 3-month observation period, but initial data indicated very low free drug levels in the rabbit vitreous. Poly(DL-lactide-co-glycolide) (PLGA), a food and drug administration (FDA)-approved biodegradable polymer, has been widely investigated for drug delivery applications due to its customizable degradation rates, its favorable mechanical properties, and its biodegradability [21–25]. We reasoned that combining the porous silicon drug delivery platform with PLGA might increase the effectiveness of the porous silicon drug delivery system for ocular application of daunorubicin. Indeed, Jie Liu et al [26] fabricated a series of DNR-loaded PLGA nanoparticles using a modified double-emulsion solvent evaporation/diffusion method and achieved the sustained release of DNR for > 2 weeks. D Fan et al [27] investigated PLGA/porous silicon composite microspheres, synthesized by a solid-in-water (S/O/W) emulsion method for a 30-day delivery of bovine serum albumin (BSA) for orthopedic tissue engineering applications. These prior studies demonstrated that both PLGA and pSi contribute to the control of release of a payload. We hypothesized that a PLGA coating would reduce the initial burst release of DNR from pSiO2, and extend the therapeutic duration. In the current study, we loaded DNR into oxidized porous silicon microparticles by infiltration and then coated the drug-loaded pSiO2 particles with PLGA. We aimed to investigate the release properties of pSiO2 and PLGA composites with the goal of identifying an effective means for intravitreal delivery of DNR.
Materials and Methods
1. Synthesis of Porous Silicon Oxide Microparticles
Porous silicon oxide (pSiO2) microparticles were prepared by electrochemical etch of highly doped, (100)-oriented p-type silicon wafers (boron-doped, 0.99 mΩ·cm resistivity; Siltronix Inc., Archamps, France) in a 3:1 (v:v) solution of 48% aqueous hydrofluoric acid to ethanol (Thermo Fisher Scientific, Pittsburg, PA) as described previously [10]. A silicon wafer with an exposed area of 8.04 cm2 was contacted on the backside with a strip of aluminum foil and mounted in a Teflon etching cell fitted with a platinum counter-electrode. The wafer was etched at a constant current density of 90.2 mA/cm2 for 200 seconds. The resulting porous layer was then lifted off by electropolishing in a 1:29 solution of 48% aqueous hydrofluoric acid to ethanol (Thermo Fisher Scientific) for 120 seconds at a current density of 6.2 mA/cm2. The etching and electropolishing procedure was repeated 20 times per wafer. The resulting porous layers were ultrasonicated (model FS5 dual-action ultrasonic cleaner; Thermo Fisher Scientific) in ethanol for 30 minutes to form the microparticles. These pSi microparticles were converted to pSiO2 by oxidation in a furnace chamber (Thermo Fisher Scientific). The pSi particles were placed in a ceramic boat and the temperature was ramped from room temperature to 800°C at a rate of 10°C /min and then maintained at 800°C for 2 hours. The furnace was allowed to cool to room temperature for an additional 3 hours prior to removal of the samples. Thereafter, the particles were dispersed into ethanol and filtered through nylon filtration membranes with the sizes of 5 μm, 20 μm and 40 μm, respectively. Finally, two samples with the size population of 6–20 μm and 21–40 μm (marked as pSiO2_6/20 and pSiO2_21/40, respectively) were collected and dried in a vacuum oven overnight. The particles showed uniform nanostructure with a pore size of 15–20 nm as measured from scanning electron microscopic (SEM) images.
2. Drug loading using physical adsorption route
10 mg pSiO2 microparticles were added into a 1.5mL eppendorf tube containing 0.5 mL of 10 mg/mL daunorubicin hydrochloride (Tocris Biosciences, Minneapolis, MN) in DPBS. The particles were then vortexed for 2 hours at room temperature and rinsed briefly with water three times. The resulting particles were dried and stored in a sealed vial at 4°C.
The drug loading of pSiO2 was determined by thermogravimetric analysis (TGA). The DNR-loaded samples (~3 mg) were placed in a 90 μL alumina sample cup. Samples were heated at a constant rate of 10 °C/min up to 900°C in a nitrogen atmosphere with a purge rate of 10 mL/min using a Q600 simultaneous TGA/DSC apparatus (TA Instruments, Newcastle, DL). As determined by TGA, the mass loading of DNR for pSiO2_6/20 and pSiO2_21/40 was 32.5μg/mg and 41 μg/mg.
3. Preparation of DNR-loaded pSiO2 Particles coated with PLGA (PLGA-pSiO2-DNR) and DNR-loaded PLGA microspheres (PLGA-DNR)
pSiO2 particles coated with PLGA were prepared by a modified S/O/W emulsion method [28] (see supplemental figure 1). Briefly, 10mg DNR-loaded pSiO2 was mixed with 1 mL 10% PLGA (75:25) (Sigma Chemicals Co. St. Louis, MO) solution (dissolved in dichloromethane) with vortex for 20 min. The mixture was added drop by drop into 50 mL 2.0% poly (vinyl alcohol) (PVA, Mw: 89,000–98,000) aqueous solution and stirred with a homogenizer (T18 Ultra Turrax, IKA) at 6,000 rpm to form an emulsion (oil in water, O/W). The emulsion was then transferred into 1% PVA aqueous solution (50 mL) and stirred at 1200 rpm for 2h to evaporate the organic solvent. The suspension was centrifuged at 10,000 rpm (Allegra® 25R Centrifuge, Beckman Coulter, Inc.) for 10 min and the supernatant was collected for determination of drug loss. The PLGA-pSiO2-DNR microspheres were washed with distilled water for 3 times, 1 min each time. Finally, the product was lyophilized and stored at 4 °C. PLGA-DNR particles were prepared using a similar procedure as in the fabrication of PLGA-pSiO2-DNR microsphere fabrication, except that DNR instead of DNR-loaded pSiO2 particles was mixed with PLGA/DCM.
