

Abstract
B lymphocytes producing high affinity antibodies (Abs) are critical for protection from extracellular pathogens, such as bacteria and parasites. The process by which high affinity B cells are selected during the immune response has never been elucidated. Though it has been shown that high affinity cells directly outcompete low affinity cells in the germinal center (GC)2, whether there are also intrinsic differences between these cells has not been addressed. It could be that higher affinity cells proliferate more rapidly or are more likely to enter cell cycle, thereby outgrowing lower affinity cells. Alternatively, higher affinity cells could be relatively more resistant to cell death in the germinal center. By comparing high and low affinity B cells for the same antigen, we show here that low affinity cells have an intrinsically higher death rate than cells of higher affinity, even in the absence of competition. This suggests that selection in the GC reaction is due at least in part to the control of survival of higher affinity B cells and not by a proliferative advantage conferred upon these cells compared to lower affinity B cells. Control over survival rather than proliferation of low and high affinity B cells in the GC allows greater diversity not only in the primary response but also in the memory response.
Introduction
High affinity B cells develop in GCs. Early in immune responses, most responding B cells have low affinity for antigen (Ag) and their V gene repertoire is very diverse (1–4). As the GC reaction progresses, somatic hypermutation of the B cell receptor (BCR) generates relatively rare higher affinity variants (5–7). Through processes that are poorly understood, these rare B cells with higher affinity BCRs are selected and their progeny increase, eventually populating the high affinity memory and plasma cell pools (8). As important as this process—known as affinity maturation—is for the generation of adaptive immunity, the mechanism for selecting higher affinity clones out of the diverse collection of V regions and subsequent mutants has never been elucidated.
For selection of B cells with high affinity BCRs to occur, a low affinity BCR must function differently than a higher affinity BCR, via its signaling function or its ability to capture Ag for subsequent presentation on MHC II or both. These affinity-dependent functions of the BCR could either differentially promote the activation or prevent the death of higher affinity B cells. Indeed, there is both extensive proliferation and death occurring in the GC (9, 10). It has been suggested that T cell signals participate in selection in the GC (11). T cell signals in the GC include CD40L (12), which can also rescue GC B cells from death in vitro (10); however, CD40L is a potent mitogen for B cells in addition to any pro-survival effects (13, 14). Similarly, in vitro, T cells promote B cell proliferation rather than rescue them from cell death (15), in contrast to signals from BAFF, a myeloid cell product (16) that prevents cell death and is important for GC development (17). Ectopic overexpression of Bcl-2-family antiapoptotic proteins does inhibit apoptosis in the GC, along with a number of other perturbations of B cell development and immune response (17). With bcl-xL Tg overexpression, affinity maturation of AFCs was subverted (18), but this was not observed in bcl-2 Tg mice, in which it seemed there was premature differentiation into memory cells instead (19). Thus, there is not agreement on the effects of preventing normal B cell and GC death by overexpression of anti-apoptotic genes. In any case, though these experiments partly support cell death as an important selective mechanism, they do not show the relative physiologic roles of death and proliferation in overall GC selection.
Shih et al elegantly showed that when placed in direct juxtaposition, high affinity cells will dramatically outcompete low affinity cells in the GC (20). However, whether there is an intrinsic difference between high and low affinity B cells in the GC, apart from influences of competition, is much less clear. The separate contributions of proliferation and death in the positive selection process have never been directly measured as a function of affinity. Such measurements would provide fundamental insights into the dynamics of GCs and how high affinity B cells are generated, as well as shed light on the differential signals that are used to discriminate low from high affinity B cells.
This issue cannot be addressed in normal mice because the B cell immune response is very heterogeneous and it is difficult to follow a single B cell. Even if one could track a single B cell, it is impossible to study the effect of affinity on selection since somatic hypermutation can change the affinity of the BCR during the course of an immune response. To address these issues and determine a cell-intrinsic basis for positive selection in the GC, we have used conventional IgH transgenic (Tg) mice to freeze the repertoire and affinity of a defined population of B cells. These transgenes do not mutate or isotype switch, in contrast to site-directed transgenes (21). We made two Tg mice with VH genes that, when paired with endogenous λ1 light chains, encode BCRs of moderate or very low affinities for the nitrophenyl (NP) antigen (22, 23). We crossed the medium affinity (B1-8, ~Ka of 9.64×105 M−1) and the very low affinity (V23, ~Ka of <5.0×104 M−1) IgH Tg mice onto a Jh knockout background, which ensures that all BCRs use the heavy chain Tg exclusively. Immune responses in these two Tg mice thus represent the fates of moderate and very low affinity B cells in GCs that would be difficult to observe in a wild-type GC. We were therefore able to assess the roles of proliferation and death in the selection of low and medium affinity B cell clones by tracking the λ-light chain bearing population during an immune response to the same antigen.
Materials and Methods
Mice and Immunizations
V23 and B1-8 IgH transgenic mice (22, 23) were backcrossed with Jh KO/Balb mice (22, 24) for nine or more generations. All mice were maintained under SPF conditions and used at 6–10 weeks of age. Mice were immunized intraperitoneally with 50ug of NP25-chicken gamma globulin (CGG) precipitated in alum or precipitated alum alone as a control. All animal experiments were approved by the Yale Institutional Animal Care and Use Committee.
Histology
Freezing, sectioning and staining were performed essentially as described (22). 5uM spleen sections from V23 and B1-8 mice were cut and thaw mounted onto Poly-L Lysine-coated slides. Sections were stained with PNA-biotin (Vector Labs), using streptavidin-horseradish peroxidase (Molecular Probes) as the secondary reagent, and goat anti-lambda-alkaline phosphatase (Southern Biotech). Sections were developed with Fast Blue BB and 3-amino-9-ethyl-carbazole (Sigma).
Flow cytometry
Single cell suspensions of spleens were made at the indicated days post immunization. Cells were stained with the appropriate antibodies for 25 minutes on ice in PBS containing 3% calf serum, 0.05% sodium azide. Live/dead discrimination was done using ethidium monoazide (EMA) according to the manufacturer’s protocols (Molecular Probes). The following antibodies were used: fluorescein-PNA (Vector Labs), goat anti-λ and goat anti-κ (Southern Biotech), which were conjugated to AlexaFluor™ 647 and AlexaFluor™ 405 (Molecular Probes), respectively. Samples were analyzed on either a BD FACSCalibur or FACSAria (Becton Dickinson).
BrdU Detection and Apoptosis Assays
For BrdU labeling, mice were given an intraperitoneal injection of 3mg of BrdU (Sigma) 13 days post immunization and at the times indicated, were sacrificed. For the 24-hour time point, two injections of 3mg of BrdU were given 12 hours apart to ensure that BrdU was not limiting. Cells were stained for surface markers as described above and then fixed in ethanol (added dropwise to a 0.15M NaCl solution to a final concentration of 70% ethanol) for 30 minutes on ice. After washing once, cells were further treated with 1% paraformaldehyde, 0.1% Tween-20 for 30 minutes at room temperature and then overnight at 4C. Fixed cells were then treated with 100 Kunitz units of DNAse (Sigma) in 0.15M NaCl/4.2mM MgCl2 for 30 minutes at room temperature prior to the addition of anti-BrdU-biotin (Phoenix Flow). Streptavidin-phycoerythrin (Molecular Probes) was used as the secondary. For detection of apoptosis in situ, cells were incubated with zVAD-FMK-Fluorescein (Casp-Glow, Biosource) in RPMI for 45 minutes at 37C and washed according to the manufacturer’s protocol. Cells were then stained as described above for surface markers and/or for detection of BrdU label.
Sequencing
λ+ GCs were microdissected from stained spleen sections at the indicated days post immunization essentially as described (22) or using a Leica laser capture microdissection instrument. Cell clusters (typically 10–50 cells) were digested overnight at 37 C in 10μL of digestion buffer (50 mM Tris pH 8, 50 mM KCL, 0.63 mM EDTA, 0.22% NP-40, 0.22% Tween-20) containing 0.8 mg/ml proteinase K (Novagen). Vλ1 sequences were amplified by nested PCR using Pfu Turbo® polymerase (Stratagene) using external primers 5′-GCACCTCAAGTCTTGGAGAG-3′ and 5′-ACTCTCTCTCCTGGCTCTCA-3′, and internal primers 5′-CTACACTGCAGTGGGTATGCAACAATGCG-3′ and 5′-GTTCTCTAGACCTAGGACAGTCAGTTTGG-3′. Amplified DNA was cloned directly into pCR®4 Blunt-TOPO® vector using the Zero Blunt® TOPO® PCR Cloning Kit for Sequencing (Invitrogen). Vλ1 DNA was further amplified by placing colonies directly into PCR reactions containing the following primers: M13 fwd 5′-GTAAAACGACGGCCAG-3′ and M13 rev 5′-CAGGAAACAGCTATGAC-3′. DNA was purified from the PCR reaction mixture with the QIAquick® PCR Purification kit (Qiagen), mixed with sequencing primer, T3 5′-AATTAACCCTCACTAAAGGG-3′, and sequenced by the Keck Biotechnology Resource Laboratory at Yale University School of Medicine. We typically recovered 8 sequences per microdissection. Sequences were aligned to a rearranged germline Vλ1/Jλ1 sequence using Lasergene DNA analysis software and all mutation data were compiled into a database program written in Filemaker® Pro.
