

SUMMARY
On the lupus-prone MRL-lpr/lpr (MRL-lpr) background AM14 rheumatoid factor (RF) B cells are activated, differentiate into plasmablasts, and undergo somatic hypermutation outside of follicles. Using multiple strategies to impair T cells, we found that such AM14 B cell activation did not require T cells, but could be modulated by them. In vitro, the signaling adaptor MyD88 is required for IgG anti-chromatin to stimulate AM14 B cell proliferation when T cells are absent. However the roles of Toll-like receptors (TLRs) in AM14 B cell activation in vivo have not been investigated. We found that activation, expansion and differentiation of AM14 B cells depended on MyD88; however, mice lacking either TLR7 or TLR9 displayed partial defects, indicating complex roles for these receptors. T-independent activation of certain autoreactive B cells, which instead can gain stimuli via endogenous TLR ligands, may be the initial step in the generation of canonical autoantibodies.
INTRODUCTION
Systemic autoimmune diseases such as Lupus Erythematosus (SLE) and Rheumatoid Arthritis (RA) are characterized by the production of canonical diagnostic autoantibodies, such as anti-nuclear antibodies and RFs (Tan, 1989). It has become increasingly clear that B cells play important pathogenic roles beyond autoantibody secretion. The significance and importance of the multiple roles of B cells and in particular their ability to interact with T cells has recently been dramatically emphasized by the responsiveness of human and murine systemic and organ-specific autoimmune diseases to therapeutic B cell depletion. T cell-mediated pathology was modulated in these individuals, and clinical remissions often occurred prior to and not in correlation with reductions in autoantibody levels (Browning, 2006).
The importance of T-B interaction has been demonstrated in lupus prone MRL-lpr animals lacking CD4+ or αβ T cells, as these animals have delayed, less-severe disease and reduced autoantibody titers (Connolly et al., 1992; Peng, 1998; Peng et al., 1996b). Conversely, the addition of autoreactive T cells to pre-autoimmune or non-autoimmune mice has led to autoreactive B cell activation and autoantibody secretion (Adams et al., 1991; Ando et al., 1987; Fields et al., 2005b), and even loss of anergic features (Seo et al., 2002). Although the nature of these B-T interactions is still not clarified, it is reasonable to think that the same types of B cells that generate cardinal autoantibodies are also providing activation signals to T cells that recognize cognate autoantigens. For example, a dsDNA-specific B cell might be stimulated by chromatin, take it up, and present histone peptides to histone-peptide specific CD4+ T cells, resulting in proliferation and differentiation of the T cells as well as delivery of T help signals to the B cell.
Indeed, autoantibodies reflect the hallmarks of T cell-dependent responses—they are often isotype-switched, somatically mutated, clonally expanded and affinity matured. Hence, germinal centers (GCs) were presumed to be the source of these autoantibodies. However, recent data from our lab, coupled with data from several other studies, has questioned whether GCs are obligatory sites of affinity-based selection and mutation of autoreactive B cells. Using mice with an Ig-Tg that increases the frequency of B cells that recognize self-IgG2a (the rheumatoid factor or “RF” specificity), we found that spontaneous, autoantigen-specific responses in the spleen were largely taking place extrafollicularly, at the T zone-red pulp border. At this location actively dividing B cells and plasmablasts were observed, and microdissection experiments demonstrated that somatic hypermutation (SHM) was taking place in situ (William et al., 2002). Anti-DNA responses have also been observed to take place at a similar site (Jacobson et al., 1995). It is notable that T-independent responses to foreign Ags also occur at this site. The roles of T cells in activating autoreactive B cells in the extrafollicular reaction have been little explored (Fields et al., 2005a; Fields et al., 2005b).
In addition to the potential effects of T cells on the autoantibody response, a second important signal, transduced via Toll-like receptors (TLRs) that recognize endogenous Ags, has been recognized. This was first observed using the AM14 Tg mouse system. It was demonstrated that IgGs that recognized chromatin and presumably formed immune complexes (ICs) with it were highly mitogenic for AM14 B cells in vitro, whereas control ICs were not; moreover, this mitogenic activity was dependent upon MyD88 and eventually pinned to a large extent to TLR9 (Leadbetter et al., 2002; Viglianti et al., 2003). The in vivo relevance of this signal for autoreactive B cell activation was supported by the phenotype of MyD88-, TLR7- and TLR9-deficient autoimmune-prone mice (Christensen et al., 2005; Christensen et al., 2006; Lau et al., 2005; Sadanaga et al., 2007). Anti-nuclear Abs and RFs were absent in MyD88-deficient mice, while anti-chromatin was lost in TLR9-deficient mice and anti-RNA was missing in TLR7-deficient animals. Interestingly, however, concentrations of serum RF were not reduced in either TLR7- or TLR9-deficient lupus-prone MRL-lpr mice (unpublished observations). Because TLR signaling can occur in many cell types and at many stages of activation, exactly how TLRs control autoreactive B cell activation and autoantibody production in vivo remains to be fully elucidated.
Here we have used the AM14 Tg system, along with a newly described method for inducing the extrafollicular RF B cell reaction in vivo by administering IgG anti-chromatin (Herlands et al., 2007), in order to study the roles of both T cells and TLRs in the activation of autoreactive B cells. Administration of anti-chromatin to induce the extrafollicular AM14 response allowed us to visualize its initiation and thereby to disentangle primary and secondary effects. We have used a combination of inhibition and genetic approaches in the context of both the spontaneous and induced RF responses. These studies have provided surprising insights into how autoreactive extrafollicular B cell responses are initiated and controlled.
RESULTS
Spontaneous activation of AM14 B cells is T-dependent
AM14 H chain Tg MRL-lpr mice (Tg WT mice) spontaneously undergo an abrupt expansion of RF-secreting B cells which can be identified with the anti-idiotype antibody 4-44. This process of “conversion” to autoimmunity occurs between 10 and 20 weeks of age (William et al., 2005a). To determine whether T cells were required for conversion, we backcrossed the AM14 H transgene to αβ T cell-deficient MRL-lpr Tcrb−/− mice (Tg Tcrb−/−). At 8 weeks of age, we began tracking the concentration of AM14 Ab in the serum of Tg WT and Tg Tcrb−/− mice. There was no detectable serum AM14 Ab in Tg Tcrb−/− mice at any time, while we saw increasing amounts in the Tg WT as the mice aged (Figure 1A). To ensure that this was not a delayed response in the T-deficient animals, we extended the tracking to 30 weeks, well beyond the time by which most T-sufficient animals have elevated serum AM14 Ab, yet none of the Tg Tcrb−/− animals developed detectable amounts of serum AM14. In addition, there were few AM14 antibody-forming cells (AFCs) in spleens of 30-week-old Tg Tcrb−/− mice, in contrast to high numbers in Tg WT mice (Figure 1B). We also identified splenic AM14 idiotype-expressing (4-44+) cells in situ by immunofluorescent staining. The morphology of these brightly staining cells with ample cytoplasm suggests that they are AFCs. There was a complete absence of AM14 AFCs in the spleens of 30-week-old Tg Tcrb−/− mice, though they were clearly seen in Tg WT animals at the T-zone-red-pulp border (Figure 1C and D; note also the absence of most CD4+ T cells with remaining cells being DCs).
Figure 1. Spontaneous activation of AM14 B cells is T-dependent.

(A) Serum levels of AM14 protein were measured by ELISA analysis of samples collected every two weeks from Tg WT (n=5, grey diamonds) and Tg Tcrb−/− mice (n=6, black squares).
