

Abstract
Importance
Several innovative disease-modifying treatments (DMTs) for relapsing remitting multiple sclerosis (RRMS) have been licensed recently, or are in late-stage development. The molecular targets of several of these DMTs are well defined. All affect at least one of four properties: (1) immune cell trafficking, (2) cell depletion, (3) immune cell function, or (4) cell replication. In contrast to β-interferons and glatiramer acetate, the first generation DMTs, several newer therapies are imbued with safety issues. In addition to efficacy, understanding the relationship between the mechanism of action (MOA) of the DMTs and their safety profile is essential for decision-making in patient care.
Objective
In this article, we relate safety issues of newer DMTs to their pharmacological characteristics, including molecular targets, MOA, chemical structure, and metabolism. Some newer DMTs also represent repurposing or modifications of previous treatments used in other diseases. Here, we describe how identification and understanding of adverse events (AEs) observed with these established drugs within the same class, provide clues regarding safety and toxicities of newer MS therapeutics.
Conclusions and relevance
While understanding mechanisms underlying DMT toxicities is incomplete, it is important to further develop this knowledge to minimize risk to patients, and to ensure future therapies have the most advantageous risk-benefit profiles. Recognizing the individual classes of DMTs described here may be beneficial when considering use of such agents sequentially and possibly in combination.
Keywords: Multiple sclerosis, disease-modifying treatments, safety, mechanism of action, metabolism
Introduction
Multiple sclerosis (MS) is a chronic central nervous system (CNS) inflammatory demyelinating disease,1 involving both genetic and environmental factors. MS pathology is characterized by focal white and grey matter lesions with myelin, oligodendrocyte and neuroaxonal loss;2 the latter is thought to be responsible for irreversible accumulation of disability.3
There is excitement in MS therapeutics as new disease modifying treatments (DMTs) are rapidly becoming available. However, some of this enthusiasm is tempered by risks engendered by certain newer agents. To optimally manage patients that may use these DMTs, it is important to understand and relate their MOAs to benefits and potential safety risks. The first DMTs, interferon-β (IFN-β) and glatiramer acetate (GA), reduce risk of new attacks and are generally well tolerated and safe. While activity of these agents was originally attributed to their influence on T cells, it now appears these drugs also influence innate immunity.4,5 Although potentially more effective and convenient, recent DMTs have been associated with risks of potentially serious adverse events (AEs), altering risk-to-benefit ratios. Consequently, treatment decisions have become more complex and require detailed information regarding drug properties.6 For many reasons, understanding risks associated with novel treatments is imperfect: (1) data collected during pre-clinical development is limited and extrapolation from animal to humans can be unreliable; (2) clinical studies often recruit insufficient patients to detect less common AEs, recruit highly selected patients and may be too short to detect AEs that only appear after prolonged exposure; (3) identifying causal relationships between treatment and an AE may be difficult. Safety issues are identified after approval for around one quarter of pharmaceutical treatments.7
While adverse events often represent unwanted pharmacological responses related to MOA, some are idiosyncratic.8 Some newer therapies also represent repurposing (e.g. rituximab, alemtuzumab) or modifications (e.g. Fumaderm, leflunomide) of previous treatments used in other diseases. Identifying and understanding AEs observed with other members of the same class, or with use of the same drug in other populations, can provide clues regarding safety and toxicities.
Here, the safety profile of DMTs for MS is reviewed from the perspective of their molecular targets, chemical structure, MOA and metabolism. As first generation therapies GA and IFN-β present few, well-defined safety issues that have been described previously,9 these medications are not discussed. Instead, we focus on more recently approved therapies, or those in late-stage clinical development. They are grouped into four categories based on their presumed target or MOA: (1) immune cell trafficking, (2) depletion, (3) function and (4) cell replication (Table 1). Better understanding of these properties should assist physicians when choosing such therapies.
Table 1.
Categories of DMTs for MS.
| Purpose | DMT |
|---|---|
| Inhibit immune cell trafficking | Natalizumab |
| Fingolimod | |
|
| |
| Promote immune cell depletion | Alemtuzumab |
| Rituximab | |
| Ocrelizumab | |
|
| |
| Influence immune cell function | Laquinimod |
| BG-12 | |
| Daclizumab | |
|
| |
| Inhibit cell replication | Mitoxantrone |
| Teriflunomide | |
DMTs Inhibiting Immune Cell Trafficking
Acute focal CNS inflammation is triggered, particularly at early stages of disease, by influx of activated lymphocytes across the blood-brain barrier (BBB). Two types of treatment that impede lymphocyte migration have been developed and are currently licensed. These treatments prevent activated immune cells from crossing the BBB into the CNS (natalizumab) or from exiting lymph nodes into the circulation (fingolimod). While these therapies may offer substantial efficacy, as a consequence of their MOAs, they alter lymphocyte distribution, which may influence immune surveillance.