Site directed mutagenesis and Ab expression
We designed sets of 2 complementary primers matching a template in the Vλ1 gene cloned to pSV2Neo and containing the desired mutation(s). We used the quickChange ™ XL – site directed mutagenesis kit (Stratagene) and PCR to generate mutant template, followed by Ultra-competent XL-10 Gold cell transformation. Mutations were confirmed by sequencing in both directions (Keck DNA sequencing facility). pSV2-Neo germline or mutated Vλ1 was co-transfected with B1.8 or V23 pEVH-Cγ1 (3) into the SP2/0 cell line by electroporation at 960MF and 240 mV using a BioRad Gene Pulser. Transfected cells were grown in RPMI1640 media with 10% FBS in the presence of Geneticin (G418). Positive colonies were screened for secretion of functional antibody by ELISA. Plates were coated with anti-mouse IgG overnight at 4°C, then washed, blocked, and incubated with cell supernatants. The presence of IgG1λ was detected by alkaline phosphatase conjugated anti-mouse λ (Southern Biotech) and pNPP as substrate. IgG1λ in cell supernatant from transfected cells was purified on protein G columns and the Ab concentration was adjusted to 5μg/ml. We performed SDS-PAGE to check purified Abs for integrity and purity.
NP-binding ELISA
ELISA was used to test binding of IgG1 germline and mutant Vλ1-expressing Abs. Immulon-2 (Fisher) plates were coated with 10μg/ml NP27-BSA or NP20-CGG overnight in PBS at 4°C. Serial dilutions of purified IgG1 with germline or mutated Vλ were added with starting concentration of 5 or 2 μg/ml. Germline antibody was added to each plate for direct comparison. After incubation and washing, bound Ab was detected by alkaline phosphatase conjugated anti-mouse IgG and pNPP substrate using an ELISA reader with detection at 405 as a ratio to absorbance at 630 nm.
Modeling methods
The GC population dynamics were modeled by the set of ordinary differential equations shown in Supplementary Equations. It is reasonable to assume that the population is at steady-state, since we have observed that the antigen-specific GC population remains approximately constant from days 9–15 post-immunization (data not shown), and we are modeling observations on day 13. The basic model does not include inflow (sin) or outflow (sout), while the in/out-flow model allows for either inflow or outflow of cells from each mouse strain. To achieve steady-state in the basic model or in the model including outflow, the rate at which cells return to the dividing compartment (r) is calculated to maintain a constant population size (see list of constraints in Supplementary Table 1). The initial size of the compartments are set so that A+B+C=1. Models including inflow (sin> 0) naturally achieve steady-state. In this case, the parameter r is chosen as part of the optimization and the rate of inflow (sin) is calculated so that the total size of the germinal center population is one (i.e., A+B+C=1). BrdU labeling is modeled by allowing dividing cells (subset A) to transition from the unlabeled (AU) to the labeled compartment (AL) (see Supplementary Equations). Integration of the model equations was performed with the LSODA algorithm (25) with algorithmic enhancements and Matlab interface developed by The BioAnalytics Group LLC. Global optimization of parameter values was performed using differential evolution with modified sampling (26) to minimize the sum-squared relative error between model and experiment.
Construction of B cell lineages
To construct B cell lineages we first identified sets of sequences that were clonally related using a set of computer algorithms we developed to handle the specific circumstances of Ig hypermutation analysis (manuscript in preparation). By combinatorial matching of the end regions of Vλ1 and Jλ1, the algorithm determined all possible V-J junctions, including those potentially generated by P-nucleotides. This allowed us to differentiate between junctional diversity and somatic hypermutation in the region of the V-J junction, as those bases that could not be accounted for by any combination of the germline sequences were considered to be mutations. Junctional diversity was then used to separate independent clones that may have been found in the same microdissection. Since there is relatively little diversity at V-J junctions and virtually no N-region addition, there are multiple independent examples of the same junction among Vλ1 sequences. Therefore, sequences that shared one of a few very common junctions were considered independent unless they also shared at least one mutation, in which case they were considered clonally related. The computer algorithm was also used to identify independent parallel mutations, which in most cases were attributed to hybridization in the PCR amplification process and were thus discarded. Since isolated independent parallel mutations are known to occur, particularly in hotspots, commonly observed single mutations seen in parallel were not discarded unless other evidence indicating PCR hybridization was found. Sequences were divided into CDR and FW regions according to our published methodology (27) based on Kabat and IMGT (27–29). Once clonality was determined a lineage was constructed of the clonally related mutants using maximum parsimony criteria (30).
Statistics
Simple statistical tests used are indicated adjacent to p-values in the text. We used a Monte Carlo approach to estimate the probability of finding a mutation in X trees out of Y independent lineages. 10,000 sets of Y lineages were generated by randomly assigning mutations to each branch, while maintaining the overall shape and mutation counts of each lineage. The p-value was calculated by determining the number of times that any mutation appeared in X or more trees for each set. A one-tailed Fisher’s Exact Test based on a 2×2 contingency table was used to determine whether a mutation appeared on non-terminal branches more frequently than expected. To carry out the analysis, the particular mutation being tested was compared to the set of all other mutations appearing in lineages with non-terminal branches. Each mutation was categorized as terminal (appearing on a branch with no children) or non-terminal (appearing on a branch with children). Selection of key mutations between days 10 and 16 was determined using a one-tailed Fisher’s exact test comparing the number of lineages with and without such mutations at each day.
Results
Very low affinity B cells do participate in GC reactions, though the low affinity GC population is smaller in size than that of the medium affinity GC population
In response to immunization with NP-CGG, an increase in λ+ (i.e. Ag-specific) GC B cells could readily be observed in V23 mice, though there was a lower frequency of GCs that contained λ+ B cells in the V23 mice compared to the B1-8 mice (Fig. 1) and a lower frequency of λ+ GC B cells overall as assessed by FACS (Fig. 2). In contrast to GCs in B1-8 mice, which were homogenously filled with λ+ cells, the GCs in V23 mice contained mixtures of both λ+ and κ+ cells. It is likely that most of the V23 κ+ B cells in the GC were specific for the carrier or environmental Ags rather than NP, as immunization with alum alone produced λ− (i.e. κ+) GCs (Fig. 1E, F, (22, 23)). This interpretation is supported by the fact that, on FACS analysis, κ+ cells did not co-stain with NIP-conjugated phycoerythrin (data not shown). Consistent with the histology, the frequency of λ+PNA+ B cells in the V23 mice was 5 to 10-fold lower than in the B1-8 mice as determined by FACS on days 10, 13 and 16 post immunization (Fig. 2, all differences between V23 and B1-8 mice p < 10−6). The fraction of the total λ+ population that was in GCs was also lower in the V23 mice on these days (Fig. 2). GC B cells demonstrated the expected reduction in surface Ig expression (as measured by levels of λ, Fig. 3). The identity of these λ+ cells as GC cells was further confirmed in some experiments by their characteristic increased expression of CD95 (31, 32), as revealed by multi-color flow cytometry (data not shown). These data suggested that although V23 λ+ B cells could participate in GCs, these cells were not being efficiently selected and/or recruited and this prevented them from accumulating to the same extent as the B1-8 B cells.
Figure 1. GCs in V23 mice contain fewer antigen-specific, λ+ B cells than GCs in B1-8 mice.

Immunohistological analysis of splenic sections from V23 and B1-8 mice immunized with NP-CGG 16 days before. Anti- λ (blue) identifies antigen-specific B cells and PNA (red) identifies GC B cells. (A and B, 40x magnification) (C and D) Higher power magnification (100x). (C) The composition of GCs in immunized V23 mice is a mixture of both λ+ and κ+ B cells. (E and F, 40x magnification) GCs are present in alum immunized mice, but there are very few λ+ B cells in these GCs.
Figure 2. Decreased frequency of λ+ GC B cells in V23 mice compared to B1-8 mice.