(B) AM14 (4-44+) AFCs as detected by ELISpot. Bars are 1 SEM. p= 0.0022.
(C–D) Immunofluorescent staining shows red pulp macrophages (green), CD4+ T cells (blue) and AM14 (4-44+) B cells (red) in splenic sections of 18 week old mice. Original magnification 200x. Note the lack of AM14 B cells in the T-deficient mice (D).
Anti-chromatin driven activation of AM14 B cells is αβ and γδ T cell independent
AM14 B cells are specific for IgG2a, a class-switched immunoglobulin. Because T-deficient MRL-lpr mice have markedly reduced concentrations of IgG2a autoantibodies (Peng et al., 1996b), we wondered if the absence of AM14 B cell activation in Tg Tcrb−/− mice was due to a lack of Ag for BCR stimulation (i.e. IgG2a or IgG2a-containing ICs) rather than lack of a T-B interaction required for direct RF B cell stimulation. Recently, we have described the ability of the IgG2a anti-chromatin PL2-3 (via intrapeitoneal [i.p.] injection of PL2-3 hybridoma cells or purified protein) to activate AM14 B cells to proliferate, differentiate, and hypermutate at extrafollicular sites in the spleen (Herlands et al., 2007). Neither IgG2b anti-chromatin nor IgG2a anti-hapten or anti-self that is not chromatin had this property (Herlands et al., 2007). We used this system to bypass the potential role of T cells in generating the isotype-switched BCR ligand for AM14 by providing it exogenously. PL2-3 or control PL2-8 (IgG2b anti-chromatin) hybridomas were injected into Tg WT and Tg Tcrb−/− MRL-lpr animals. Surprisingly, T-deficient animals receiving PL2-3 developed AM14 CD22lo plasmablasts at frequencies comparable to T-sufficient mice (Figure 2A and data not shown). Furthermore, there were indistinguishable numbers of AM14 AFCs in the spleens of both T-deficient and T-sufficient animals (Figure 2B). The localization of activated cells in the spleen was also similar in the two strains. Immunofluorescence analysis showed clustering of AM14 B cells at the T-zone-red pulp border, outside of B cell follicles in Tg WT (Figure 2C) and Tg Tcrb−/− mice (Figure 2D) that received PL2-3, but not PL2-8 (data not shown).
Figure 2. Anti-chromatin drives AM14 B cell activation in a T-independent manner.

(A) Representative FACS plots of live cells from PL2-3 and PL2-8 treated Tg Wt (“WT”) and Tg Tcrb−/− mice. Gates identify AM14 B cells (4-44+, CD22lo) and plasmablasts (4-44+, CD22hi). Note the induction of plasmablasts in both types of mice by PL2-3 but not PL2-8.
(B) AM14 (4-44+) AFCs determined by ELISpot analysis of Tg WT and Tg Tcrb−/− mice treated with PL2-3 hybridoma (n=33 and 30, respectively), PL2-8 hybridoma (n=9 and 6, respectively), or immunized with IgG2a anti-NP/NP-CGG ICs in alum (n=3 and 3, respectively). For Tg WT and Tg Tcrb−/− mice, treatment with PL2-3 was statistically different compared to PL2-8 or protein IC, p<0.0001. The values for Tg WT and Tg Tcrb−/− mice treated with PL2-3 were not statistically different.
(C–D) Micrographs of immunofluorescent staining of splenic sections from Tg WT and Tg Tcrb−/− mice showing red-pulp macrophages (green), CD4+ T cells (blue) and AM14 (4-44+) B cells (red).
(E) Histograms showing PNA binding on live, 4-44+, CD22hi cells from Tg WT and Tg Tcrb−/− mice immunized with IgG2a anti-NP/NP-CGG ICs in alum.
(F–G) Micrographs of immunofluorescent staining showing AM14 (4-44+) B cells (red), red-pulp macrophages (blue) and PNA+ GC cells (green) in splenic sections of mice described in (E). In (F) note the large 4-44+ GC, as recognized by the yellow color of the light zone.
As a further control, to ensure that the Tg Tcrb−/− mice were indeed unable to respond to T-dependent antigens, we tested the ability of foreign protein-containing ICs to elicit an AM14 GC response. We previously reported that IgG2a anti-TNP+TNP-KLH ICs and IgG2a anti-NP+NP-CGG ICs can stimulate an RF GC response in Tg WT animals (Herlands et al., 2007; William et al., 2002; William et al., 2006). Immunization with IgG2a anti-NP+NP-CGG ICs generated AM14 PNA+ GC cells detected by both FACS (Figure 2E) and immunofluorescence (Figure 2F) in Tg WT mice. However, Tg Tcrb−/− mice produced neither AM14 PNA+ cells (Figure 2E) nor AM14+ GCs (Figure 2G). Consistent with previous observations (William et al., 2002; William et al., 2006), this immunization with protein ICs produced an exclusively GC response, and did not generate AM14 AFCs in either Tg Tcrb−/− or Tg WT mice (Figure 2B, F, and G). Together, these data demonstrate that Tg Tcrb−/− mice are incapable of generating T-dependent responses, but are fully able to support the extrafollicular AM14 B cell response elicited by IgG2a anti-chromatin.
Tg Tcrb−/− mice have increased numbers of γδ T cells, which could potentially provide help for AM14 B cells in the absence αβ T cells (Peng et al., 1996a). To exclude this, a γδ-depleting antibody, UC7 (Houlden, 1989), was used to eliminate the γδ T cells during PL2-3 treatment (Hiromatsu, 1992). Figure 3A shows successful depletion of γδ T cells to less than 1% of live cells in mice treated with UC7, but not rat IgG. Despite effective γδ cell depletion, there was no effect on splenic AM14 ELISpots in PL2-3 treated animals (Figure 3B). Thus activation of AM14 B cells by PL2-3 anti-chromatin is both αβ and γδ T cell-independent, suggesting that the lack of spontaneous activation in Tg Tcrb−/− MRL-lpr mice was due to the lack of IgG2a autoantigen rather than the absence of T cell help.
Figure 3. Anti-chromatin activation of AM14 B cells is αβ and γδ T cell-independent.

(A) Representative FACS plots of live cells from spleens of Tg WT and Tg Tcrb−/− mice injected with either γδ–depleting antibody UC-7 or control rat Ig during PL2-3 hybridoma treatment. Gates identify αβ and γδ T cell populations.
(B) AM14 (4-44+) AFCs determined by ELISpot. Mice were injected with either control rat Ig (black bars) or UC-7 (grey bars) during PL2-3 hybridoma treatment (n=4 and 4, respectively, for each of WT and Tcrb−/−). Mice that received neither PL2-3 nor antibody treatment were included as negative controls (n=4 for each of WT and Tcrb−/−, white bars). * p<0.05.
CD4 T Cells Potentiate but are not Required for Sustained Activation of AM14 B cells
Though mice lacking αβ and γδ T cells clearly generated a robust RF response within one week of exposure to IgG2a anti-chromatin, it remained possible that T cells could promote or facilitate the activation of AM14 B cells in the spontaneous RF response. To address this issue, we treated autoimmune AM14 mice with the CD4 monoclonal antibody GK1.5. Consistent with previous reports (Wofsy and Seaman, 1987), we achieved partial depletion (40–70%) of circulating CD4+ T cells in lupus-prone mice, in contrast to 90–99% depletion in BALB/c mice (Supplemental Figure 1A–D). The remaining CD4+ cells were apparently rendered functionally inactive, as evidenced by their complete opsonization with GK1.5 in vivo, marked surface CD4 downregulation, and most importantly by the inability of GK1.5-treated mice to mount a T-dependent humoral response (Supplemental Figure 1E–G). This ability of GK1.5 to inhibit T cell function in the absence of complete depletion has been previously documented in autoimmune mice (Carteron et al., 1989).