Natalizumab
Natalizumab is a humanized monoclonal antibody (mAb; see Fig. 1) that has demonstrated robust reductions in clinical and radiological outcomes in RRMS.10,11 Natalizumab is directed against the α4 subunit of the cell adhesion molecule VLA-4 expressed on the surface of lymphocytes and monocytes. Binding of VLA-4 to its receptor, VCAM-1, on vascular endothelium is required for transmigration of immune cells across the BBB. As binding of α4 integrin is required for immune cell transmigration into the gut, and after successful testing, natalizumab was also approved for treatment of Crohn's disease.12
Figure 1.

Classes of therapeutic antibodies. Green: protein sequences of murine origin; yellow: protein sequences of human origin. MS: multiple sclerosis
Due to blockade of leukocyte migration from blood, natalizumab treatment leads to mild leukocyte elevation13 and concomitant lymphocyte reduction in cerebrospinal fluid (CSF).14 Upon treatment discontinuation, the CSF lymphocyte population reconstitutes within 6-12 months.14
The principal safety issue with use of natalizumab is the increased risk of progressive multifocal leukoencephalopathy (PML),15 which can be fatal or result in permanent disability. The risk for PML became evident shortly after approval of natalizumab. Two patients in the SENTINEL trial, which tested addition of natalizumab to weekly i.m. IFN-β, developed PML after 28 infusions and 37 infusions, respectively. These observations underscored the need to evaluate treatments for sufficiently long durations, and for carefully-designed Phase IV trials. In this regard, measuring duration of therapy may be more relevant than simply reporting “patient-years” of exposure.
The incidence of PML for MS patients treated ≥2 years is 5.05/1,000 (February 2013).16 PML may result from reactivation of JC virus within the CNS or possibly mobilization of peripheral viral reserves to the CNS.17 Three risk factors are recognized for development of PML: evidence of prior JC virus exposure, duration of natalizumab exposure and previous use of immunosuppressants.16 Recently, a test to detect serum anti-JC virus antibodies was developed and serves as a useful biomarker for risk stratification in natalizumab treatment. This test should be repeated in JC virus-negative patients every six months due to the annual 1-2% seroconversion rate.18 Similarly, a high incidence of PML (1 in 500) was reported with efalizumab, which was developed for treatment of psoriasis, but later withdrawn. Efalizumab is a mAb directed against another adhesion molecule, CD11a on T and B cells, which binds to ICAM-1.19 Thus, this elevated PML risk may be a class effect of selective adhesion molecule (SAM) inhibitors.
Chimeric and humanized antibodies contain murine sequences (Fig. 1), which increases their immunogenicity. Use of mAbs can be associated with infusion reactions and persistent neutralizing antibodies; for natalizumab, neutralizing antibodies are associated with loss of therapeutic response and increased risk of hypersensitivity reactions.20 Recrudescence of disease activity occurs approximately 3-5 months after natalizumab discontinuation and corresponds to desaturation of VLA-4 binding. In some cases, natalizumab discontinuation has been associated with a rebound (“overshoot”) beyond baseline activity, and was fatal in one case.21-23 Unfortunately, predisposing risk factors for rebound after natalizumab withdrawal have not been identified.
Fingolimod
Fingolimod (Fig. 2) is an oral medication approved for treatment of RRMS that has demonstrated superior activity to interferon-β1a i.m.24,25 Fingolimod is a sphingosine-1-phosphate (S1P) agonist that binds to four of the five members of the S1P receptor family (S1P1, 2, 3 and 5). However, following binding and activation of S1P1 receptors, fingolimod acts as a functional antagonist and prevents CCR7+ lymphocytes, including naïve and central memory T cells, from exiting lymph nodes.26 Consequently, lymphopenia occurs within hours of administration. Since S1P receptors are present on both neurons and glia, and fingolimod penetrates the CNS,27 fingolimod may exert direct CNS effects.26
Figure 2.