The drug loading of PLGA-pSiO2-DNR microparticles or PLGA-DNR microspheres was calculated as following:
The drug total was the total amount of drug used to fabricate the microsphere composite; and the drug loss was the drug detected from the aqueous phase after remove of the particulate.
The drug loading of PLGA-pSiO2_6/20-DNR, PLGA-pSiO2_21/40-DNR, and PLGA-DNR was 1.34μg/mg, 1.52 μg/mg and 2.25μg/mg, respectively. Drug-loading efficacy = actual drug-loading / total drug used × 100%. Different formulation of particulate had different drug loading efficiency (Table 1).
Table 1.
Drug loading efficiency for different formulations
| Formulations* | Drug-loading efficacy |
|---|---|
| pSiO2_6/20-DNR | 6.5% |
| pSiO2_21/40-DNR | 8.2% |
| PLGA-DNR | 2.74% |
| PLGA-pSiO2_6/20-DNR | 3.08% |
| PLGA-pSiO2_21/40-DNR | 4.64% |
4. Morphological characteristics of pSiO2 particles, PLGA-pSiO2-DNR microspheres, and PLGA-DNR microspheres
The shape and surface morphology of the microparticles were characterized by scanning electron microscopy (Phillips XL30 ESEM, Philips Corp, Netherlands). Microparticles were mounted on an aluminum stub using adhesive carbon tape and sputter-coated with a mixture of gold and palladium (60:40) in an argon atmosphere under low pressure using a Dynavac Mini Coater. In addition, to observe the cross-section of the different microsphere formulation, the microspheres (1 mg for each) were embedded in Neg-50 Frozen Section Medium (Thermo Scientific, Waltham, MA), sectioned with a thickness of 6μm using a cryostat (Cryostat HM550, Thermo Scientific, Waltham, MA). The cross-sectional images of the microspheres were analyzed by light microscopy (EVOS FL Auto, Life Technologies, Carlsbad, CA).
5. In vitro release profiles and determination of DNR concentration
The rate of the DNR release from the microparticles was evaluated in Hank’s Balanced Salt Solution (HBSS). Briefly, 3 mg of dried DNR-loaded pSiO2 particles, PLGA-pSiO2-DNR microspheres and PLGA-DNR microspheres were suspended in 1.2 ml of HBSS, respectively. The samples were placed in an incubator at 37°C and continuously stirred with a mini lab roller. At various pre-determined time points, 1 mL of the supernatant was withdrawn after centrifugation at 10,000 rpm for 5 min, and replaced with an equal volume of fresh release medium.
The amount of DNR released was measured by spectrofluorometer (SpectraMax M5, Molecular Devices Corp. USA.). The fluorescence intensity of DNR in the solution was measured at a 590 nm emission wavelength and a 470 nm excitation wavelength. The concentration of DNR was calculated from a standard curve, prepared by measuring the fluorescence intensity of known concentrations of free DNR. The linear range was determined to be from 50 ng/mL to 1500 ng/mL. The samples with the drug concentration higher than 1500ng/mL were diluted before detection. Similarly, some samples with the drug concentration lower than 50ng/mL was concentrated by evaporation before detection.
6. Cytotoxicity study with the in vitro DNR release samples
A human endothelial-like immortalized cell line EA-HY926, derived from the fusion of human umbilical vein endothelial cells (HUVEC) with the lung carcinoma cell line A549 (CRL-2922; ATCC) was used to assess the cytotoxicity of DNR using a water-soluble tetrazolium salt (WST-1) assay (Roche Diagnostics Corp., Indianapolis, IN))[29]. Briefly, EA-HY926 cells were seeded in a 96-well plate at a cellular density of 5000 cells per well in DMEM media (ATCC, Manassas, VA) with 10% FBS (ATCC, Manassas, VA) and antibiotics (ATCC, Manassas, VA). After 1 day of incubation at 37°C in 5% CO2 to allow the cells to attach, the cell culture medium was replaced with a mixture of 75% (by volume) culture medium and 25% (by volume) of the supernatant from in vitro release studies of PLGA-pSiO2_21/40-DNR microspheres. 100uL of test sample was added into each well with EA-HY926 cells and the plate was allowed to incubate for 5 days at 37 °C 5% CO2. On day 5 of incubation, 10uL of the WST-1 compound (premixed by the manufacturer) was added into each well and the plate was placed back in the incubator for 2 hours. The plate was read at an absorbance of 440 nm every 30 minutes for 2 hours, with the plate being placed back in the incubator between readings. The optical density readings (OD) were calculated as % cell viability of the controls, which had culture medium without DNR. The positive controls were created using fresh prepared commercial DNR with the equivalent concentrations.