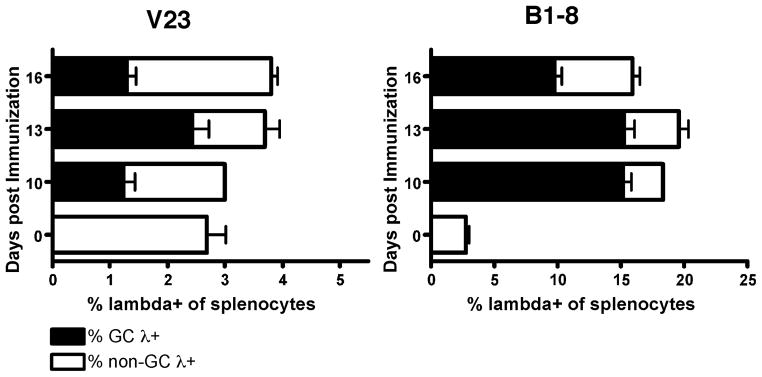
The frequency of live spleen cells that are λ+PNA+ or λ+PNA− as determined by FACS is shown. Numbers of spleen cells that are λ+PNA+ and PNA− correlate with the frequencies in the spleen (data not shown). Error bars represent SEM and n=8–16 mice per strain from at least 3 independent experiments for each day. All differences between V23 and B1-8 mice, p < 10−6.
Figure 3. V23 λ+ GC B cells proliferate to at least the same extent as B1-8 GC B cells.
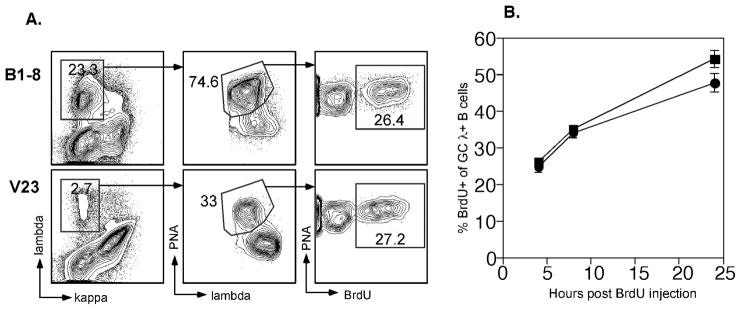
Mice were injected with BrdU 13 days post immunization and sacrificed at the indicated time points post injection. (A) Detection of BrdU-labeled GC B cells in V23 and B1-8 mice 4 hours post-injection. BrdU+ cells (third column) were gated from the PNA+λ+ cells (second column) that were originally gated on live λ+/κ − spleen cells (first column). (B) Summary of the percentage of BrdU+ cells among PNA+ λ+ cells in V23 (λ) and B1-8 (ν) spleens. Error bars represent SEM and n=6–9 mice per strain for each time point from at least 2 experiments. The differences between the two groups were not significant (p=0.47 and 0.64 for 4 and 8 hrs, but approached significance at 24 hrs (p=0.08) as assessed by two-tailed Student’s t test.
Direct measurement of proliferation and death in very low and medium affinity GCs
To determine the intrinsic mechanism of differential selection, we assessed the proliferation rate of the B1-8 and V23 B cells in the GC using bromodeoxyuridine (BrdU) incorporation (Fig. 3, which shows data from d13). Surprisingly, considering the differences in numbers of Ag-responsive GC cells, among λ+PNA+ cells, there were similar frequencies of V23 and B1-8 B cells that had incorporated BrdU after either 4 or 8 hours of labeling. Similar BrdU incorporation curves were obtained 10 and 16 days post immunization (data not shown). These findings were contrary to our expectation that lower affinity B cells would not proliferate as well as higher affinity B cells. Instead, the similar rates of BrdU incorporation for B1-8 and V23 cells suggested that above a low threshold for activation, the rate of division was maintained independently of BCR signal strength. Interestingly, the frequency of BrdU+ GC cells at 24 hours in V23 mice was marginally, yet not significantly, lower than in B1-8 mice (Fig. 3B). We reasoned that the trend toward a decrease at 24 hours in accumulated BrdU+ cells in the low affinity GCs could have been due to greater cell death in V23 mice. To address this, we measured the frequency of dying λ+PNA+ cells (Fig. 4), using an established assay for apoptosis that measured activated caspases (33–35). Strikingly, we found a three-fold increase in V23 GCs as compared to B1-8 GCs in the fraction of antigen-specific cells with detectable caspase activation, and therefore undergoing death. This was true at 10, 13 and 16 days post immunization (Fig. 4). Notably, many of these cells with elevated levels of active caspases were also permeable to ethidium monoazide (data not shown), again as expected from apoptotic cells (34). In addition, the bright signal of caspase activation is not consistent with the weaker levels of activation sometimes seen in proliferating cells (36). Interestingly, and in keeping with the notion that the κ+ cells in the GCs are specific for the carrier and likely of more average affinity, κ+ GC cells had approximately a 10% frequency of apoptotic cells in the V23 mice (Supplementary Fig. 1, average value of 9.3 +/− 1.2 %, n=3 mice), a value similar to the λ+ cells in the B1-8 and 3-fold lower than the λ+ cells in the same V23 mice; these cells thus served as an internal control in the V23 mice. Finally, we observed that both BrdU+ and BrdU− cells were undergoing cell death; after 4 hours of labeling, amond λ+ cells, V23 mice demonstrated caspase positivity in 30% of BrdU+ vs 23% of BrdU− cells and B1-8 mice had 15% caspase positive BrdU+ cells vs 9% of BrdU−. Taken together, these results thus demonstrate that the underlying mechanism of selection in the GC is the control of survival of lower affinity B cells and not control of cell division.
Figure 4. There is a higher frequency of V23 GC B cells undergoing apoptosis than B1-8 GC B cells.

(A) Representative flow cytometric profiles of spleen cells stained with PNA, λ, and zVAD-FMK-FITC. (B) The percentage of zVAD-FMK binding cells of the PNA+ λ+ population was determined at 10, 13 and 16 days post immunization in V23 (λ) and B1-8 (ν) mice. Error bars represent SEM from n=3 mice at days 10 and 16 and n>=18 mice per strain at day 13. Day 13 data were compiled from 5 individual experiments. V23 and B1-8 mice were significantly different overall (p < 10−9). P-values for individual days were: day 10: 0.03; day 13: 8.5 × 10−8; day 16: 0.06.
Analysis of somatic mutation
In order to gain further insight into the nature of selection and affinity maturation in GCs that began with fixed low and higher affinity B cells specific for the same Ag, we conducted an extensive survey of endogenous rearranged Vλ1 sequences obtained from small numbers of cells microdissected from individual GCs (see Supplementary Table 1 for a summary) at days 10 and 16 post-immunization. An average of 9.8 and 8.3 sequences per GC from a total of 37 and 25 GCs were recovered from sections of 18 different V23 and 13 different B1-8 spleens, respectively. Lineage trees were constructed from mutation patterns as described in the Methods. As reported (37), analysis of the mutation patterns in sequences grouped according to day and mouse type show an excess of replacement (R) mutations in the CDR, indicating positive selection (p<0.05). Analyzing the frequency of non-conservative mutations among R mutations in the FW region revealed significant negative selection for such mutations in both V23 and B1-8 mice (27).
The most striking indication of Ag-selection is that certain key mutations occurred repeatedly for both mice; interestingly, these differed between V23 and B1-8. At day 10 in V23 mice, mutation 349 (A to T, Asn to Tyr) was found in 7 out of 37 lineages (p=0.003, likelihood of having 7 or more repeated mutations in 37 lineages). In addition, a very rare junction, CT(T/C)TGG, which is the equivalent of a non-conservative mutation (at position 353 from A to T producing His to Leu) was found in 8 out of 37 lineages (Fig. 5A). As there is no obvious way to create this junction from the germline sequence without the introduction of non-templated nucleotides, we considered that it was actually a somatic mutation. However, these junctions were readily observed in V23 mice that had been crossed to aicda-deficient mice (data not shown), which do not undergo mutation (38), confirming the origin as a junctional variation. This junction was not found among 21 sequences that were PCR amplified from lambda positive cells from non-immune V23 mice and hence probably occurs naturally at a frequency <5%. Importantly, at day 16 both of these features were further enriched among the surviving clones. 349 (A to T) was found in 18 out of 41 lineages in V23 mice (p=0.016, Fisher’s exact test) and the CTT/CTGG junction was found in 29 of 41 lineages (p= 1.6 × 10−5, Fisher’s exact test). At day 16 in V23 there was also an additional overrepresented mutation at position 170 (A to C or G, Asn to Ser or Val); this mutation was not found frequently at day 10.
Figure 5. VJ junctions differ dramatically between V23 and B1-8 mice.