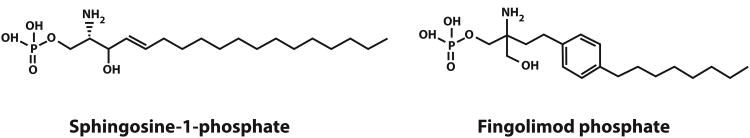
Chemical structure of sphingosine-1-phosphate and fingolimod.
Few opportunistic infections have been documented in fingolimod-treated patients. Two deaths occurred from viral infections during phase III trials testing fingolimod, one from herpes simplex virus (HSV) encephalitis and one from disseminated varicella, although both patients were treated with a higher dose (1.25 mg) than was approved (0.5 mg). Since approval, there has been one reported case of varicella encephalitis at 0.5 mg.28 Currently, a trial is underway to determine whether 0.25 mg may be efficacious and pose less risk of viral infection.29 VZV vaccination is recommended for patients with no history of chickenpox or prior vaccination.30 Viral infections associated with use of fingolimod are presumably linked to lymphopenia from lymphocyte sequestration. Persistent lymphopenia after drug withdrawal has been observed,31 and may also pose concern when considering initiating another therapy soon after fingolimod discontinuation. S1P receptor subtypes are found in other tissues, and may contribute to AEs associated with fingolimod, notably bradycardia, dyspnea, and macular edema. For example, S1P3 receptors are found in cardiac smooth muscle, vascular endothelium and airways.32 More selective S1P1 agonists are under development with the aim of eliminating certain AEs such as macular edema, a consequence from binding retinal S1P2 receptors.33-35
DMTs Producing Immune Cell Depletion
While attention has focused primarily on the role of T cells in MS pathogenesis, recent successes using B cell-depleting agents have provided greater appreciation of the importance of this lymphocyte subset. Several mAbs, originally developed for treatment of hematological malignancies, targeting B and T cells or B cells alone, are being evaluated for potential use in MS. These antibodies are IgG1 and cause cell depletion.
Alemtuzumab
Alemtuzumab, a humanized mAb (Fig. 1), originally developed for treatment of B cell chronic lymphocytic leukemia demonstrated dramatic and sustained reductions in relapses and MRI markers of disease activity in Phase II36 and Phase III37,38 studies versus high-dose interferon.
Alemtuzumab is directed against CD52, a surface glycoprotein present on several mature leukocyte subpopulations, including T, B and NK cells.36 Binding of alemtuzumab to these leukocytes leads to elimination via complement and antibody dependent cellular cytotoxicity (ADCC). However, reconstitution of leukocyte subpopulations varies;39 B cells recover in approximately six months whereas T cells require more than one year.
Treatment-induced humoral autoimmunity is a major concern associated with alemtuzumab. Grave's disease, idiopathic thrombocytopenic purpura (ITP) and Goodpasture's syndrome have been observed following treatment and may be life-threatening without appropriate clinical management. Grave's disease is the most common iatrogenic autoimmunity and occurs in up to one-quarter of alemtuzumab-treated patients,36,40,41 most frequently arising 12-18 months after starting treatment.41 These humoral autoimmune disorders may relate to differences in reconstitution dynamics of B and T cells. Development of autoimmunity may be driven by interleukin-21.42 Besides autoimmunity, alemtuzumab-treated patients experienced significantly higher infection rates.
Rituximab and ocrelizumab
Rituximab and ocrelizumab have shown robust reduction in MS disease activity in phase II MS trials.43,44 Rituximab is a chimeric mAb (Fig. 1) approved for treatment of B cell lymphoma and rheumatoid arthritis (RA).45 Ocrelizumab is a humanized mAb. Rituximab and ocrelizumab are directed against CD20, a glycoprotein primarily found on B cells, with the exception of early progenitor (pro-B) cells and plasma cells. Binding of rituximab and ocrelizumab leads to rapid B cell elimination that persists for 6-8 months, without significant IgG reduction. Reduced MS activity has been attributed to loss of B cell-mediated cellular immunity, namely B cell antigen presentation.46,47
Severe infections have been observed in lymphoma patients receiving rituximab. Further, development of ocrelizumab in RA and lupus was discontinued due to occurrence of fatal opportunistic infections.48 In addition, PML has occurred in a small number of patients with RA or lupus treated with rituximab,49 and in rituximab-treated lymphoma patients.50 So far, no PML cases have been associated with rituximab or ocrelizumab treatment in MS, where these agents are tested in monotherapy.