7. Statistical Analysis
For the size comparison between the different microspheres, t-test was used. For in vitro drug release, the data were normalized using the initial drug loading dose. The drug release from the different formulations was compared using Kruskal-Wallis one-way analysis of variance. For the cytotoxicity study, OD values were normalized by the OD value of MediumHBSSCells (control) and expressed as the percent of the control. The normalized OD values were compared across the samples using all pairs Tukey-Kramer HSD. All analyses were performed using JMP statistical software (version 11; SAS Institute Inc, Cary, NC) and p-value smaller than 0.05 was considered to be significant.
Results
1. Characterization of pSiO2 and pSiO2/PLGA composite particles
Figure 1 shows the SEM image of pSiO2_21/40 (Figure 1A) and pSiO2_6/20 (Figure 1B). These microparticles were fabricated using ultrasonication of fresh prepared pSi film. Both the film and particles, displayed a similar nanostructure with pore size of 15–20 nm (Figure 1C, 18.67±4.23 nm) before and after ultrasonication (see supplemental figure 2), indicating that the nanostructure was largely preserved during ultrasonication. The overall aspect and the morphology of the PLGA-pSiO2-DNR and PLGA-DNR microspheres were characterized by optical and scanning electron microscopy. Figure 2 shows the optical microscope image of PLGA microspheres without any drug (2A), PLGA-DNR microspheres (2B) and the composite PLGA-pSiO2_21/40-DNR microspheres (2C). These images indicate that the drug or the drug-loaded pSiO2 particles were fully encapsulated in the transparent PLGA spheres. Figures 2D–2F show SEM images of the corresponding microspheres in Figures 2A–2C. PLGA-pSiO2_6/20-DNR displayed a similar size distribution to that of PLGA-pSiO2_21/40-DNR (Figure 3), with the mean diameter of 52.33±16.37 μm for PLGA-pSiO2_21/40-DNR and the mean diameter of 49.31±8.87 μm for PLGA-pSiO2_6/20-DNR. However, the mean size of PLGA-DNR was significantly smaller (26.00±8 vs. 49.31±8.87 μm, p<0.0001; and 26.00±8 vs. 52.33±16.37 μm, p<0.0001; Figure 4) though the PLGA microsphere fabrication procedure was the same.
Figure 1.

Scanning electron microscope (SEM) images of the two different pSiO2 microparticle sizes used in this study: (A) pSiO2_21/40 and (B) pSiO2_6/20. (C) Plan-view image of the pore structure on the (100) surface of the particles.
Figure 2.

Physical characterization of PLGA-DNR and PLGA-pSiO2. Optical microscope images of: (A) PLGA microspheres without DNR, (B) PLGA-DNR microspheres, (C) PLGA-pSiO2_21/40-DNR microspheres. SEM images of: (D) PLGA microspheres without DNR, (E) PLGA-DNR microspheres, (F) PLGA-pSiO2_21/40-DNR microspheres.
Figure 3.

SEM images of PLGA-pSi_6/20-DNR (left panel) and PLGA-pSi_21/40-DNR (right panel). PLGA-pSiO2_6/20-DNR displayed a similar size distribution to that of PLGA-pSiO2_21/40-DNR. For these two types of composite, The fabricating condition was the same for those two types of PLGA-p SiO2 composite microspheres: 10mg DNR-loaded pSiO2 (pSiO2_21/40 or pSiO2_6/20) was mixed with 1 mL 10% PLGA (75:25). The ratio of polymer versus pSiO2 was both 1: 10 (w/w).
Figure 4.
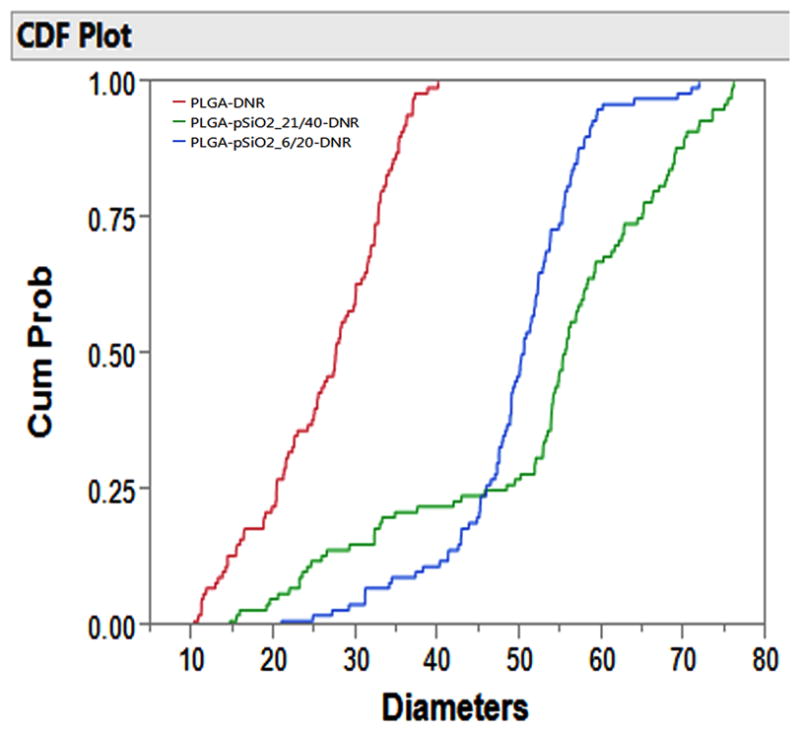
Cumulative distribution plot comparing the size distribution of PLGA-DNR, PLGA-pSiO2_6/20-DNR, and PLGA-pSiO2_21/40-DNR microparticles. Cumm Prob=Cummulative Probability.