We then used GK1.5 to functionally impair CD4+ T cell help for 2–4 weeks immediately following spontaneous conversion in AM14 Tg mice. In Tg WT mice treated with GK1.5 for 2 weeks, in the frequencies of AM14 AFCs were reduced by half, compared to mice that received control antibody (p = 0.04, Figure 4A). Thus T cells do contribute to an ongoing spontaneous RF response. Because the AM14 plasmablast response is rapidly turning over (William et al., 2005b), we extended the treatment duration to determine if this enhanced the effect. However, in mice treated for 4 weeks, there was still the same reduction by half of the frequency of AM14 AFCs (p = 0.0052 compared to control, Figure 4A). Despite 4 weeks of GK1.5 treatment, the number of splenic AM14 AFCs was more than five-fold greater than in age-matched, non-converted Tg WT mice (p = 0.0006, Figure 4A). Thus, the ability of AM14 B cells to differentiate into AFCs was not completely abrogated, and a significant autoantibody response was maintained in the absence of functional CD4+ T cell help.
Figure 4. Reduced but persistent AM14 activation in mice treated with anti-CD4.

(A) ELISpot analysis was used to quantify splenic AM14 (4-44+) AFCs in Tg WT mice treated with control Ig for 2 weeks (black triangles, n = 9) or 4 weeks (black circles, n = 9), or GK1.5 for 2 weeks (white triangles, n = 9) or 4 weeks (white circles, n = 7). Age-matched non-converted Tg WT mice are shown in black triangles (n = 7).
(B) Identification by FACS of splenic AM14 B cells (4-44+, CD22hi) and plasmablasts (4-44+, CD22lo) in control and GK1.5-treated mice.
(C) Percentage of 4-44+ CD22lo plasmablasts in control (black diamonds, n = 9) and GK1.5-treated (white diamonds, n = 7) mice.
(D) Percentage of CD22hi and CD22lo 4-44+ cells expressing elevated amounts of CD44 and CD69 in mice treated with control Ig (black bars, n = 9) or GK1.5 (white bars, n = 7).
(E) BrdU incorporation in CD22hi and CD22lo 4-44+ cells from mice treated with control Ig (black bars, n = 9) or GK1.5 (white bars, n = 7) and given a single dose of BrdU 12 hrs earlier. For C–E, results after 4 weeks of GK1.5 treatment are shown; similar results were seen after 2 weeks of treatment.
*, p<0.05; **, p<0.01 by Mann-Whitney.
Conversion was previously shown to include activation of CD22hi B cells as well as development of CD22lo plasmablasts (William et al., 2005b). As predicted by the ELISpot analysis, GK1.5-treated mice had reduced frequencies of AM14 CD22lo cells compared to control mice (p = 0.0081 for 2 week and p = 0.0052 for 4 week treatment), although a residual population of phenotypic plasmablasts persisted in the absence of T cell help (Figure 4B and C). We then examined activation marker expression and proliferation of AM14 B cells and plasmablasts after GK1.5 treatment. Expression of CD44 and CD69 was decreased in CD22hi B cells (p = 0.0164 and 0.0033, respectively), but a high level of CD44 expression was maintained and CD69 was partially but significantly reduced (p = 0.0115) in CD22lo plasmablasts (Figure 4D). To assess the effect of GK1.5 treatment on proliferation, the incorporation of 5-bromo-2′-deoxyuridine (BrdU) was measured after a 12 hour labeling period in mice that had been treated with GK1.5 or control antibody. Proliferation was decreased in CD22hi B cells (p = 0.0164), but was not significantly reduced (p=ns) in CD22lo plasmablasts after 4 weeks of GK1.5 treatment (Figure 4E). Taken together, these results suggest that while CD4+ T cells may enhance spontaneous AM14 B cell activation, a substantial degree of B cell activation and antibody formation can be maintained in the absence of T cell help.
Chromatin Antibodies Drive T-independent Somatic Hypermutation at Extrafollicular Sites
SHM is classically associated with T-dependent GC responses, but we recently found that SHM occurs robustly in AM14 B cells at extrafollicular sites of the spleen in Tg WT mice (Herlands et al., 2007; Kleinstein et al., 2003; William et al., 2002). The discovery that the extrafollicular AM14 response could initiate in the absence of T cells raised the important question of whether SHM occurred in the absence of T cells. To examine this, small clusters of 4-44+ B cells from splenic sections of PL2-3 treated Tg MRL-lpr or Tg Tcrb−/− MRL-lpr mice were microdissected (Figure 5A). The endogenous Vκ8 L chains were amplified, cloned and sequenced (William et al., 2002). Remarkably, mutations were occurring in Tg Tcrb−/− mice (Table 1) as well as in Tg WT [data shown in (Herlands et al., 2007)]. As previously reported, all 6 picks from Tg WT mice showed mutation with an average of 1.8 mutations per sequence (Herlands et al., 2007). Similarly, all 7 picks from Tg Tcrb−/− mice had mutation, with 0.94 mutations per sequence (Table 1). We previously calculated a PCR error rate using Pfu Turbo of <0.1 mutations per sequence (William et al., 2002); these current sequences were generated with Pfu Ultra, which has one-third the error rate (Stratagene). Thus, the observed mutation rates are far above PCR error. To account for the possibility of generating multiple copies of a single sequence that do not represent different cells, we also calculated the number of mutations per unique sequence (Table 1). We found 2.8 and 1.11 mutations per unique sequence for picks from Tg WT (Herlands et al., 2007) and Tg Tcrb−/− mice, respectively (p<0.005, Student’s t test with equal variance). Thus, although T cells are clearly not required for mutation to occur, T cells promote the accumulation of mutations, either by increasing the inherent mutation rate or by promoting longer survival of mutating cells.
Figure 5. Somatic hypermutation occurs at extrafollicular sites of Tg WT and Tg Tcrb−/− mice given IgG2a anti-chromatin.

(A) Immunohistochemistry showing AM14 B cells stained with 4-44 anti-idiotype antibody (blue) in a splenic section from a Tg Tcrb−/− mouse treated with PL2-3 protein every other day for 2 weeks. Images are from before and after laser microdissection. Typical picks contained 10–30 4-44+ B cells.
(B) Representative genealogical trees from two Tg WT mice (tree A2 and B3) and two Tg Tcrb−/− mice (trees K1 and L2) treated with PL2-3 protein as described in (A). The trees demonstrate ongoing mutation. Circles at the base of trees represent germline sequences, and non-mutated sequences found in a pick will be listed in this circle. Every other open circle, or “node”, represents one or more clones with the same sequence. Tree “branches” are the lines between nodes. The positions of nucleotide mutations are listed alongside branches.
Table 1.