Plotted is the number of B cell lineages that have specific junctions at day 10 (projecting upwards) and at day 16 (projecting downwards) in (A) V23 and (B) B1-8 mice.
In B1-8 GCs at day 10 only one mutation is overrepresented, at position 158 (G to A, Ser to Asn), which is found in 9 out of 51 B cell lineages (p<10−4, likelihood of having 9 or more repeated mutations in 51 lineages). This key mutation is not accompanied by the selection of a rare junction. At both day 10 and 16 all but two trees exhibit the canonical junction CA(T/C)TGG (Fig. 5B), which in all cases encoded the same amino acids. At day 16 this key mutant is even more prevalent (p=0.037, Fisher’s exact test), being found in 13 out of 35 lineages. In addition, as in V23 GCs, this key mutation is joined by secondary mutations at day 16, the most common of which is the mutation at position 164 from A to T (Fig 2A). Notably, the primary mutations in V23 and B1-8 GCs do not occur at mutational hotspots, as shown in Fig. 6B, and hence we attribute their initial enrichment and subsequent further purification to selection. This interpretation is further supported by the fact that the two mutations are mainly found associated only with their respective Vh’s, which would not be expected if their appearance were due to inherent high mutability.
Figure 6. Distribution of nucleotide substitions in V23 and B1-8 sequences recovered from GCs and days 10 and 16.

The number of B cell lineages in (A) V-23 and (C) B1-8 mice that have specific nucleotide mutations at days 10 (projecting upwards) and 16 (projecting downwards). The colors indicate distribution of these mutations amongst terminal (red/dark) and non-terminal (yellow/light). Statistical significance of mutation enrichment at the non-terminal branch is indicated by an asterisk (p < 0.05). Double asterisk (q < 0.05) reflects significance after correction for multiple testing. (B) shows the relative mutability scores (37) at each mutated position in (A) and (C). Regions corresponding to CDRs are indicated by the “C”. The black dotted lines indicate the average mutability in the different CDRs and FWRs.
Though it has been suggested that selection is ongoing rather than in a single step (39), there have been only a few and somewhat limited sequence analyses that track dynamic selection of defined B cells specific for a single Ag within the primary GC (40, 41). We find that, although the cell numbers in the GCs we studied do not change dramatically between days 10 and 16 in both of these strains, the clonal composition does, with substantially greater purification of rare mutants, a direct indication of ongoing and stepwise selection.
To test more directly for the continued evolution of clones, we analyzed the placement of the above recurrent mutations in B cell clonal lineages. Mutations that are important for clonal expansion or survival should preferentially be selected early in the life of successful clones. Therefore, such beneficial mutations should be found in trunks of lineage trees more often than expected at random; more generally, they should be found in so-called “non-terminal” branches, which include trunks and higher level main branches that have sub-branches above them. Amongst trees with multiple branch levels at day 16, we found that both the mutations at positions 158 in B1-8 (p = 3.1 × 10−6) and 349 in V23 (p = 6.8 × 10−5) are found on non-terminal branches significantly more frequently than expected (p<0.05). We further saw that the secondary mutations at position 164 in B1-8 (p = 7.2 × 10−3) and position 170 in V23 (p = 2.3 × 10−3) also appear on non-terminal branches significantly more than expected (Fig. 6A and C). Taken together, increased mutation frequency, progressive and substantial enrichment of key R mutations in CDRs, and the appearance of recurrent mutations in non-terminal branches, are all consistent with strong and ongoing selection both prior to day 10 and between days 10 and 16.
Effect of mutations on Ag binding
Despite the evident selection for certain mutations, it is less clear how they affect the phenotype and function of the B cells that harbor them. The premise of the systems we are using is to limit competition and affinity maturation by fixing the Vh region, which is thought to dominantly control affinity to NP (42, 43). Indeed, the persistently small size of the V23 relative to B1-8 GC reactions indicate that the V23 B cells, despite mutations in Vλ, do not evolve to become phenotypically similar to the B1-8 B cells. Nonetheless, mutations in Vλ could promote relative improvements within a restricted realm of affinities that was determined by the Vh. To test this, we reconstructed some of the key mutations, singly or in some combinations, and expressed them as IgG1 Abs with either V23 or B1-8, and then tested their binding to NP substrates in an ELISA assay. While such assays are not as sensitive a test of affinity as solution-based methods (23), they can distinguish gross differences. Perhaps in keeping with the very frequent usage of variant VJ junctions, IgG1 V23 combined with germline Vλ containing a “canonical” VJ join showed no detectable binding to NP, even with plates coated at 10μg/ml of NP27-BSA or NP20-CGG. However, when tested on NP20-CGG, the 349 (Asn to Tyr) mutant, either alone or in combination with other common mutations, showed markedly enhanced binding (Fig. 7A and data not shown). Most of the other mutations, in combination with 349 (Asn to Tyr), did not detectably improve binding further, with the exception of the variant junctional amino acid Leu (a product of the enriched rare junction), which had a modest enhancing effect (Fig. 7A and data not shown). This variant also had a modest but detectable effect by itself. Two mutations common in B1-8 but not in V23 GCs, 158 (Ser to Asn) and 233 (Ala to Val) had little effect in the context of V23. Although clearly selected for in vivo, these recurrent mutations found in the context of B1-8 provided little or no observable improvement in binding when tested with the B1-8 Vh on various concentrations and conjugation ratios of NP-BSA in vitro (data not shown). This likely reflects a lack of high-end sensitivity of the assay rather than that these recurrently selected mutants actually do not affect affinity. Most importantly, the improved avidity of the Asn to Tyr and His to Leu variants in the context of V23 did not nearly approach that of the germline B1-8 Ab, even when assayed on NP27-BSA at 10μg/ml (Fig. 7B). Hence, in the context of low starting affinity, recurrent and early amino acid changes did improve affinity, but even V23 cells harboring these retained markedly lower affinity than the germline starting cells in B1-8 mice.
Figure 7. Effects of recurrent Vλ1 mutations on NP-binding.

The indicated mutations were made in Vλ1 by site-directed mutagenesis and proteins expressed in combination with the V23 H chain expressed as an IgG1 (see Materials and Methods). Germline Vλ1 was expressed with either V23 or B1-8 H chains also as IgG1 for comparison. These were tested for binding to NP20-CGG by ELISA. OD405 is shown with points being averages of duplicates for each concentration on a single plate. (A) Comparison of various Vλ1 mutants and the germline in combination with V23. (B) Comparison of the germline Vλ1 in context of B1-8 or V23 with the best of the Vλ1 mutants in context of V23.
Mathematical model of low and high affinity GC cell kinetics
Given the evidence for selection but restricted effect on global affinity, we wanted to synthesize the BrdU and cell death data to develop a comprehensive model of how intrinsic affinity affects GC B cell turnover. We estimated the relative proliferation and death rates in V23 and B1-8 GCs by fitting a mathematical model to the day 13 BrdU labeling data, and the fraction of dying cells in V23 and B1-8 GCs (Figs. 3 and 4). To develop a better data set for fitting to the model, we also performed additional labeling experiments in which BrdU was administered twice daily for 48 or 72 hr (Fig. 8). Previous models of BrdU labeling (44) cannot be used to fit our data for two reasons. First, equating cell labeling with mitosis, as prior models do, will underestimate the fraction of labeled cells, because the time during which BrdU is active in our system (≤2 hours) is much less than a cell division time. Second, previous models did not explicitly track apoptotic cells. This subset constitutes a large fraction of the GC population in our system and is an important constraint.
Figure 8. Mathematical modeling of GC cell turnover.

(A) Depiction of model scheme and parameters. Estimated parameter values are shown in parenthesis (B1-8, V23), as are the steady-state relative population sizes for each of the compartments. As discussed in the text, this basic model assumes that inflow and outflow of cells from the GC is negligible at day 13. (B) Optimal fit of the model to the experimental BrdU and Casp-Glow labeling data reproduces well the observed BrdU staining (as well as the apoptotic fraction, 13% for B1-8 and 26% for V23 GC B cells at day 13 (see Supplementary Table 2 for parameter values). The optimization minimizes a sum of squares error function where the two sources of data (i.e., BrdU and Casp-Glow labeling) are weighted equally. Red squares are the B1-8 experimental data, while blue circles are the V23 experimental data (from Figure 4). The total population size was constrained to be constant (i.e., the population is at steady-state).