The majority of the microspheres had sizes between 10 and 80 μm, as shown in Figures 2 and 3. A close-up examination of the microspheres revealed many micron-sized pores on the surface for all the samples, ranging from 0.5 to 2 μm (Figure 5). There was no evidence of DNR aggregates on the surface of the microspheres from the SEM analysis. The cross-sectional images of these particles revealed that multiple porous silicon particles were present in a PLGA microsphere though in general pSiO2 particles were larger in PLGA-pSiO2_21/40-DNR (Figure 6D) than in PLGA-pSiO2_6/20-DNR composite microsphere (Figure 6C). In PLGA-DNR (Figure 6B) and PLGA microspheres (Figure 6A), daunorubicin aggregates were seen in the former and the latter were much smaller but uniform in the cross-sectional view.
Figure 5.

SEM image of PLGA-pSiO2_21/40-DNR microspheres, showing the presence of micron-scale voids at the surface of the microspheres.
Figure 6.

Cross-sectional views of microspheres of (A) PLGA microspheres without DNR, (B) PLGA-DNR microspheres, (C) PLGA-pSiO2_6/20-DNR microspheres, and (D) PLGA-pSiO2_21/40-DNR microspheres.
2. In vitro drug release
The release profiles of DNR from the different microparticle formulations are given in Figures 7. DNR release from pSiO2 particles of either size showed a large burst release, with the drug concentration in the eluent solution dropping from 20 μg/mL at the initial sampling to < 100ng/mL on day 14, with over 90% of the total drug payload leached out within the first 2 days. The size of the pSiO2 particles seems not to be a significant factor. In contrast, DNR release from PLGA-DNR as well as from the pSiO2-PLGA composite microspheres (PLGA-pSiO2_6/20-DNR and PLGA-pSiO2_21/40-DNR) was much slower. All three formulations showed a much smaller burst release within the first two days, with approximately 50% of the payload released (Figure 7). After the first 2 days, the PLGA-DNR formulation maintained a steady state of DNR release for up to three weeks, which then tapered off as the material approached 100% release (Figure 7). By contrast, DNR release from the pSiO2-PLGA composite microspheres (PLGA-pSiO2_6/20-DNR and PLGA-pSiO2_21/40-DNR) was gradual and sustained for a period of >60 days. Around 90% of the payload leached out over the course of 70 days. The DNR release characteristics were similar for these two formulations of composite microspheres, with DNR release from the PLGA-pSiO2_6/20-DNR formulation a bit faster in the first 5 days (Figure 7).
Figure 7.

Cumulative amount of DNR released from the indicated microsphere formulations as a function of time.
3. Cytotoxicity assay in vitro
To confirm the bioactivity of DNR released from the PLGA-pSiO2 composite microspheres, PLGA-pSiO2_21/40-DNR eluents containing the released DNR were added to cultured cells. An equivalent concentration of commercial DNR was used as a positive control and cell culture medium as well as medium:HBSS (75:25) were used as negative controls. HBSS was used because the in vitro drug release experiments were performed in HBSS. The OD values from each well were divided by the mean of the controls, which were the wells of medium:HBSS (75:25) plus cells and expressed as percent of cell viability of the controls. The data are presented in Figure 8, which demonstrates that the cell viability for medium + cells was 111% and the cell viability for medium + HBSS was slightly lower at 100%. This is to be expected, as cells are expected to be less viable in diluted medium. The data are summarized in Table 2. For the positive controls and the test samples, the expected trend of lower cell viability with higher DNR concentration was observed. Comparing the positive controls with the test samples, sample with DNR concentrations of 71.5 ng/mL displayed equivalent cytotoxicity as the commercial DNR equivalent. The cytotoxicity of 228.5 ng/mL of DNR sample from the pSiO2-PLGA composite showed greater cytotoxicity than the commercial DNR equivalent (Table 2). The positive controls were set up according to the available sample DNR concentration and the concentration was escalated up by half-logs. So the highest positive control concentration was 731.25 (actually measured by mass spectrometry) which had the most cytotoxicity and was significantly different from all the other samples (Table 2)
Figure 8.

Viability of EA-HY926 cells (quantified by WST-1) after exposure to the samples with different concentrations of DNR released from PLGA-pSiO2_21/40-DNR for 5 days. Medium_Cells=medium plus cells; MediumHBSSCells=culture medium 75% plus HBSS 25% plus cells; pctrl=positive control. The concentration unit is ng/mL. In the box plot, the box represents the middle 50% of the data sample. The upper 25% of the data was represented by the upper whisker while the lower 25% of the data represented by the lower whisker. The median line was within the box. The outliers were denoted by the dots above or below the whiskers of the box.
Table 2.