Summary of microdissections and mutations in T cell deficient MRL/lpr mice treated with PL2-3 hybridomaa
| Mouse | Pick | Tree name | Total sequences | Total mutations | Mutations/sequence | Total unique sequences | Total mutations among unique sequences | Mutations/unique sequence | Trunk mutations |
|---|---|---|---|---|---|---|---|---|---|
| 11-8-05 G | 1 | I1 | 12 | 11 | 0.92 | 2 | 1 | 0.50 | 1 |
| 11-8-05 G | 2 | I2 | 12 | 5 | 0.42 | 3 | 3 | 1.00 | 1 |
| 11-8-05 H | 1 | J1 | 12 | 14 | 1.6 | 2 | 4 | 2.00 | 1 |
| 11-8-05 H | 2 | J2 | 10 | 2 | 0.20 | 3 | 2 | 0.67 | 0 |
| 11-8-05 E | 1 | K1 | 10 | 6 | 0.60 | 3 | 3 | 1.00 | 1 |
| 11-8-05 E | 3 | K2 | 11 | 32 | 2.91 | 2 | 5 | 2.50 | 0 |
| 10-18-06 E | 2 | L2 | 14 | 6 | 0.43 | 3 | 2 | 0.67 | 0 |
|
| |||||||||
| Total | 81 | 76 | 0.94 | 18 | 20 | 1.11 | 4 | ||
Sequences recovered from 4 Tg Tcrb−/− MRL/lpr mice treated with PL2-3 were grouped by pick and assigned to a lettered genealogical tree. Identical sequences in a pick were conservatively treated as one clone, instead of as the products of multiple cells, and were assigned to one ‘unique sequence.’
Using sequences recovered from each microdissection, we created genetic lineage trees that reflect the inferred clonal evolution; representative trees are shown in Figure 5B (William et al., 2002). Five out of 6 trees from Tg WT mice (Herlands et al., 2007) had at least one branch, while 8 of 9 trees from Tg Tcrb−/− mice had at least one branch. On average, each pick from Tg WT mice had 1.33 branches, and each pick from Tg Tcrb−/− mice had 1.65 branches. Branching patterns, though not expected to be extensive after only 7 or fewer days of mutation, nonetheless are indicative of authentic mutation and clonal expansion. Both the branching and the number of mutations per sequence confirm that mutation was occurring at a high rate at extrafollicular sites in both types of mice treated with PL2-3.
Stimulation of AM14 B cells by Anti-chromatin Depends on MyD88 and the Combination of TLR7 and TLR9
Although PL2-3 causes AM14 B cells to proliferate in vitro in a MyD88-dependent fashion (Leadbetter et al., 2002), the roles of TLRs in activation and differentiation of RF B cells have not been well-defined in vivo. Considering that T cell signals are dispensable, we hypothesized that BCR/TLR dual-engagement was required to drive AM14 B cell activation and differentiation in PL2-3 treated mice. To test this hypothesis, PL2-3 and PL2-8 hybridomas were injected i.p. into Tg WT, Tg Tlr9−/−, Tg Tlr7−/−, Tg Tlr7−/− Tlr9−/−, and Tg Myd88−/− mice (all on the MRL-lpr background).
PL2-3 treated Tg Myd88−/− mice had numbers of AM14 AFCs indistinguishable from those of Tg WT mice treated with control PL2-8 and significantly lower than those of PL2-3 treated Tg WT mice (Fig 6A). Interestingly, the generation of RF AFCs was only partially TLR9- or TLR7- dependent, as AM14 AFC numbers, though reduced significantly in each knockout strain, were not completely eliminated. To determine if there was redundancy between TLR7 and TLR9 in providing essential signals, we treated AM14 Tg Tlr7 Tlr9 double-mutant mice. AM14 AFCs were almost absent in these mice after PL2-3 treatment (Figure 6A), indicating that either TLR7 or TLR9 can contribute to the response to PL2-3. This was somewhat unexpected considering that PL2-3 is thought to be an anti-chromatin-DNA (Losman et al., 1992) and only TLR9, not TLR7, is thought to recognize DNA.
Figure 6. PL2-3-Driven Activation of AM14 B cells is TLR-dependent.

(A) AM14 (4-44+) AFCs detected by ELISpot from spleens of mice treated for 1 week with PL2-3 hybridoma (black bars, n=35 for WT, n=19 for Tlr9−/−, n=10 for Tlr7−/−, n=5 for Tlr7−/− Tlr9−/−, and n=10 for Myd88−/−) or PL2-8 hybridoma (striped bars, n=10 for WT and n=5 for Tlr9−/−).
(B) Representative FACS plots gated on live cells from Tg WT, Tg Tlr9−/−, Tg Tlr7−/−, Tlr7−/− Tlr9−/− and Tg Myd88−/− mice given PL2-3 hybridoma.
(C) Summary of FACS data from multiple experiments showing percent of live cells that are 4-44+ CD22lo. Bars show means.
(D) Ratio of plasmablast phenotype cells (assessed by FACS as in Fig. 6B) to actual ELISpots. Data derived from Fig. 6A and 6C.
(E–G) Micrographs of immunofluorescent staining showingAM14 (4-44+) B cells (red), red pulp macrophages (green) and CD4+ T cells (blue). PL2-3 treated Tg WT mouse (E), Tg Tlr9−/− mouse (F), and Tg Myd88−/− mouse (G) are displayed. Original magnification 100x.
*p<0.05; **p<0.001 by Mann-Whitney test.
As expected, Tg Myd88−/− and Tg Tlr7−/− Tlr9−/− mice had lower frequencies of 4-44+ CD22lo cells than Tg WT mice (Figure 6B,C). Surprisingly, Tg Tlr9−/− mice had increased frequencies of 4-44+ CD22lo cells compared to Tg WT (Figure 6B, C). Similarly, Tg Tlr7−/− had frequencies of 4-44+ CD22lo cells similar to WT. This was not anticipated since Tg Tlr9−/− and Tg Tlr7−/− mice had substantially fewer AM14 ELISpots than Tg WT mice, and thus reduced numbers of fully-fledged AFCs (Figure 6A). Together these data suggested that in the absence of either TLR7 or TLR9, initial activation could proceed normally but complete differentiation to a functional AFC was inefficient. To further substantiate this, we calculated a ratio that reflects efficiency of complete differentiation: the frequency of CD22lo cells identified by FACS (potential AFCs) to the number of ELISpots (actual AFCs, Figure 6D). This ratio in TLR-deficient animals was almost 10-fold lower than that of WT, indicating a marked deficit in completion of AFC differentiation. Consistent with the flow cytometry results, AM14 cells were identified by in both Tg WT and Tg Tlr9−/− mice (Figure 6E, F) and Tg Tlr7−/− mice (data not shown). In contrast, AM14 B cells were not identified in splenic sections of PL2-3 treated Tg Myd88−/− mice (Figure 6G) or PL2-3 treated Tlr7 Tlr9 double-mutant mice (data not shown).
In trying to reconcile these data, we first investigated whether TLRs have an impact on cell survival. However, we found that Tlr9−/− AM14 cells did not die at a different rate than WT AM14 cells ex vivo, arguing against death as a cause for the lower ELISpot counts (data not shown). To examine whether TLR9 had an effect on proliferation in AM14 CD22lo cells, we pulsed mice with BrdU 12hrs before sacrifice. However, preliminary data (3 mice per group) did not reveal differences in BrdU incorporation, making this explanation unlikely (data not shown).
If TLR9 (and TLR7) were important for late stage differentiation into AFCs, the large number of AM14 CD22lo cells could be indicative of a build up of pre-plasmablast cells that cannot fully differentiate (Culton et al., 2006), thus explaining the excess of CD22lo cells in the face of reduced numbers of bona fide AFCs. To address this theory, we assessed expression of differentiation markers on CD22lo AM14 cells from Tg Tlr9−/−, Tlr7−/−, and WT cells. No differences were found in levels of MHCII, CD38, CD80 or CD86 (data not shown). However, some surface molecules tested did demonstrate different expression in the TLR-deficient vs WT strains, including CD43, CD45, and CD138 (Supplementary Fig. 2).