To model GC population dynamics, we divided the B cell population into three subsets (Fig. 8A): A (dividing cells, ~S/G2/M phases), B (non-dividing cells, ~G0/G1 phases) and C (Casp-Glow+, dying cells). Dividing cells proliferate at rate p per hour and die at rate dA per hour. During the labeling period (a fixed interval of Lt hours following each BrdU injection) these cells are labeled at rate Lr per hour. Following each division, both daughter cells enter the non-dividing compartment, where each can independently either die with rate dB per hour or re-enter the dividing subset at rate r per hour. Dying cells are cleared from the GC at rate α per hour. Equations predicting the fraction of BrdU-labeled and Casp-Glow+ cells were derived from this model (Supplementary Fig. 2). Global optimization using differential evolution with modified sampling (26) was applied to search for parameter values that produced the best (i.e., least-squares) fit between model and experiment. We included several constraints on the possible parameter values. First, based on published data (45–47) we restricted the doubling time for a GC B cell to be at least 6 hours (i.e., p ≤ log(2)/6 = 0.12). Second, the maximum BrdU labeling period (Lt) was set to 30 minutes since we and others previously observed that the fraction of BrdU+ cells does not increase significantly between 30 minutes and one hour (47, 48). Third, the minimum BrdU labeling rate (Lr) was set to 2.0 per hour since we and others previously observed that virtually all cells in S phase were labeled within 30 minutes (47, 48). Finally, the clearance rate for apoptotic cells (α), the BrdU labeling rate (Lr) and labeling time (Lt) were assumed to be equivalent for B1-8 and V23 GCs.
As shown in Fig. 8B, the basic model produces good fits with the observed BrdU labeling curves and fraction of Casp-Glow+ cells. Parameter values providing optimal fits are given in Supplementary Table 2. The labeling period (Lt) is predicted to be 25 minutes, which is consistent with experimentally observed labeling periods (47, 48). As might be expected from the steady-state activated caspase measurements, the overall death rate is substantially higher in V23 GCs. Despite higher death rates, the population size of V23 GCs remains constant because this is balanced by a higher proliferation rate (p). Thus, the model results are consistent with the conclusion that affinity maturation is driven by death, since both the division and death rates in V23 are faster than those predicted for B1-8 mice. Interestingly, these considerations imply that GCs are self-correcting or homeostatic in the sense that when filled with low affinity B cells there will be much more proliferation, whereas when affinity matures, proliferation will naturally slow down.
An interesting prediction of the model is the relatively long half-life (4.3 hours) for clearance of apoptotic cells. Indeed, a simple calculation illustrates why this is necessary in any event to account for the high fraction of observed Casp-Glow+ cells seen here, especially in V23 mice, and in other studies (49, 50). In order to maintain a constant population size, cleared cells must continually be replaced by newly apoptotic cells. If C% of the GC is Casp-Glow+ and we assume that the rest of the GC population is actively dividing, then the necessary proliferation rate to maintain a constant population is given by: p = [C/(1−C)]*α. Thus, if 30% of the GC is Casp-Glow+ and the half-life for dying cells is one hour, then the required doubling time is 2.3 hours, much faster than is thought possible. To achieve a minimum doubling time of 6 hours requires that dying cells have a half-life of at least 2.5 hours. The half-life predicted by the model is longer than this since not all cells in the GC are actively dividing. This conclusion is an inevitable consequence of the observation of high frequencies of activated caspase positive cells in the GC, and may reflect the fact that this assay identifies cells at the earliest stages of apoptosis as well as that, due to the high load of apoptotic cells, overall clearance in the GC may be relatively slow.
The model predicts that B cells in V23 GCs are dividing at their maximal rate of around every 6 hours. If the upper bound on proliferation rate were removed, then shorter doubling times would be predicted (6.2h for B1-8 and 1.7h for V23). While such high proliferation rates for cells in V23 mice are clearly unrealistic, doubling times slightly shorter than 6 hours are not unreasonable. Nevertheless, these results, even without a physiological constraint on doubling time, are consistent with the conclusion that affinity maturation is driven by death, since without this constraint death rates in V23 remain faster than those predicted for B1-8 mice. Thus, while the exact parameter values estimated by the model should be interpreted cautiously, the broad trends presented here were true across all of the simulations we ran and lead to the conclusion that lower affinity GC B cells have higher death rates with either similar or even faster proliferation rates compared to higher affinity GC B cells.
In comparing B1-8 and V23 GCs, a conundrum requiring explanation is how both populations can be at steady-state with similar BrdU labeling curves, but very different apoptosis rates (since the most basic model would predict that proliferation exactly balances death to achieve a constant population size). The optimal solutions to the model have a higher proliferation rate in V23 mice, which balances death, but also a smaller steady-state proportion of cells in S phase that limits the speed of labeling (47% A, 40% B and 13% C in B1-8 versus 37 % A, 37% B and 26% C in V23). However, an alternate explanation for these observations might be differential outflow of B cells from B1-8 GCs and/or inflow of B cells to V23 GCs. To explore these possibilities, we extended the basic model to allow for either an inflow of unlabeled cells into the dividing compartment at a rate of sin per hour or an outflow of cells from the non-dividing compartment at a rate of sout per hour. In order to limit the number of parameters in the model, for each mouse strain only inflow or outflow was allowed, but not both. The fit produced by these models was only slightly better than the basic model above and the main conclusions were unchanged (Supplementary Fig. 3 with parameter values in Supplementary Table 2). In these models, an outflow of cells is predicted for B1-8 GCs, while inflow is predicted for V23 GCs. Consistent with this, limited observations show far more memory phenotype cells in B1-8 than V23 mice >8 weeks post-immunization (unpublished observations). However, several points suggest that inflow is not a significant factor at this stage of the GC reaction. For example, B cell lineage trees showing persistently evolving clones with increasing numbers of somatic mutations, as we observe (see below), are unlikely to be generated under such conditions. Furthermore, FACS analysis at these time points indicates that most of the Ag-specific B cells have a mature GC phenotype, suggesting that inflow is not a significant contributor. Finally, it has recently been reported that new cells are only successful in joining the GC reaction when their affinity is similar to or higher than existing clones (51); since the existing clones in mature GCs at day 10 and 16 are already highly evolved compared to the germline (Fig. 6), this report suggests there should be little joining by new cells at the time points we investigated. Nevertheless, even accounting for inflow and outflow, the model predicts equivalent proliferation rates, along with higher death rates in V23 GCs compared with B1-8 GCs. Thus, as with all the other model variants we investigated, the main conclusions remain unchanged: selection is driven by a survival advantage.
Surface markers that may distinguish dividing cells
The existence of compartment B (quiescent, see Fig. 8 and Supplementary Fig. 2) can clearly be inferred by the labeling kinetics of the GC after multiple doses of BrdU; a flattening of the curve compared to predicted exponential labeling is evident by 24 hr (Fig. 3B and data not shown) and even after 48 hr of ongoing labeling there is a notable population that still has not taken up BrdU (Fig. 8B). However, it is difficult to distinguish cells in vivo that would be in compartment A (actively dividing) vs. compartment B. BrdU labeling could be used to approach this issue. However, since labeled cells can differentiate from compartment A to B (at rate p) and since cells in compartment B can return to the dividing pool (at rate r), the BrdU+ cells are not necessarily indicative of one compartment or another (Supplementary Fig. 2). Nonetheless, with shorter labeling times, compartment A cells should be enriched in the BrdU+ population. To test for marker expression that might correlate with proliferation, we assessed the expression of a series of relevant surface proteins on the GC cells of both V23 and B1-8 mice as a function of BrdU labeling (Table I). A number of markers were expressed at similar levels on both BrdU+ and BrdU− cells, including CD95 and CD38, which are known to be modulated on GC cells as a whole. Interestingly, two surface markers, CD86 (Fig. 9A, B) and CD23 (Fig. 9A and data not shown), were expressed at lower levels in BrdU+ cells. Though these observations further support the notion of heterogeneity within the GC and compartmentalization with respect to proliferation, it is possible that expression of these two proteins is simply regulated in a cell-cycle specific manner, rather than that they reflect distinct compartments of cells.
Table I.
Surface markers tested to determine differences between BrdU+ and BrdU− cells in the GC.
| Marker | Result from V23 and B1-8 mice |
|---|---|
| CD86 | BrdU+ cells have lower expression |
| CD23 | BrdU+ cells have lower expression |
| CD62L | No Difference |
| CD38 | No Difference |
| CD80 | No Difference |
| CD95 | No Difference |
| BLA-1 | No Difference |
Figure 9. CD86 and CD23 are expressed at lower levels on BrdU+ GC B cells as compared to BrdU-negative GC cells.

Mice were injected with BrdU 13 days post immunization and sacrificed 8 hours later. (A) Levels of CD86 (top row) and CD23 (bottom row) were compared between BrdU+ (black lines) and BrdU-negative (shaded histograms) GC B cells from V23 (left column) and B1-8 (right column) mice. Data is representative of 7–9 mice from 3 independent experiments. (B) Summary of the Mean Fluorescence Intensity (MFI) of CD86 expression on BrdU+ and BrdU-negative GC B cells. Results are shown for both the V23 and B1-8 strains from a representative experiment out of 2 independent replicates. Each symbol is an independent mouse from that experiment and the bar is the mean value.