Cytotoxicity of the in vitro drug release samples and positive controls
| Levels | Mean % of the Control | ||||
|---|---|---|---|---|---|
| Medium_Cells | A | 111 | |||
| MediumHBSSCells | A | 100 | |||
| pctrl_71.5 | B | 82 | |||
| Sample_71.5 | B | 79 | |||
| pctrl_228.5 | C | 58 | |||
| Sample_228.5 | D | 35 | |||
| pctrl_731.25 | E | 18 |
Levels not connected by same letter are significantly different. (Comparisons for all pairs using Tukey-Kramer HSD, Alpha=0.05).
Sample: Sample from in vitro release; pctrl: Positive control
Medium_Cells=cell plus 100% medium; MediumHBSSCells=cell plus 75% medium and 25% HBSS; pctrl_71.5=cell plus 75% medium and 25% commercial DNR sample, having a resultant DNR concentration of 71.5 ng/mL; Sample_71.5= cell plus 75% medium and 25% in vitro DNR release sample, having a resultant DNR concentration of 71.5 ng/mL; pctrl_228.5, Sample_228.5, and pctrl_731.25 were made in a same paradigm. HBSS was used as the in vitro drug release medium therefore MediumHBSSCells was used as a control to calculate cell viability for the other test samples.
Discussion
In this study, pSiO2-PLGA composite microspheres were fabricated with the goal of developing an ocular drug delivery system. It is encouraging that DNR released from pSiO2-PLGA composite particles retains its full biological function in inhibiting cell proliferation. Though the concentration of 228.5 ng/mL from in vitro release sample showed more potent cytotoxicity than its positive control, the difference magnitude was within the ranges between its equivalent control and the next higher control concentration. Due to the limited sample size and spread of data variance, this difference needs to be further investigated in a future in vivo study. The current results suggest that a single intravitreal injection of pSiO2-PLGA-DNR microspheres containing 5 μg of DNR should not be toxic and can provide better vitreous pharmacokinetics than the single intravitreal bolus injection of 5 μg of DNR that is typically administered [4, 6] [4] to inhibiting PVR in experimental studies. One study has shown that splitting one 5 ug intravitreal injection into two smaller doses administered at different times to extend the vitreous drug presence can enhance the therapeutic effect. Machemer and colleagues reported that a single intravitreal dose of 15 nmol per eye on the 3rd day following intravitreal cell injection was not effective in preventing retinal detachment. However splitting the dose into 10 nmol and 5 nmol injected 4 hours apart was effective [4]. This suggests that sustained drug exposure at the disease site can drive the minimum effective concentration of a drug much lower. Indeed, it has been reported that exposure to 700nM of DNR for 1 hour inhibits 50% of fibroblast proliferation, but cell proliferation is completely inhibited by exposure to lower concentrations (500 nM) for longer times (5 hours) [6]. Therefore, a delivery vehicle such as PLGA-pSiO2 composite microspheres that can sustain the release of daunorubicin should improve the therapeutic effect.
From the in vitro drug release studies, the DNR-loaded PLGA-pSiO2 composites demonstrated sustained release of DNR for a 70 day period. Composites made with larger pSiO2 particles (size range 21–40 microns) released drug more slowly and for a longer period of time. Stable sustained release was observed between days 14 and 70 (Figure 7). However, between days 0–14 there were two phases of release, one immediate burst release within day 1 and then a much less pronounced period of high drug release between days 2–14. We assign the initial burst release to loosely bound daunorubicin at or near the surface of the PLGA-pSiO2 composite, as a consequence of the two-step emulsification method used in this work. The initial DNR burst release from all three types of microspheres was similar because they were fabricated in the same way. The loosely bound daunorubicin is expected to be released quickly into a dissolution medium and generate a massive burst release. Indeed, nearly 50% of the loaded drug was released during the first day. More thorough washing of the composite microspheres after the drug loading step may reduce this initial burst release and improve the release profile. The second phase of release from the PLGA-pSiO2 composite microspheres (between days 2–14) was moderate compared with the initial burst release. We hypothesize that this phase of release was a typical first-order kinetic process. Though a thicker PLGA coating may extend the drug release profile to longer times, such engineering will not achieve a zero-order release [30]. However, compared with pSiO2 alone or PLGA alone, the PLGA-pSiO2 composite provides a more extended daunorubicin release profile with a more consistent steady state drug concentration. Comparison of the PLGA-pSiO2 composite particles with different size of pSiO2 particles, the one with smaller pSiO2 particles had a slightly faster drug release which may be contributed to the merely difference of pSiO2 particle size because daunorubicin-loaded pSiO2 particles were small and numerous in the cross-sectional view of PLGA-pSiO2_6/20-DNR composite microsphere (Figure 6). At the end of the release period, the concentration of drug in the release medium containing the PLGA-DRN microspheres dropped 10 fold, from ~ 500 ng/mL to ~50 ng/mL within a week. This is attributed to the rapid degradation of PLGA at the end stage of dissolution [31, 32]. It has been reported that PLGA experiences rapid degradation when its molecular weight reduces from 51 to 8 kDa, which usually develops between 19 and 33 days of incubation [33].
In the current study, micron-scale voids were found on the surface of PLGA-pSiO2 composite microspheres. These voids on the surface of the microspheres may be responsible for the increased rate of release seen in days 2–14, acting as outlets for the first-order release of drug trapped in the pSiO2 particles. Therefore, engineering these pores may provide another layer of control to optimize the drug release profile and warrants further exploration.