B cell Intrinsic Expression of MyD88 is Required for Initial Proliferation of AM14 B cells
Multiple cell types are capable of MyD88-dependent TLR signaling. To determine whether such signaling was specifically required in B cells, we performed transfers of MyD88-deficient or WT B cells into WT While transferred MyD88-intact AM14 H Tg B cells differentiated into AFCs in WT recipient mice, MyD88-deficient B cells generated no detectable AFCs (Fig. 7A). This demonstrates an absolute requirement of MyD88 signaling in B cells for differentiation into AFCs. As differentation into AFCs is typically preceded by proliferation, MyD88 could be required for proliferation, differentiation, or both. To examine this, AM14 H Tg B cells were labeled with CFSE prior to transfer and then splenocytes from recipients given PL2-3 were analyzed by FACs. 4-44+ cells from MyD88-deficient donors proliferated little, if at all, whereas WT 4-44+ cells proliferated and expanded robustly (Fig. 7B and Supplemental Fig. 3). Thus, for the activation of the AM14 extrafollicular response MyD88 signaling is required within the B cells at the earliest stages of activation.
Figure 7. B cell intrinsic MyD88 is required for AM14 B cell differentiation and proliferation.
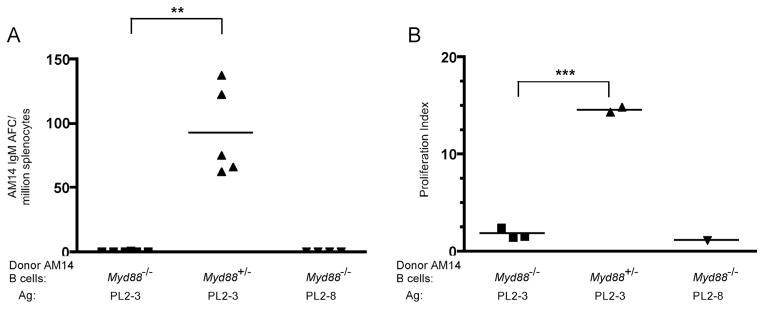
Myd88+/− or Myd88−/− AM14 Tg B cells were labeled with CFSE and transferred into Balb/c mice that were then treated with anti-chromatin. (A) 4-44 ELISpot assay of splenocytes. Data compiled from two experiments. ** p < 0.005 (Mann-Whitney test).
(B) Proliferation of 4-44+ splenocytes. CFSE labeling and flow cytometry was used to detect division (Supplemental Figure 3). Proliferation index was calculated by dividing the number of 4-44+ B cells which had divided by the number of undivided 4-44+ B cells.
*** p = 0.0001 (unpaired t-test).
DISCUSSION
It is becoming appreciated that autoreactive B cell responses often occur extrafollicularly, generating short-lived plasmablasts in dynamic anti-self responses (Weisel et al., 2007). Yet, knowledge is relatively limited concerning the requirements for initiation and regulation of this response. Here we have used the well-characterized AM14 system and our newly developed system for triggering extrafollicular RF responses to begin to dissect this mode of autoreactive B cell response. We report two important findings: that T cells are not required for initiation of RF responses but that TLR signals are essential. We also identify overlapping yet important roles for TLR7 and TLR9, along with qualitative alterations that occur in the absence of either receptor. Finally, we show a B cell-intrinsic requirement for MyD88 signaling, and that these signals are needed to initiate proliferation.
It has been thought that autoantibody responses in systemic autoimmunity require T cells (Peng et al., 1996a). Our results suggest that the role of T cells is considerably more complex than previously believed. Most strikingly, we found that T cells are dispensable for activation and differentiation of RF B cells to plasmablasts, provided that the proper antigen is available. Surprisingly, somatic hypermutation at extrafollicular sites also does not require T cells. On the other hand, we found that T cells do have a role in ongoing spontaneous responses, as anti-CD4 treatment of recently-converted mice caused a rapid, 50% reduction in the numbers of plasmablasts. Nonetheless, the residual RF B cell response was sustained for at least 4 weeks without further decay. It is interesting that although SHM could take place in the absence of T cells, the number of mutations per cell was higher in their presence. Whether this is due to direct effects of T cells in induction of higher amounts of AID or other required factors, or the sustenance of mutation-competent cells for more generations is not clear. These definite, albeit limited, consequences of CD4+ T cell inhibition--coupled with the fact that CD4+ T cells were not required for a complete extrafollicular plasmablast response to IgG2a anti-chromatin administration--suggests that T cells can amplify a response once initiated, but are not required for the response per se. The notion that T cells do regulate an ongoing response would reconcile our data with results from a variety of systems (Peng et al., 1996a; Peng et al., 1996b; Wofsy, 1993).
A potential caveat to this interpretation is that anti-CD4 treatment did not completely deplete CD4+ T cells. We believe that CD4+ cells that remained were functionally inhibited, as reported by others who demonstrated inhibition with nondepleting Abs (Carteron et al., 1989; Zelenika et al., 2001); residual CD4+ T cells in treated animals were completely opsonized with GK1.5 in vivo, and, most importantly, these cells had lost the ability to promote a classic T-dependent humoral response. Until there are methods to fully eliminate CD4+ T cells in lupus-prone mice in an inducible fashion, we cannot fully exclude that CD4+ T cells are absolutely (rather than relatively) necessary for propagation of an ongoing response.
That T cells do influence the response is consistent with data indicating that B cells play important roles in activating autoreactive CD4+ T cells, most likely via direct Ag presentation. Our data suggests that the extrafollicular autoreactive reaction is a relevant site of B-T interaction. Previously we found that some but not most RF B cells engaged in the spontaneous extrafollicular reaction were in contact with T cells (William et al., 2002; William et al., 2005a). This is consistent with the concept that extrafollicular plasmablast development is only partially T cell-dependent. In normal mice, the extrafollicular reaction can be involved in both T-independent and T-dependent responses to model antigens and pathogens (MacLennan et al., 2003). Indeed, an emerging theme is that T cells, if present, can promote such responses. For example, activated T cells that can directly collaborate with anti-DNA B cells are critical in stimulating these cells to differentiate at extrafollicular sites (Seo et al., 2002). Thus, together with data from others, our results suggest that the extrafollicular activation of autoreactive B cells could also result in productive B-T interactions. Presumably these interactions in turn lead to T cell activation, though the direct demonstration of this will likely require in vivo imaging. Exactly how T cells modify the extrafollicular autoreactive B cell response is much less clear and will require further investigation using a combination of inhibitors and genetic manipulation. Secretion of cytokines such as IL-4 and IL-21, T cell products which are known to promote isotype switch as well as B cell proliferation and differentiation (Good et al., 2006), are good candidates. Both CD40L and ICOS are parts of costimulatory pathways that are important for GC responses (Tarlinton, 2006), but their role in B-T interactions elsewhere are little investigated.
In the absence of a requirement for T cell help in the initiation of RF B cell extrafollicular plasmablast differentiation and SHM, we found that TLR signals were required. Importantly, the lack of response in TLR7, TLR9 double-deficient mice identifies these two as the critical (although potentially redundant) receptors and excludes other MyD88-dependent receptors. Our in vivo findings extend previous in vitro data showing that RF B cells must express MyD88 in order to proliferate in response to IgG2a anti-chromatin. The transfer studies demonstrated a requirement for MyD88 in B cells in vivo. Thus, while the need for direct TLR-mediated stimulation of B cells in the T-dependent GC response remains controversial (Gavin et al., 2006; Meyer-Bahlburg et al., 2007; Pasare and Medzhitov, 2005), it is clearly required for the extrafollicular plasmablast response to anti-chromatin ICs, providing support for the notion that B cells themselves must express relevant TLRs for at least some types of response (Pasare and Medzhitov, 2005).