DMTs Targeting Immune Cell Function
These DMTs, also called immunomodulators, correspond to treatments that primarily influence functional characteristics of both innate and adaptive immunity. They may affect multiple signaling pathways that alter cytokine production or effector cell functions, or both. This class includes two small molecules, dimethyl fumarate (DMF) and laquinimod, and a mAb, daclizumab. A preparation of DMF, BG-12, was recently approved and laquinimod is in late-stage development. BG-12 and laquinimod may have direct central effects due to passive entry into the CNS.
BG-12
BG-12, an oral treatment, has demonstrated efficacy in two Phase III RRMS trials.51,52 BG-12 was developed from the fumaric acid ester (FAE) preparation Fumaderm®, containing a mixture of dimethylfumarate (DMF) and monoethylfumarate (MEF), used for psoriasis treatment in Germany. BG-12 contains only DMF and is rapidly converted to monomethylfumarate (MMF).53
DMF and MMF activate the antioxidant transcription factor nuclear factor (erythroid-derived 2)-related factor 2 (Nrf2) pathway,54,55 leading to expression of detoxifying enzymes, glutathione S-transferase A2 (GSTA2), heme oxygenase-1 (HO-1) and NADPH quinone oxidoreductase 1 (NQO1).56 Fumarates, which are electrophilic, conjugate to glutathione57,58 and can covalently link to essential thiol groups (nucleophiles) on macromolecules, including Keap1 (Fig. 3A), the inhibitor of the Nrf2 pathway.55,59,60 Thus, DMF and its metabolite, MMF activate the Nrf2 pathway by “inhibiting the inhibitor” (Figure 3B,C).
Figure 3.

Methylfumarates promote activation of the Nrf2 pathway via regulation of Keap1, the Nrf2 inhibitor. (A) Methylfumarates are electrophiles that covalently bind the nucleophilic thiol group (-S-H) of Keap1 residue Cys151.55 Two products can be generated depending upon which carbon of the π bond is conjugated. (B) In the absence of MMF, Keap1 binds Nrf2, promoting its ubiquitylation and consequent degradation.107 (C) Upon covalent binding of MMF to Keap1, interaction between Keap1 and Nrf2 is disrupted, stabilizing Nrf2, which permits it to bind the anti-oxidant response element (ARE), and promote gene transcription.
DMF preserves neurons and glial cells in EAE, while MMF protects murine neurons and human astrocytes from oxidative insult in vitro.55 In contrast, others have reported a neuroprotective effect in vitro with DMF, but not with MMF.61 Treatment of mice with DMF induces anti-inflammatory “type II” dendritic cells,54 which drive anti-inflammatory T cell polarization.54 Similar effects have been observed with MMF.54,62 DMF has anti-proliferative effects.63 While potential neuroprotective effects of DMF are attributed to Nrf2 activation, whether its anti-inflammatory and immunomodulatory properties are dependent upon triggering Nrf2 is unknown. In contrast, some animal studies suggest DMF may promote renal tubular hyperplasia and oncogenic activity, also possibly related to Nrf2 activation.64
Safety data are available from two BG-12 phase III RRMS clinical trials51,52 and their combined extension study.65 AEs included flushing, diarrhea, nausea, upper abdominal pain, decreased lymphocyte counts and elevated liver aminotransferases.51,52 Renal AEs ranged from 4-14% and proteinuria (<5%) was the most common.65 Lymphopenia was observed in 4-5% of BG-12-treated patients versus <1% in the placebo group.65 Although no opportunistic infections were reported in the BG-12 Phase III trials, several PML cases have been reported using FAEs in psoriasis, including two cases using FAE monotherapy where PML was associated with lymphopenia that developed after initiating FAE treatment.66-68
Some of these side effects may relate to the MOA of DMF and/or its metabolites, which may be increased at higher doses.51,52 Following administration, DMF undergoes rapid hydrolysis to MMF and methanol.55,69 Interestingly, abdominal pain is a common symptom associated with methanol exposure.70,71 Further metabolism of MMF occurs through the tricarboxylic acid (TCA) cycle, without involvement of cytochrome p450.64 Exhalation of CO2 is the primary route of elimination, accounting for approximately 60% of the DMF dose.64 Drug-protein (e.g. Keap1) adducts,72 may be responsible for liver enzyme elevations that have been reported for BG-12.52 Flushing is thought to be attributed to release of prostaglandins causing local vasodilation.73 Recently, bardoxolone methyl, an Nrf2 activator, was being advanced for treatment of chronic diabetic nephropathy.74 However, its development was halted due to deaths in the phase III trial testing its efficacy. Whether bardoxolone methyl toxicity is related to its activation of Nrf2, its structure or its metabolites is not clear.