A PLGA coating layer is known to act as an effective barrier in preventing the premature release of drugs into aqueous media [30] [34–36] [37]. In the PLGA-pSiO2 composites of the present study, the drug-loaded pSiO2 particles act as drug reservoirs in the core of a PLGA microsphere, and drug leaching is slowed compared with PLGA-only microspheres.
The drug release profile may also depend on the nature of the pSiO2 particles within the PLGA microspheres [38–40]. A relatively large drug-containing pSiO2 particle will have a smaller surface area and tend to degrade and leach drug more slowly than multiple smaller pSiO2 particles, and this is observed in the data. For the PLGA-pSiO2_6/20-DNR samples, the smaller drug-loaded pSiO2 microparticles (of nominal sizes between 6 and 20 microns) were more numerous in a given PLGA microsphere, and they therefore presented a larger specific surface area and a thinner PLGA coating compared with the 21–40 micron sized pSiO2 particles of the PLGA-pSiO2_21/40-DNR formulation.
The faster DNR release characteristic seen with the PLGA-only microspheres can also be due to the formation of phase-segregated aggregates of DNR in PLGA which were shown in figure 6B. Larger aggregates of DNR cannot form in the nanoscale pores of pSiO2 particles due to the small physical size of these pores. Burst release from PLGA microspheres is a common phenomenon [27, 34, 36, 41, 42]. The initial burst release is hard to completely eliminate but could be reduced by using larger drug molecules and increased mass fraction of PLGA [26, 27]. With the current PLGA microsphere technique, the higher drug loading usually leads to a higher initial burst release [26], which might be attributed to the high accumulation of drug on the surface or near the surface of the microspheres [43].
5. Conclusion
In the current study, pSiO2/PLGA microspheres were fabricated by an S/O/W emulsion method to combine the advantages of a biodegradable polymer with the drug release characteristics of biocompatible porous silica for extended release of DNR. Compared with the infiltrative DNR- loaded pSiO2 particles or the PLGA-DNR microspheres, the PLGA-pSiO2-DNR composite microsphere system demonstrated a 5-fold and 2-fold longer duration of DNR release, respectively. This system is encouraging and may be of value in managing unwanted ocular proliferation through a single intravitreal injection.
Supplementary Material
Suplemental Figure 1. Schematic showing the fabrication of PLGA-pSiO2-DNR microspheres using the S/O/W emulsion method.
Suplemental Figure 2. The SEM images of fresh prepared pSi film (left panel) before ultrasonication and the given particles after the ultrasonication. The corresponding insert demonstrated similar nano-scaled pore structure.
Acknowledgments
Financial support: This study was supported by the National Institutes of Health under grant number NIH EY020617 and a NIH P30 core grant P30EY022589 (Histology Module).
Footnotes
Disclosure: W.R. Freeman, Spinnaker Biosciences (C); M.J. Sailor, Spinnaker Biosciences (I); L. Cheng, Spinnaker Biosciences (C)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pastor JC. Proliferative vitreoretinopathy: an overview. Survey of ophthalmology. 1998;43:3–18. doi: 10.1016/s0039-6257(98)00023-x. [DOI] [PubMed] [Google Scholar]
- 2.Wiedemann P, Hilgers RD, Bauer P, Heimann K. Adjunctive daunorubicin in the treatment of proliferative vitreoretinopathy: results of a multicenter clinical trial. Daunomycin Study Group. Am J Ophthalmol. 1998;126:550–9. doi: 10.1016/s0002-9394(98)00115-9. [DOI] [PubMed] [Google Scholar]
- 3.Shinohara K, Tanaka M, Sakuma T, Kobayashi Y. Efficacy of daunorubicin encapsulated in liposome for the treatment of proliferative vitreoretinopathy. Ophthalmic surgery, lasers & imaging: the official journal of the International Society for Imaging in the Eye. 2003;34:299–305. [PubMed] [Google Scholar]
- 4.Khawly JA, Saloupis P, Hatchell DL, Machemer R. Daunorubicin treatment in a refined experimental model of proliferative vitreoretinopathy. Graefes Arch Clin Exp Ophthalmol. 1991;229:464–7. doi: 10.1007/BF00166311. [DOI] [PubMed] [Google Scholar]
- 5.Chen EP, Steinhorst UH, Samsa GP, Saloupis PT, Hatchell DL. The effect of combined daunorubicin and triamcinolone acetonide treatment on a refined experimental model of proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1992;33:2160–4. [PubMed] [Google Scholar]
- 6.Wiedemann P, Sorgente N, Bekhor C, Patterson R, Tran T, Ryan SJ. Daunomycin in the treatment of experimental proliferative vitreoretinopathy. Effective doses in vitro and in vivo. Invest Ophthalmol Vis Sci. 1985;26:719–25. [PubMed] [Google Scholar]
- 7.Santana M, Wiedemann P, Kirmani M, Minckler DS, Patterson R, Sorgente N, et al. Daunomycin in the treatment of experimental proliferative vitreoretinopathy: retinal toxicity of intravitreal daunomycin in the rabbit. Graefes Arch Clin Exp Ophthalmol. 1984;221:210–3. doi: 10.1007/BF02134142. [DOI] [PubMed] [Google Scholar]