Discussion
The GC is a site of active proliferation, cell death, somatic hypermutation, and cellular selection (52, 53). The outcome of this process is the generation of high affinity memory B cells and plasma cells, both of which are critical for long term and recall immunity. Yet, the underlying method by which GCs select higher affinity mutants and indeed how affinity affects the fate of B cells in the GC in general has not been elucidated, as recently pointed out (54). This is in part because GCs are a heterogeneous mix of cells that are undergoing active mutation, making it impossible to track fates of cells with defined affinity in normal mice. In addition, although competition between clones specific for the same Ag is clearly one factor that drives selection (20), this factor does not account for potential intrinsic differences in fates of B cells receiving strong vs. weak signals in the GC. To address how affinity affects B cell fate in the GC, we have used two H chain Ig Tg mouse models, both with specificity for the NP antigen but with markedly different affinities. Because these H chains do not mutate and because L chain mutations can have only limited effect on affinity, to a first approximation we were able to study the differential fates of high and low affinity B cells in the GC in the absence of substantial interclonal competition for the immunizing Ag.
Our work thus provides direct evidence of the intrinsic mechanism of selection in the GC. Most notably, we demonstrate that an increased rate of cell death, rather than a decreased proliferation rate, is a characteristic of lower affinity clones. Moreover, mathematical modeling of these data revealed that a model allowing for death and division of both dividing and non-dividing cells provided an excellent fit for these BrdU labeling and death data. The signals that control death and division in the GC are not well-defined. Mice with suboptimal CD19 signaling showed mainly proliferative defects—though death was not quantitated (55)—indicating that affinity for antigen provides qualitatively different inputs to GC cells than accessory signals provided via CD19, which transduces signals from the CD21/35 complex (56). While cross-linking of both BCR and CD40 of human tonsillar GC cells partially rescues cell death in vitro (10), these stimuli are also clearly mitogenic for B cells (57); thus, it has not been clear how to extrapolate these in vitro data to in vivo situations in which GC B cells are developing in the context of their normal milieu including FDCs and T cells.
Our data on the fates of high and low affinity B cells in the GC in vivo suggests new insights into the overall design of the B cell immune response and in particular the role of low affinity B cells that comprise the majority of the antigen-specific repertoire. Because low affinity B cells divide at least as quickly as high affinity B cells, they contribute to the initial GC response by diversifying the repertoire of antigen receptors. Critically, low affinity cells can increase their affinity for antigen by undergoing multiple rounds of mutation (6, 39). This would not occur if proliferation were differentially controlled by affinity; in that case, very low affinity cells would divide little if at all and thus would be unable to contribute to the mutated repertoire. Yet, despite the high division rates implied by our BrdU labeling data and the model fit to it, the low affinity cells do not accumulate because of their high death rate. The size of the dividing compartment remains small in the case of low affinity cells and thus utilizes minimal resources in the GC. Nonetheless, rare mutants with improved affinity will have a lower death rate and will begin to accumulate. Thus via constant cycling and mutation without accumulation, a large number of mutations can be tested in a short time. We believe that this is an important element of how GCs can so efficiently generate and select higher affinity mutants that was not previously appreciated (58–60).
Most importantly, the design of the B cell immune response that is suggested by our data and analysis allows low affinity precursors, in mutated form, to contribute to the memory population. While certain model antigen systems have been selected for study precisely because a relatively high affinity precursor clonotype happened to exist in the germline repertoire, we believe that the great majority of responding B cells in a normal situation will be of low affinity, as low affinity cells are undoubtedly more numerous. It would seem critical that such cells could ultimately contribute to the memory and long-term antibody compartment. Indeed, it has been long known that highly mutated clones that were only rarely, if ever, observed in the primary response often dominate the memory response, a phenomenon termed “clonal shift” (2, 61). This is likely the hallmark of an important contribution of what were initially very low affinity cells to the mutated memory compartment.
Our detailed analysis of mutations in multiple discrete GCs of both types of mice shed some further light on these processes. This analysis was unique in comparing B cells responding to the same Ag with different starting affinities, yet using the same Vl; it also largely filtered out the effects of interclonal competition to better reveal the potential of intraclonal affinity maturation. The use of microdissection allowed for reconstruction of many small local clones, permitting mapping of mutations to different branch levels. This in turn permitted assessment of when recurrent mutations were occurring. Coupled with reconstruction of these mutations, the approach unambiguously linked selection with actual improvements in affinity. The results are a direct demonstration that low affinity cells do indeed improve their affinity and survive and expand, in the absence of high affinity competition. Shih et al have already shown that high affinity cells will very effectively suppress low affinity ones in trans (20), a conclusion further bolstered by Schwickert et al (51). It is worth noting that the mechanisms for this have not yet been defined, and could differ from how intrinsically low affinity B cells fare in the GC, as shown in our current study.
These data revealed remarkably high R/S ratios, with significant enrichment of R in CDR; this was not unexpected although efficiency or R enrichment was rather striking. This indicates, even for low affinity B cells, without interclonal competition from intrinsically higher affinity cells, that selection is intense. Furthermore, the data provide three types of clear evidence that selection is an iterative process. First, there is ongoing enrichment of repeated (and in some cases proven affinity-enhancing) mutations between days 10 and 16. Second, at day 16 there are additional repeated mutations that emerge that were not (as) evident at day 10. Third, we developed a method to analyze the location in genealogic trees of key mutations, and found that these occurred non-randomly in the non-terminal branches, i.e. in the early rather than late phases of clonal evolution. This is in agreement with the view of the GC as cycling through a large amount of “mutational space” owing to high proliferation rates regardless of affinity, then selecting via death.
One of the most unexpected conclusions we reached based on mathematical modeling of GC BrdU labeling and apoptosis data was that, within the dividing compartment, V23 B cells divided at least as quickly and most likely more quickly than higher affinity B1-8 cells. Although this conclusion is an inference from the model, it is interesting to consider potential reasons for this. Higher division rates could be an inherent feature of B cells receiving low affinity signals, although this seems paradoxical. Alternatively, since low affinity GCs are in fact smaller, with many fewer proliferating cells, more resources in the GC niche would be available per cell, resulting in overall higher rates of proliferation. Since competition occurs mostly or exclusively between clones specific for the same Ag, rather than between separate Ags (40, 62, 63), at least some of these resources should be Ag-specific; this could include Ag itself, as well as FDC contacts, and T cells specific for the Ag; non-specific resources such as cytokines, and cellular nutrients could also contribute (64–66). If this is the reason for faster proliferation, then competition by higher affinity B cells in the same GC would suppress the proliferation rate of low affinity B cells, and indeed there is evidence for this (20, 23, 40). The present system minimizes competition between clones of different affinity for the same Ag, allowing the measurement of the parameters inherent to B cell affinity without this additional variable.
The observation that affinity–which in turn presumably affects signal strength and/or duration—has differential effects on proliferation and death also has implications for BCR signaling in GC cells. We propose that fates of GC B cells are controlled by (at least) two signals: one signal for proliferation that has a low threshold and which is not further sensitive to increases in affinity but which must remain above a minimum level, and one signal for sustaining cells that have initiated proliferation that is tuned according to the affinity of the BCR (i.e. the signal strength). Both of these signals must be transduced through the same receptor, the BCR. It is well known in fact that there are multiple signaling pathways downstream of the BCR and that, at least in mutants, they can be partially uncoupled from each other (67, 68). There is evidence for differential signaling for death, survival or proliferation through the T cell receptor as well (69, 70).
Immunologists typically refer to “GC B cells” as if they were a discrete and homogenous population. However, unlike almost every other cell type in the body, GC B cells have great heterogeneity in the nature of their key stimulatory receptor, the BCR. That cells of different affinities can have intrinsically different death and proliferation rates, as we show here, implies that there could be heterogeneity within the GC in addition to apoptosis and cell division. There could be other fates or functions that are controlled by BCR affinity; for example, selection into the memory or plasma cell differentiation pathways could be a function of BCR signal strength itself (71, 72). Surface marker analysis of GC B cells does indeed demonstrate phenotypic heterogeneity, the significance of which is poorly understood, but could reflect the outcomes of differential BCR-mediated signals (73, 74). Our data (Fig. 9) does indeed show some phenotypic differences between GC cells that have recently taken up BrdU and those that did not, though the functional significance of these differences is unknown. Similarly, the observation that cell death occurs in both the BrdU+ and BrdU− fractions indicates that both “centroblasts” (defined here as proliferating cells) and “centrocytes” (non-proliferating cells) are subject to selection. This is counter to some classsical notions of GC function (75) that envision only centrocytes being selected and thus subject to death. Given these unresolved issues, mouse models like ours that constrain BCR affinity should be very useful in efforts to further elucidate how B cell fate is governed by its ability to bind antigen.