Taken together, experiments presented here link the in vitro response to in vivo differentiation at the site that yields both anti-DNA and RF responses in autoimmune mice (Jacobson et al., 1995; Mandik-Nayak et al., 1999; William et al., 2002; William et al., 2005a). The findings establish a physiologic in vivo context for how TLRs promote activation of certain autoreactive B cells. Notably, prior in vitro cultures were harvested after two days and never progressed to differentiation (Leadbetter et al., 2002), so whether the TLR signal was required for differentiation to AFC function was unknown. Mechanistic studies revealed that, in vivo, AM14 cells lacking MyD88 failed to proliferate, indicating that MyD88-mediated signals are required for initial activation. Of significance, we show that, even in the presence of T cells, TLR-mediated activation is a non-redundant signal in vivo. In vitro cultures did not have T cells (or other accessory cells) (Leadbetter et al., 2002) and so it was only shown that TLRs were required when other potentially important cells were absent in the culture dish. The lack of clonal expansion, activation or differentiation in the absence of MyD88 indicates that therapeutic approaches to TLR inhibition should expect to block the earliest steps of autoreactive B cell activation.
It was somewhat surprising that responses were only modestly reduced in the TLR7 or TLR9 single-deficient mice. This finding could provide insight into the natural chromatin autoantigen in vivo as it is recognized by an authentic disease-associated autoantibody (e.g. PL2-3). It is possible that PL2-3 directly binds both DNA and RNA, allowing it to capture ligands for both TLR7 and TLR9. However, PL2-3 is reported to bind only chromatin or nucleosomes (Losman et al., 1993). Similarly, given the well-documented specificity of TLR7 for RNA and TLR9 for DNA (Heil et al., 2004; Hemmi et al., 2000; Lund et al., 2004), it is unlikely that TLR7 directly recognizes the DNA in chromatin. Rather, we suggest that the partial dependence on TLR7 indicates that the autoAg recognized by PL2-3 in vivo has both DNA and RNA components. This could be RNA in the process of transcription or RNA that associates with DNA-binding proteins or becomes associated during the processes of cell death and release of chromatin.
One unexpected result was that deficiency of TLR7 or TLR9 had a much more profound effect on fully-fledged AFCs than it did on the proliferating precursors of such cells. This was reflected in an almost ten-fold alteration in the ratio of these two cell types. We investigated a number of possible explanations for this result. Certain differentiation markers were consistently altered in the absence of either TLR7 or TLR9, and these differed between the two TLRs. Though the reason for this is unclear, this result does indicate that signaling by both TLRs is required for completion of differentiation, whereas it seems that signals through either one are sufficient to initiate proliferation and to permit some, albeit inefficient, differentiation to the terminal plasmablast. Unexpected dichotomies between TLR7 and TLR9 were also observed in lupus-prone MRL-lpr mice lacking one or the other TLR, in which TLR7-deficiency protected against but TLR9-deficiency promoted autoimmune disease (Christensen et al., 2006). Further work will be required to discover the details of the differential regulation of both autoantibody production and disease by these two related molecules.
Taken together, our findings suggest an order in which self-tolerance mechanisms break down, eventually leading to autoimmunity. B-T collaboration is currently envisioned as a positive feedback loop that amplifies autoreactive immune activation (Shlomchik et al., 2001; Weisel et al., 2007). However, there have been few clues as to whether autoreactive T or B cells are activated first and where these interactions take place. The finding that T cell activation is not required to generate autoantibodies directly supports the proposal that autoreactive B cell activation is an initial step in systemic autoimmunity, followed by subsequent activation of autoreactive T cells by B cells. However, we also find that T cells modify the extrafollicular reaction, suggesting that subsequent productive B-T interactions can occur at this site. This notion links to findings of others that autoreactive B cells can activate autoreactive T cells in vitro and in vivo (Lin et al., 1991; Mamula et al., 1994; Singh and Hahn, 1998). Finally, stimulation of TLR7 or TLR9 within RF B cells appears to be a critical requirement for activation of these cells whether or not T cell help is available. The central role of the autoreactive B cell in this paradigm could help to explain why B cell depletion is proving so effective in a wide array of autoimmune diseases (Browning, 2006).
EXPERIMENTAL PROCEDURES
Mice
The AM14 heavy chain Tg was backcrossed at least 10 generations onto the MRL-lpr background (William et al., 2002). Tcrb−/− MRL-lpr mice (Peng et al., 1996a) were obtained from Joseph Craft. Tlr9−/− MRL-lpr and Tlr7−/− MRL-lpr mice were described (Christensen et al., 2006). The MyD88 mutant allele (Kawai et al., 1999) was provided by Shizuo Akira and was backcrossed to MRL-lpr for >5 generations and then to Tg MRL-lpr mice before being made homozygous. The MyD88-deficient BALB/c mice were obtained from Dr. Ruslan Medzhitov (Yale) and were backcrossed at least 10 times. All mice were housed under specific pathogen-free conditions. PCR to genotype for heavy chain was as described (Shlomchik et al., 1993). For experiments involving anti-chromatin injection we used mice 7 weeks old and younger; it is rare for mice this age to have spontaneous RF B cell activation (William et al., 2005a). All experimental procedures were approved by the Yale Animal Care and Use Committee.
ELISA analysis
ELISA assays were performed as described (Wang and Shlomchik, 1999). Briefly, plates were coated with 10 μg/ml of anti-IgM (Chemicon) overnight in PBS, washed, and blocked. Serially diluted samples were incubated in duplicate for 1 hour, followed by washing, detection with anti-idiotype antibody, 4-44-biotin (0.25 μg/ml in 1% BSA/PBS), washing, incubation with streptavidin-alkaline phosphatase (Southem Biotechnology), and finally development with p-nitrophenylphosphate. Concentrations were determined using Deltasoft software (Biometallics) by comparison to standard curves on each plate.
ELISPOT Analysis
ELISpot analysis was performed as previously described (Hannum et al., 1996; Wang and Shlomchik, 1999).
Depletion of CD4+ T cells
For CD4+ T cell depletion, GK1.5 was produced and purified as described (Shlomchik et al., 1993). Mice were injected i.p. with GK1.5 or control rat gamma globulin (Rockland). For most experiments, mice were injected with 2.0 mg of antibody twice per week. For inhibition of the T-dependent response to NP-CGG, mice were treated with 4.0 mg/week for one week then immunized with 100 μg NP-CGG emulsified in alum, as previously described (Rossbacher and Shlomchik, 2003). Mice were then treated with GK1.5 (4.0 mg/week) for an additional 10 days, at which time T cell depletion and antibody production were determined.
Anti-Chromatin Hybridomas
The IgG2a and IgG2b anti-chromatin hybridomas, PL2-3 and PL2-8 (Losman et al., 1993), were obtained from Mark Monestier. For in vivo hybridoma growth, mice were first injected with 0.25ml of pristane (Sigma) on day 0 and 7. Hybridoma cells were injected (107 cells) i.p. on day 10, and mice were sacrificed on day 17 or 18. For cell transfer experiments ascites grown in BALB/c Rag-deficient mice was used as a source of PL2-3 or PL2-8, with dosages determined by quantitating IgG concentrations in the ascites by ELISA.