- 8.Mietz H, Heimann K. Onset and recurrence of proliferative vitreoretinopathy in various vitreoretinal disease. The British journal of ophthalmology. 1995;79:874–7. doi: 10.1136/bjo.79.10.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tzur-Balter A, Gilert A, Massad-Ivanir N, Segal E. Engineering porous silicon nanostructures as tunable carriers for mitoxantrone dihydrochloride. Acta Biomater. 2013;9:6208–17. doi: 10.1016/j.actbio.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Chhablani J, Nieto A, Hou HY, Wu EC, Freeman WR, Sailor MJ, et al. Oxidized Porous Silicon Particles Covalently Grafted with Daunorubicin as a Sustained Intraocular Drug Delivery System. Invest Ophthalmol Vis Sci. 2013;54:1268–79. doi: 10.1167/iovs.12-11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson SHC, Elliott H, Wallis DJ, Canham LT, Powell JJ. Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions. Phys Status Solidi A. 2003;197:331–5. [Google Scholar]
- 12.Nieto A, Hou H, Sailor MJ, Freeman WR, Cheng L. Ocular silicon distribution and clearance following intravitreal injection of porous silicon microparticles. Experimental Eye Research. 2013;116:161–8. doi: 10.1016/j.exer.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart MP, Buriak JM. Chemical and biological applications of porous silicon technology. Adv Mater. 2000;12:859–69. [Google Scholar]
- 14.Hartmann KI, Nieto A, Wu EC, Freeman WR, Kim JS, Chhablani J, et al. Hydrosilylated porous silicon particles function as an intravitreal drug delivery system for daunorubicin. Journal of ocular pharmacology and therapeutics: the official journal of the Association for Ocular Pharmacology and Therapeutics. 2013;29:493–500. doi: 10.1089/jop.2012.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng L, Anglin E, Cunin F, Kim D, Sailor MJ, Falkenstein I, et al. Intravitreal properties of porous silicon photonic crystals: a potential self-reporting intraocular drug-delivery vehicle. The British journal of ophthalmology. 2008;92:705–11. doi: 10.1136/bjo.2007.133587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anglin EJ, Schwartz MP, Ng VP, Perelman LA, Sailor MJ. Engineering the Chemistry and Nanostructure of Porous Silicon Fabry-Pérot Films for Loading and Release of a Steroid. Langmuir. 2004;20:11264–9. doi: 10.1021/la048105t. [DOI] [PubMed] [Google Scholar]
- 17.Andrew JS, Anglin EJ, Wu EC, Chen MY, Cheng LY, Freeman WR, et al. Sustained Release of a Monoclonal Antibody from Electrochemically Prepared Mesoporous Silicon Oxide. Adv Funct Mater. 2010;20:4168–74. doi: 10.1002/adfm.201000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Low SP, Williams KA, Canham LT, Voelcker NH. Evaluation of mammalian cell adhesion on surface-modified porous silicon. Biomaterials. 2006;27:4538–46. doi: 10.1016/j.biomaterials.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Anglin EJ, Cheng L, Freeman WR, Sailor MJ. Porous silicon in drug delivery devices and materials. Advanced drug delivery reviews. 2008;60:1266–77. doi: 10.1016/j.addr.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu EC, Andrew JS, Cheng LY, Freeman WR, Pearson L, Sailor MJ. Real-time monitoring of sustained drug release using the optical properties of porous silicon photonic crystal particles. Biomaterials. 2011;32:1957–66. doi: 10.1016/j.biomaterials.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mundargi RC, Srirangarajan S, Agnihotri SA, Patil SA, Ravindra S, Setty SB, et al. Development and evaluation of novel biodegradable microspheres based on poly(D,L-lactide-co-glycolide) and poly(epsilon-caprolactone) for controlled delivery of doxycycline in the treatment of human periodontal pocket: In vitro and in vivo studies. Journal of Controlled Release. 2007;119:59–68. doi: 10.1016/j.jconrel.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 22.Panyam J, Labhasetwar V. Sustained cytoplasmic delivery of drugs with intracellular receptors using biodegradable nanoparticles. Mol Pharmaceut. 2004;1:77–84. doi: 10.1021/mp034002c. [DOI] [PubMed] [Google Scholar]
- 23.Yoon KR, Chi YS, Lee KB, Lee JK, Kim DJ, Koh YJ, et al. Surface-initiated, ring-opening polymerization of p-dioxanone from gold and silicon oxide surfaces. J Mater Chem. 2003;13:2910–4. [Google Scholar]
- 24.Chan JM, Zhang LF, Yuet KP, Liao G, Rhee JW, Langer R, et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30:1627–34. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Jensen DMK, Cun D, Maltesen MJ, Frokjaer S, Nielsen HM, Foged C. Spray drying of siRNA-containing PLGA nanoparticles intended for inhalation. Journal of Controlled Release. 2010;142:138–45. doi: 10.1016/j.jconrel.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, Qiu ZY, Wang SQ, Zhou L, Zhang SM. A modified double-emulsion method for the preparation of daunorubicin-loaded polymeric nanoparticle with enhanced in vitro anti-tumor activity. Biomed Mater. 2010;5 doi: 10.1088/1748-6041/5/6/065002. [DOI] [PubMed] [Google Scholar]