Supplementary Material
Acknowledgments
We thank Michelle Horniak and the Yale Animal Resources Center husbandry staff for excellent work and attention to detail that made these studies possible.
Footnotes
Supported by grant R01-AI43603 from the NIH to M.J.S. The work of S.H.K. was supported in part by NSF IGERT grant DGE-9972930.
Abbreviations: GC: germinal center; Tg: transgene or transgenic; NP: nitrophenyl; CGG: chicken gamma globulin; EMA: ethidium monoazide; R: replacement (mutations); S: silent (mutations).
References
- 1.Chua MM, Goodgal SH, Karush F. Germ-line affinity and germ-line variable-region genes in the B cell response. J Immunol. 1987;138:1281–1288. [PubMed] [Google Scholar]
- 2.Clarke SH, Staudt LM, Kavaler J, Schwartz D, Gerhard WU, Weigert MG. V region gene usage and somatic mutation in the primary and secondary responses to influenza virus hemagglutinin. Journal of Immunology. 1990;144:2795–2801. [PubMed] [Google Scholar]
- 3.Dal Porto JM, Haberman AM, Shlomchik MJ, Kelsoe G. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J Immunol. 1998;161:5373–5381. [PubMed] [Google Scholar]
- 4.Eisen HN, Siskind GW. Variations in Affinities of Antibodies During the Immune Response. Biochemistry. 1964;155:996–1008. doi: 10.1021/bi00895a027. [DOI] [PubMed] [Google Scholar]
- 5.Jacob J, Przylepa J, Miller C, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B cells. Journal of Experimental Medicine. 1993;178:1293–1307. doi: 10.1084/jem.178.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kocks C, Rajewsky K. Stepwise intraclonal maturation of antibody affinity through somatic hypermutation. Proc Natl Acad Sci U S A. 1988;85:8206–8210. doi: 10.1073/pnas.85.21.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3- nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J Exp Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camacho SA, Kosco-Vilbois MH, Berek C. The dynamic structure of the germinal center. Immunol Today. 1998;19:511–514. doi: 10.1016/s0167-5699(98)01327-9. [DOI] [PubMed] [Google Scholar]
- 10.Liu YJ, Joshua DE, Williams GT, Smith CA, Gordon J, MacLennan ICM. Mechanism of antigen-driven selection in germinal centres. Nature. 1989;342:929–931. doi: 10.1038/342929a0. [DOI] [PubMed] [Google Scholar]
- 11.Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foy TM, Laman JD, Ledbetter JA, Aruffo A, Claassen E, Noelle RJ. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J Exp Med. 1994;180:157–163. doi: 10.1084/jem.180.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tangye SG, Avery DT, Hodgkin PD. A division-linked mechanism for the rapid generation of Ig-secreting cells from human memory B cells. J Immunol. 2003;170:261–269. doi: 10.4049/jimmunol.170.1.261. [DOI] [PubMed] [Google Scholar]
- 14.Dadgostar H, Zarnegar B, Hoffmann A, Qin XF, Truong U, Rao G, Baltimore D, Cheng G. Cooperation of multiple signaling pathways in CD40-regulated gene expression in B lymphocytes. Proc Natl Acad Sci U S A. 2002;99:1497–1502. doi: 10.1073/pnas.032665099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawakami K, Parker DC. Antigen and helper T lymphocytes activate Blymphocytes by distinct signaling pathways. European Journal of Immunology. 1993;23:77–84. doi: 10.1002/eji.1830230113. [DOI] [PubMed] [Google Scholar]
- 16.Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. 2002;2:465–475. doi: 10.1038/nri844. [DOI] [PubMed] [Google Scholar]
- 17.Rahman ZS, Manser T. B cells expressing Bcl-2 and a signaling-impaired BAFF-specific receptor fail to mature and are deficient in the formation of lymphoid follicles and germinal centers. J Immunol. 2004;173:6179–6188. doi: 10.4049/jimmunol.173.10.6179. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi Y, Cerasoli DM, Dal Porto JM, Shimoda M, Freund R, Fang W, Telander DG, Malvey EN, Mueller DL, Behrens TW, Kelsoe G. Relaxed negative selection in germinal centers and impaired affinity maturation in bcl-xL transgenic mice. J Exp Med. 1999;190:399–410. doi: 10.1084/jem.190.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 20.Shih TA, Meffre E, Roederer M, Nussenzweig MC. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat Immunol. 2002;3:570–575. doi: 10.1038/ni803. [DOI] [PubMed] [Google Scholar]
- 21.Taki S, Meiering M, Rajewsky K. Targeted insertion of a variable region gene into the immunoglobulin heavy chain locus. Science. 1993;262:1268–1271. doi: 10.1126/science.8235657. [DOI] [PubMed] [Google Scholar]
- 22.Hannum LG, Haberman AM, Anderson SM, Shlomchik MJ. Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J Exp Med. 2000;192:931–942. doi: 10.1084/jem.192.7.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dal Porto JM, Haberman AM, Kelsoe G, Shlomchik MJ. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J Exp Med. 2002;195:1215–1221. doi: 10.1084/jem.20011550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, Huszar D. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int Immunol. 1993;5:647–656. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- 25.Hindmarsh AC. ODEPACK, a systematized collection of ODE solvers. In: Stepleman RS, editor. Scientific Computing. Amsterdam: North-Holland; 1983. p. 10. [Google Scholar]
- 26.Kleinstein SH, Bottino D, Georgieva A, Sarangapani R, Lett GS. In: Perrone FPWLF, Liu J, Lawson BG, Nicol DM, Fujimoto RM, editors. Nonuniform Sampling for Global Optimization of Kinetic Rate Constants in Biological Pathways; Winter Simulation Conference.2006. [Google Scholar]
- 27.Hershberg U, Shlomchik MJ. Differences in potential for amino acid change following mutation reveals distinct strategies for κ and λ light chain variation. PNAS. 2006;103:15963–15968. doi: 10.1073/pnas.0607581103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabat EA, Wu TT, Reid-Miller M, Perry H, Gottesman K. US Govt. Printing Off. No. 165–492. 1987. Sequences of Proteins of Immunological Interest. [Google Scholar]
- 29.Lefranc MP, GV, Kaas Q, Duprat E, Jabado-Michaloud J, Scaviner D, Ginestoux C, Clement O, Chaume D, Lefranc G. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2005:593–597. doi: 10.1093/nar/gki065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Molecular ecology. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe D, Suda T, Nagata S. Expression of Fas in B cells of the mouse germinal center and Fas-dependent killing of activated B cells. Int Immunol. 1995;7:1949–1956. doi: 10.1093/intimm/7.12.1949. [DOI] [PubMed] [Google Scholar]
- 32.Smith KG, Nossal GJ, Tarlinton DM. Fas is highly expressed in the germinal center but is not required for regulation of the B-cell response to antigen. Prod Natl Acad Sci USA. 1995;92:11628–11632. doi: 10.1073/pnas.92.25.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grabarek J, Amstad P, Darzynkiewicz Z. Use of fluorescently labeled caspase inhibitors as affinity labels to detect activated caspases. Hum Cell. 2002;15:1–12. doi: 10.1111/j.1749-0774.2002.tb00094.x. [DOI] [PubMed] [Google Scholar]
- 34.Smolewski P, Grabarek J, Halicka HD, Darzynkiewicz Z. Assay of caspase activation in situ combined with probing plasma membrane integrity to detect three distinct stages of apoptosis. J Immunol Methods. 2002;265:111–121. doi: 10.1016/s0022-1759(02)00074-1. [DOI] [PubMed] [Google Scholar]
- 35.Jayaraman S. Intracellular determination of activated caspases (IDAC) by flow cytometry using a pancaspase inhibitor labeled with FITC. Cytometry A. 2003;56:104–112. doi: 10.1002/cyto.a.10094. [DOI] [PubMed] [Google Scholar]
- 36.O’Neill RM, Hassan J, Reen DJ. IL-7-regulated homeostatic maintenance of recent thymic emigrants in association with caspase-mediated cell proliferation and apoptotic cell death. J Immunol. 2003;170:4524–4531. doi: 10.4049/jimmunol.170.9.4524. [DOI] [PubMed] [Google Scholar]
- 37.Hershberg U, Uduman M, Shlomchik MJ, Kleinstein SH. Improved methods for detecting selection by mutation analysis of Ig V region sequences. Int Immunol. 2008;20:683–694. doi: 10.1093/intimm/dxn026. [DOI] [PubMed] [Google Scholar]