Antibody Reagents
Abs prepared in our laboratory as described (Shlomchik et al., 1993) were: 4-44 biotin (anti-AM14 idiotype), 4-44 FITC, 4-44 Alexa Fluor 647, 4-44 Alexa Fluor 488, 4-44 Alexa 568, GL1 (anti-CD86) FITC, 160A1 (anti-CD80) Alexa Fluor 488 and Pgp-1 (anti-CD44) Alexa Fluor 488. Anti-CD22.2 FITC, anti-CD22.2 PE, and anti-CD138 PE were from BD Pharmingen. BM8-Alexa647 (anti-F4/80) and A3-1 biotin (anti-F4/80) were from Caltag. Streptavidin-Alexa 647 and streptavidin-PE were from Molecular Probes. PNA-FITC was from Vector.
The anti-TCRδ depleting antibody UC7-13D5 (Houlden, 1989) was obtained from Zhinan Yin and Joseph Craft. 200μg (Hiromatsu, 1992) was administered i.p. the day before they hybridoma cell injection, and subsequently every other day until the mice were sacrificed.
Protein Immune Complexes
Immune complexes were generated by combining 125μg of 23.3 (IgG2a anti-NP) and 31.25μg of NP-CGG (per mouse) and incubating the mixture for 1 hour at 37 degrees. The ICs were then emulsified in alum as previously described (Rossbacher and Shlomchik, 2003) and injected i.p.
Flow Cytometry
Flow cytometric analysis was performed as described (Shlomchik et al., 1993). Data were collected on a FACSCalibur or an LSRII. Dead cells were excluded in all experiments using either propidium iodide or ethidium monoazide. Following surface staining with 4-44-biotin and Streptavidin-PE-Cy7, intracellular staining with 4-44 was was performed by fixation in 1% paraformaldehyde followed by permeabilization with Perm/Wash™ (BD Biosciences) and staining with 4-44 conjugated with Alexa 647 overnight.
Histology/Immunofluorescence
Sections of spleen were prepared and stained as described (Hannum et al., 2000). Nuclei were identified with 4′,6′-diamidino-2-phenylindole (Molecular Probes). Images were captured on an Olympus BX-40 microscope using a SPOT-RT Slider (Scanalytics) digital camera. 4-44-FITC or 4-44-biotin antibodies along with anti-FITC-alkaline phosphatase (Molecular Probes) and streptavidin-HRP (Southern Biotechnology Associates) were used for immunohistochemistry. Sections were developed with Fast Blue BB or 3-amino-9-ethyl-carbazole (Sigma-Aldrich), as described (Hannum et al., 1996).
Laser Microdissection
Splenic sections were cut from OCT embedded tissues onto Leica PEN-membrane 2.0μm slides and stained to identify extrafollicular AM14 B cells. AM14 B cell clusters (10–30 cells) were dissected using a Leica LMD6000 and cells were digested for 1 hr at 55°C in 10 μl of 0.8 mg/ml proteinase K, 50 mM Tris, pH 8, 50 mM KCl, 0.63 mM EDTA, 0.22% Igepal, and 0.22% Tween 20.
Sequencing
Sequencing was performed as described (William et al., 2002) except with Pfu Ultra rather than Pfu Turbo (Stratagene).
Cell Transfer
9 × 106 AM14 Balb/c B cells heterozygous or deficient for MyD88 were isolated, labeled with carboxy-fluorescein succinimide ester (CFSE, Invitrogen) according to manufacturer’s instructions, and transferred into BALB/c mice i.v. on day 0. B cells were purified from spleens using an Easy-Sep kit (StemCell Technologies) and were typically >95% pure. Ascites containing the equivalent of 500 μg PL2-3 or PL2-8 was given i.p. on days 0, 2, 4, and 6. Mice were sacrificed on day 8 and assessed as indicated.
Supplementary Material
Acknowledgments
We thank Dr. Joe Craft and Dr. Zhinan Yin for generously providing the Tcrb−/− mice and UC-7 Ab. We thank Michelle Horniak, Terrence Hunt, Melissa Baez and Denise Kent for outstanding animal husbandry. We thank the members of the Shlomchik lab autoimmunity group for many useful discussions and Ann Rothstein and Joe Craft for critical comments on the manuscript. Supported by NIH grants P01-AI36529 and P01-AR050256.
References
- Adams S, Leblanc P, Datta SK. Junctional region sequences of T-cell receptor beta-chain genes expressed by pathogenic anti-DNA autoantibody-inducing helper T cells from lupus mice: possible selection by cationic autoantigens. Proc Natl Acad Sci U S A. 1991;88:11271–11275. doi: 10.1073/pnas.88.24.11271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando DG, Sercarz EE, Hahn BH. Mechanisms of T and B cell collaboration in the in vitro production of anti-DNA antibodies in the NZB/NZW F1 murine SLE model. J Immunol. 1987;138:3185–3190. [PubMed] [Google Scholar]
- Browning JL. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nat Rev Drug Discov. 2006;5:564–576. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- Carteron NL, Schimenti CL, Wofsy D. Treatment of murine lupus with F(ab′)2 fragments of monoclonal antibody to L3T4. Suppression of autoimmunity does not depend on T helper cell depletion. J Immunol. 1989;142:1470–1475. [PubMed] [Google Scholar]
- Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Connolly K, Roubinian JR, Wofsy D. Development of murine lupus in CD4-depleted NZB/NZW mice. Sustained inhibition of residual CD4+ T cells is required to suppress autoimmunity. J Immunol. 1992;149:3083–3088. [PubMed] [Google Scholar]
- Culton DA, O’Conner BP, Conway KL, Diz R, Rutan J, Vilen BJ, Clarke SH. Early preplasma cells define a tolerance checkpoint for autoreactive B cells. J Immunol. 2006;176:790–802. doi: 10.4049/jimmunol.176.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields ML, Hondowicz BD, Metzgar MH, Nish SA, Wharton GN, Picca CC, Caton AJ, Erikson J. CD4+ CD25+ regulatory T cells inhibit the maturation but not the initiation of an autoantibody response. J Immunol. 2005a;175:4255–4264. doi: 10.4049/jimmunol.175.7.4255. [DOI] [PubMed] [Google Scholar]
- Fields ML, Nish SA, Hondowicz BD, Metzgar MH, Wharton GN, Caton AJ, Erikson J. The influence of effector T cells and Fas ligand on lupus-associated B cells. J Immunol. 2005b;175:104–111. doi: 10.4049/jimmunol.175.1.104. [DOI] [PubMed] [Google Scholar]
- Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science. 2006;314:1936–1938. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J Immunol. 2006;177:5236–5247. doi: 10.4049/jimmunol.177.8.5236. [DOI] [PubMed] [Google Scholar]
- Hannum LG, Haberman AM, Anderson SM, Shlomchik MJ. Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J Exp Med. 2000;192:931–942. doi: 10.1084/jem.192.7.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum LG, Ni D, Haberman AM, Weigert MG, Shlomchik MJ. A disease-related RF autoantibody is not tolerized in a normal mouse: implications for the origins of autoantibodies in autoimmune disease. J Exp Med. 1996;184:1269–1278. doi: 10.1084/jem.184.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Herlands RA, William J, Hershberg U, Shlomchik MJ. Anti-chromatin antibodies drive in vivo antigen-specific activation and somatic hypermutation of rheumatoid factor B cells at extrafollicular sites. European Journal of Immunology. 2007;37:3339–3351. doi: 10.1002/eji.200737752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiromatsu K. A protective role of gamma/delta T cells in primary infection with Listeria monocytogenes in mice. J Exp Med. 1992;175:49–56. doi: 10.1084/jem.175.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden BA. A TCR gamma delta cell recognizing a novel TL-encoded gene product. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):45–55. doi: 10.1101/sqb.1989.054.01.006. [DOI] [PubMed] [Google Scholar]