- 27.Fan DM, De Rosa E, Murphy MB, Peng Y, Smid CA, Chiappini C, et al. Mesoporous Silicon-PLGA Composite Microspheres for the Double Controlled Release of Biomolecules for Orthopedic Tissue Engineering. Adv Funct Mater. 2012;22:282–93. [Google Scholar]
- 28.Ho ML, Fu YC, Wang GJ, Chen HT, Chang JK, Tsai TH, et al. Controlled release carrier of BSA made by W/O/W emulsion method containing PLGA and hydroxyapatite. Journal of Controlled Release. 2008;128:142–8. doi: 10.1016/j.jconrel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 29.Garcia SN, Gutierrez L, McNulty A. Real-time cellular analysis as a novel approach for in vitro cytotoxicity testing of medical device extracts. Journal of biomedical materials research Part A. 2013;101:2097–106. doi: 10.1002/jbm.a.34507. [DOI] [PubMed] [Google Scholar]
- 30.Xue JM, Shi M. PLGA/mesoporous silica hybrid structure for controlled drug release. Journal of Controlled Release. 2004;98:209–17. doi: 10.1016/j.jconrel.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 31.Huang CL, Steele TWJ, Widjaja E, Boey FYC, Venkatraman SS, Loo JSC. The influence of additives in modulating drug delivery and degradation of PLGA thin films. Npg Asia Mater. 2013;5 [Google Scholar]
- 32.Samadi N, Abbadessa A, Di Stefano A, van Nostrum CF, Vermonden T, Rahimian S, et al. The effect of lauryl capping group on protein release and degradation of poly(D,L-lactic-co-glycolic acid) particles. Journal of Controlled Release. 2013;172:436–43. doi: 10.1016/j.jconrel.2013.05.034. [DOI] [PubMed] [Google Scholar]
- 33.Xu Q, Chin SE, Wang CH, Pack DW. Mechanism of drug release from double-walled PDLLA(PLGA) microspheres. Biomaterials. 2013;34:3902–11. doi: 10.1016/j.biomaterials.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva AL, Rosalia RA, Sazak A, Carstens MG, Ossendorp F, Oostendorp J, et al. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: low-burst release is crucial for efficient CD8(+) T cell activation. European journal of pharmaceutics and biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV. 2013;83:338–45. doi: 10.1016/j.ejpb.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Gajendiran M, Divakar S, Raaman N, Balasubramanian S. In Vitro Drug Release Behavior, Mechanism and Antimicrobial Activity of Rifampicin Loaded Low Molecular Weight PLGA-PEG-PLGA Triblock Copolymeric Nanospheres. Current drug delivery. 2013;10:722–31. doi: 10.2174/15672018113109990002. [DOI] [PubMed] [Google Scholar]
- 36.Ahmed AR, Elkharraz K, Irfan M, Bodmeier R. Reduction in burst release after coating poly(D,L-lactide-co-glycolide) (PLGA) microparticles with a drug-free PLGA layer. Pharmaceutical development and technology. 2012;17:66–72. doi: 10.3109/10837450.2010.513989. [DOI] [PubMed] [Google Scholar]
- 37.Liao YT, Wu KCW, Yu JS. Synthesis of mesoporous silica nanoparticle-encapsulated alginate microparticles for sustained release and targeting therapy. J Biomed Mater Res B. 2014;102:293–302. doi: 10.1002/jbm.b.33007. [DOI] [PubMed] [Google Scholar]
- 38.Bian XM, Liang S, John J, Hsiao CH, Wei X, Liang D, et al. Development of PLGA-based itraconazole injectable nanospheres for sustained release. International Journal of Nanomedicine. 2013;8:4521–31. doi: 10.2147/IJN.S54040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnes TJ, Jarvis KL, Prestidge CA. Recent advances in porous silicon technology for drug delivery. Therapeutic delivery. 2013;4:811–23. doi: 10.4155/tde.13.52. [DOI] [PubMed] [Google Scholar]
- 40.Aragon DM, Rosas JE, Martinez F. Relationship between the solution thermodynamic properties of naproxen in organic solvents and its release profiles from PLGA microspheres. J Microencapsul. 2013;30:218–24. doi: 10.3109/02652048.2012.717114. [DOI] [PubMed] [Google Scholar]
- 41.Ahmed AR, Dashevsky A, Bodmeier R. Reduction in burst release of PLGA microparticles by incorporation into cubic phase-forming systems. European journal of pharmaceutics and biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV. 2008;70:765–9. doi: 10.1016/j.ejpb.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 42.Mao S, Xu J, Cai C, Germershaus O, Schaper A, Kissel T. Effect of WOW process parameters on morphology and burst release of FITC-dextran loaded PLGA microspheres. International journal of pharmaceutics. 2007;334:137–48. doi: 10.1016/j.ijpharm.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 43.Cui F, Cun D, Tao A, Yang M, Shi K, Zhao M, et al. Preparation and characterization of melittin-loaded poly (DL-lactic acid) or poly (DL-lactic-co-glycolic acid) microspheres made by the double emulsion method. Journal of controlled release: official journal of the Controlled Release Society. 2005;107:310–9. doi: 10.1016/j.jconrel.2005.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suplemental Figure 1. Schematic showing the fabrication of PLGA-pSiO2-DNR microspheres using the S/O/W emulsion method.
Suplemental Figure 2. The SEM images of fresh prepared pSi film (left panel) before ultrasonication and the given particles after the ultrasonication. The corresponding insert demonstrated similar nano-scaled pore structure.