- 38.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 39.McKean D, Huppi K, Bell M, Straudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc Natl Acad Sci USA. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vora KA, Tumas-Brundage K, Manser T. Contrasting the In situ behavior of a memory B cell clone during primary and secondary immune responses. J Immunol. 1999;163:4315–4327. [PubMed] [Google Scholar]
- 41.Mehr R, Edelman H, Sehgal D, Mage R. Analysis of mutational lineage trees from sites of primary and secondary Ig gene diversification in rabbits and chickens. J Immunol. 2004;172:4790–4796. doi: 10.4049/jimmunol.172.8.4790. [DOI] [PubMed] [Google Scholar]
- 42.Allen D, Simon T, Sablitzky F, Rajewsky K, Cumano A. Antibody engineering for the analysis of affinity maturation of an anti-hapten response. EMBO J. 1988;7:1995–2001. doi: 10.1002/j.1460-2075.1988.tb03038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furukawa K, Akasako-Furukawa A, Shirai H, Nakamura H, Azuma T. Junctional amino acids determine the maturation pathway of an antibody. Immunity. 1999;11:329–338. doi: 10.1016/s1074-7613(00)80108-9. [DOI] [PubMed] [Google Scholar]
- 44.Bonhoeffer S, Mohri H, Ho D, Perelson AS. Quantification of cell turnover kinetics using 5-bromo-2′-deoxyuridine. J Immunol. 2000;164:5049–5054. doi: 10.4049/jimmunol.164.10.5049. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, I, MacLennan CM, Liu YJ, Lane PJL. Is rapid proliferation in B centroblasts linked to somatic mutation in memory B cell clones? Immunol Let. 1988;18:197–300. doi: 10.1016/0165-2478(88)90178-2. [DOI] [PubMed] [Google Scholar]
- 46.Hauser AE, Shlomchik MJ, Haberman AM. In vivo imaging studies shed light on germinal-centre development. Nat Rev Immunol. 2007;7:499–504. doi: 10.1038/nri2120. [DOI] [PubMed] [Google Scholar]
- 47.Allen CD, Okada T, Tang HL, Cyster JG. Imaging of Germinal Center Selection Events During Affinity Maturation. Science. 2006 doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- 48.Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, von Andrian UH, Shlomchik MJ, Haberman AM. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity. 2007;26:655–667. doi: 10.1016/j.immuni.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 49.Rossbacher J, Haberman AM, Neschen S, Khalil A, Shlomchik MJ. Antibody-independent B cell-intrinsic and -extrinsic roles for CD21/35. Eur J Immunol. 2006;36:2384–2393. doi: 10.1002/eji.200636172. [DOI] [PubMed] [Google Scholar]
- 50.Al-Qahtani A, Xu Z, Zan H, Walsh CM, Casali P. A role for DRAK2 in the germinal center reaction and the antibody response. Autoimmunity. 2008;41:341–352. doi: 10.1080/08916930802170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, Nussenzweig MC. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- 52.MacLennan IC. Germinal centers. Ann Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 53.Kelsoe G. The germinal center: a crucible for lymphocyte selection. Semin Immunol. 1996;8:179–184. doi: 10.1006/smim.1996.0022. [DOI] [PubMed] [Google Scholar]
- 54.MacLennan IC. Germinal centers still hold secrets. Immunity. 2005;22:656–657. doi: 10.1016/j.immuni.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Carter RH. CD19 regulates B cell maturation, proliferation, and positive selection in the FDC zone of murine splenic germinal centers. Immunity. 2005;22:749–761. doi: 10.1016/j.immuni.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 56.Fearon DT, Carter RH. The CD19/CR2/TAPA-1 complex of B lymphocytes: Linking natural to acquired immunity. Ann Rev Immunol. 1995;13:127–149. doi: 10.1146/annurev.iy.13.040195.001015. [DOI] [PubMed] [Google Scholar]
- 57.Rush JS, Hasbold J, Hodgkin PD. Cross-linking surface Ig delays CD40 ligand-and IL-4-induced B cell Ig class switching and reveals evidence for independent regulation of B cell proliferation and differentiation. J Immunol. 2002;168:2676–2682. doi: 10.4049/jimmunol.168.6.2676. [DOI] [PubMed] [Google Scholar]
- 58.Shlomchik MJ, Watts P, Weigert MG, Litwin S. “Clone”: A Monte-Carlo Computer Simulation of B Cell Clonal Expansion, Somatic Mutation and Antigen-Driven Selection. Curr Top Micro Immunol. 1998;229:173–197. doi: 10.1007/978-3-642-71984-4_13. [DOI] [PubMed] [Google Scholar]
- 59.Kepler TB, Perelson AS. Cyclic re-entry of germinal center B cells and the efficiency of affinity maturation. Immunol Today. 1993;14:412–414. doi: 10.1016/0167-5699(93)90145-B. [DOI] [PubMed] [Google Scholar]
- 60.Kepler TB, Perelson AS. Modeling and optimization of populations subject to time-dependent mutation. Proc Natl Acad Sci USA. 1995;92:8219–8223. doi: 10.1073/pnas.92.18.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wysocki L, Manser T, Gefter ML. Somatic evolution of variable region structures during an immune response. Proceedings of the National Academy of Science. 1986;83:1847–1851. doi: 10.1073/pnas.83.6.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu AH, Jena PK, Wysocki LJ. Tracing the development of single memory-lineage B cells in a highly defined immune response. J Exp Med. 1996;183:2053–2063. doi: 10.1084/jem.183.5.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vora KA, Manser T. Altering the antibody repertoire via transgene homologous recombination: evidence for global and clone-autonomous regulation of antigen-driven B cell differentiation. J Exp Med. 1995;181:271–281. doi: 10.1084/jem.181.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tew JG, Kosco MH, Burton GF, Szakal AK. Follicular dendritic cells as accessory cells. Immunol Rev. 1990;117:185–211. doi: 10.1111/j.1600-065x.1990.tb00573.x. [DOI] [PubMed] [Google Scholar]
- 65.Zheng B, Han S, Kelsoe G. T helper cells in murine germinal centers are antigen-specific emigrants that downregulate Thy-1. J Exp Med. 1996;184:1083–1091. doi: 10.1084/jem.184.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6:683–692. doi: 10.1016/s1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 67.Fruman DA, Ferl GZ, An SS, Donahue AC, Satterthwaite AB, Witte ON. Phosphoinositide 3-kinase and Bruton’s tyrosine kinase regulate overlapping sets of genes in B lymphocytes. Proc Natl Acad Sci U S A. 2002;99:359–364. doi: 10.1073/pnas.012605099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han A, Saijo K, Mecklenbrauker I, Tarakhovsky A, Nussenzweig MC. Bam32 links the B cell receptor to ERK and JNK and mediates B cell proliferation but not survival. Immunity. 2003;19:621–632. doi: 10.1016/s1074-7613(03)00275-9. [DOI] [PubMed] [Google Scholar]
- 69.Teixeiro E, Daniels MA, Hausmann B, Schrum AG, Naeher D, Luescher I, Thome M, Bragado R, Palmer E. T cell division and death are segregated by mutation of TCRbeta chain constant domains. Immunity. 2004;21:515–526. doi: 10.1016/j.immuni.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 70.Boissonnas A, Combadiere B. Interplay between cell division and cell death during TCR triggering. Eur J Immunol. 2004;34:2430–2438. doi: 10.1002/eji.200425271. [DOI] [PubMed] [Google Scholar]
- 71.O’Connor BP, Vogel LA, Zhang W, Loo W, Shnider D, Lind EF, Ratliff M, Noelle RJ, Erickson LD. Imprinting the Fate of Antigen-Reactive B Cells through the Affinity of the B Cell Receptor. J Immunol. 2006;177:7723–7732. doi: 10.4049/jimmunol.177.11.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med. 2006;203:1081–1091. doi: 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shinall SM, Gonzalez-Fernandez M, Noelle RJ, Waldschmidt TJ. Identification of murine germinal center B cell subsets defined by the expression of surface isotypes and differentiation antigens. J Immunol. 2000;164:5729–5738. doi: 10.4049/jimmunol.164.11.5729. [DOI] [PubMed] [Google Scholar]
- 74.Wolniak KL, Noelle RJ, Waldschmidt TJ. Characterization of (4-hydroxy-3-nitrophenyl)acetyl (NP)-specific germinal center B cells and antigen-binding B220- cells after primary NP challenge in mice. J Immunol. 2006;177:2072–2079. doi: 10.4049/jimmunol.177.4.2072. [DOI] [PubMed] [Google Scholar]
- 75.Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol. 2005;5:853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.