- Jacobson BA, Panka DJ, Nguyen KA, Erikson J, Abbas AK, Marshak-Rothstein A. Anatomy of autoantibody production: dominant localization of antibody-producing cells to T cell zones in Fas-deficient mice. Immunity. 1995;3:509–519. doi: 10.1016/1074-7613(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Kleinstein SH, Louzoun Y, Shlomchik MJ. Estimating hypermutation rates from clonal tree data. J Immunol. 2003;171:4639–4649. doi: 10.4049/jimmunol.171.9.4639. [DOI] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- Lin R-H, Mamula MJ, Hardin JA, Janeway CA., Jr Induction of autoreactive B cells allows priming of autoreactive T cells. J Exp Med. 1991;173:1433–1439. doi: 10.1084/jem.173.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman MJ, Fasy TM, Novick KE, Monestier M. Monoclonal autoantibodies to subnucleosomes from a MRL/Mp-+/+ mouse: oligoclonality of the antibody response and recognition of a determinant composed for histones H2A, H2B, and DNA. J Immunol. 1992;148:1561–1569. [PubMed] [Google Scholar]
- Losman MJ, Fasy TM, Novick KE, Monestier M. Relationships among antinuclear antibodies from autoimmune MRL mice reacting with histone H2A-H2B dimers and DNA. Int Immunol. 1993;5:513–523. doi: 10.1093/intimm/5.5.513. [DOI] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan ICM, Toellner K-M, Cunningham AF, Serre K, Sze DMY, Zuniga E, Cook MC, Vinuesa CG. Extrafollicular antibody responses. Immunol Rev. 2003;194:8–18. doi: 10.1034/j.1600-065x.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- Mamula MJ, Fatenejad S, Craft J. B cells process and present lupus autoantigens that initiate autoimmune T cell responses. J Immunol. 1994;152:1453–1461. [PubMed] [Google Scholar]
- Mandik-Nayak L, Seo SJ, Sokol C, Potts KM, Bui A, Erikson J. MRL-lpr/lpr mice exhibit a defect in maintaining developmental arrest and follicular exclusion of anti-double-stranded DNA B cells. J Exp Med. 1999;189:1799–1814. doi: 10.1084/jem.189.11.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Bahlburg A, Khim S, Rawlings DJ. B cell intrinsic TLR signals amplify but are not required for humoral immunity. J Exp Med. 2007;204:3095–3101. doi: 10.1084/jem.20071250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- Peng SL. Pathogenesis of autoimmunity in alphabeta T cell-deficient lupus-prone mice. Clin Exp Immunol. 1998;111:107–116. doi: 10.1046/j.1365-2249.1998.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SL, Madaio MP, Hayday AC, Craft J. Propagation and regulation of systemic autoimmunity by gammadelta T cells. J Immunol. 1996a;157:5689–5698. [PubMed] [Google Scholar]
- Peng SL, Madaio MP, Hughes DP, Crispe IN, Owen MJ, Wen L, Hayday AC, Craft J. Murine lupus in the absence of alpha beta T cells. J Immunol. 1996b;156:4041–4049. [PubMed] [Google Scholar]
- Rossbacher J, Shlomchik MJ. The B cell receptor itself can activate complement to provide the complement receptor 1/2 ligand required to enhance B cell immune responses in vivo. J Exp Med. 2003;198:591–602. doi: 10.1084/jem.20022042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadanaga A, Nakashima H, Akahoshi M, Masutani K, Miyake K, Igawa T, Sugiyama N, Niiro H, Harada M. Protection against autoimmune nephritis in MyD88-deficient MRL/lpr mice. Arthritis Rheum. 2007;56:1618–1628. doi: 10.1002/art.22571. [DOI] [PubMed] [Google Scholar]
- Seo SJ, Fields ML, Buckler JL, Reed AJ, Mandik-Nayak L, Nish SA, Noelle RJ, Turka LA, Finkelman FD, Caton AJ, Erikson J. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity. 2002;16:535–546. doi: 10.1016/s1074-7613(02)00298-4. [DOI] [PubMed] [Google Scholar]
- Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001;1:147–153. doi: 10.1038/35100573. [DOI] [PubMed] [Google Scholar]
- Shlomchik MJ, Zharhary D, Saunders T, Camper S, Weigert M. A Rheumatoid factor transgenic mouse model of autoantibody regulation. Int Immunol. 1993;5:1329–1341. doi: 10.1093/intimm/5.10.1329. [DOI] [PubMed] [Google Scholar]
- Singh RR, Hahn BH. Reciprocal T-B determinant spreading develops spontaneously in murine lupus: implications for pathogenesis. Immunol Rev. 1998;164:201–208. doi: 10.1111/j.1600-065x.1998.tb01221.x. [DOI] [PubMed] [Google Scholar]
- Tan EM. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv Immunol. 1989;44:93–151. doi: 10.1016/s0065-2776(08)60641-0. [DOI] [PubMed] [Google Scholar]
- Tarlinton D. B-cell memory: are subsets necessary? Nat Rev Immunol. 2006;6:785–790. doi: 10.1038/nri1938. [DOI] [PubMed] [Google Scholar]
- Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19:837–847. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- Wang H, Shlomchik MJ. Autoantigen-specific B cell activation in Fas-deficient rheumatoid factor immunoglobulin transgenic mice. J Exp Med. 1999;190:639–649. doi: 10.1084/jem.190.5.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel F, Wellmann U, Winkler TH. Autoreactive B cells get activated in extrafollicular sites. Eur J Immunol. 2007;37:3330–3333. doi: 10.1002/eji.200737971. [DOI] [PubMed] [Google Scholar]
- William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- William J, Euler C, Leadbetter E, Marshak-Rothstein A, Shlomchik MJ. Visualizing the onset and evolution of an autoantibody response in systemic autoimmunity. J Immunol. 2005a;174:6872–6878. doi: 10.4049/jimmunol.174.11.6872. [DOI] [PubMed] [Google Scholar]
- William J, Euler C, Primarolo N, Shlomchik MJ. B cell tolerance checkpoints that restrict pathways of antigen-driven differentiation. J Immunol. 2006;176:2142–2151. doi: 10.4049/jimmunol.176.4.2142. [DOI] [PubMed] [Google Scholar]
- William J, Euler C, Shlomchik MJ. Short-lived plasmablasts dominate the early spontaneous rheumatoid factor response: differentiation pathways, hypermutating cell types, and affinity maturation outside the germinal center. J Immunol. 2005b;174:6879–6887. doi: 10.4049/jimmunol.174.11.6879. [DOI] [PubMed] [Google Scholar]
- Wofsy D. Treatment of murine lupus with anti-CD4 monoclonal antibodies. [Review] Immunology Series. 1993;59:221–236. [PubMed] [Google Scholar]
- Wofsy D, Seaman WE. Reversal of advanced murine lupus in NZB/NZW F1 mice by treatment with monoclonal antibody to L3T4. J Immunol. 1987;138:3247–3253. [PubMed] [Google Scholar]
- Zelenika D, Adams E, Humm S, Lin CY, Waldmann H, Cobbold SP. The role of CD4+ T-cell subsets in determining transplantation rejection or tolerance. Immunological Reviews. 2001;182:164–179. doi: 10.1034/j.1600-065x.2001.1820113.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.