Laquinimod
Laquinimod, a quinolone-3-carboxamide, is an orally active immunomodulator that appears to have more pronounced beneficial effects on disease progression and brain atrophy than on clinical and radiological markers of inflammation in RRMS.75,76 Laquinimod is derived from linomide (Fig. 4), whose development in MS was abandoned after occurrence of fatal serositis and myocardial infarction.77 In evaluation of structure-activity relationship (SAR), quinolone-3-carboxamide compounds (>60) were designed, synthesized and evaluated in MS models.78 Individual modifications to the quinolone ring or carboxamide affected efficacy and safety, respectively. Laquinimod, containing one modification in the quinolone and one in the carboxamide, exhibited the best safety and efficacy profile78 and has since been developed for treatment of MS, Crohn's disease and lupus. Laquinimod affects the peripheral immune system and acts within the CNS. Its targets include innate immune cells, including monocytes and dendritic cells, which function as antigen presenting cells (APCs). In EAE, laquinimod induces anti-inflammatory APCs, which then down-regulate pro-inflammatory Th1 and Th17 T cells and promote development of regulatory T cells.79 Glial cells, including astrocytes and microglia, are CNS targets. Laquinimod treatment reduced CNS invasion of inflammatory monocytes, and prevented demyelination and subsequent axonal loss in rodents by down-regulating NF-κB signalling as well as proinflammatory cytokine and NO production in astrocytes.80-82 Laquinimod treatment of MS patients was associated with elevation of brain-derived neurotrophic factor (BDNF).83
Figure 4.

Chemical structure of linomide and laquinimod.
Laquinimod was studied in two phase III trials using annualized relapse rate reduction as its primary endpoint (PEP). Because of its more pronounced beneficial effect on disability progression, a third trial is being conducted using disability as its PEP. Safety data from the first Phase III trials demonstrated laquinimod was well-tolerated and not associated with serious AEs; notably, serositis or myocardial infarction were not observed. Laquinimod undergoes slow hepatic metabolism, which may correlate with transient transaminase elevation seen in 5% of laquinimod-treated patients in comparison to 2% in placebo-treated patients.
Daclizumab
Daclizumab is a humanized non-depleting IgG1 mAb that demonstrated promising results in small pilot MS studies,84,85 and in a phase II trial testing addition of daclizumab to interferon-β 86 Two phase IIb-III studies are underway to evaluate clinical endpoints.87
Daclizumab is directed against the high-affinity α-subunit (CD25) of the interleukin-2 receptor, which is expressed on activated T cells. Interestingly, daclizumab does not block T cell proliferation.88 Instead, beneficial clinical and radiological measures during MS treatment were associated with expansion of regulatory CD56+(bright) natural killer (NK) cells.88 No specific AEs emerged from addition of daclizumab to interferon-β,86 although liver enzyme elevations and cutaneous reactions were observed.
DMTs Targeting Immune Cell Replication
The recognized role of lymphocytes in MS pathogenesis has provided the foundation for advancing drugs that inhibit their expansion. In this class, mitoxantrone and teriflunomide are agents approved for MS treatment that target DNA.
Mitoxantrone
Mitoxantrone is an anthracenedione approved for treatment of rapidly evolving relapsing or secondary progressive MS (SPMS).89 It is an anti-neoplastic agent used for treatment of metastatic breast cancer, acute myeloid leukemia and Non-Hodgkin's lymphoma.
Mitoxantrone is an inhibitor of topoisomerase II90 and can intercalate into double-stranded DNA. Mitoxantrone affects all proliferating cells and is therefore non-selective, although it appears to inhibit B cells more than T cells. Like the related anthracycline chemotherapeutics, mitoxantrone is associated with dose-dependent cardiotoxicity.91 Initially, the recognized risk of therapy-related acute leukemia (TRAL) in MS treatment was 0.25%, but 10 years after mitoxantrone approval, this risk approached 1.0%.91 This increased risk of TRAL provides another example underscoring the importance of vigilant post-approval safety monitoring. Because of concerns for cardiotoxicity and TRAL, use of mitoxantrone for MS is generally confined to second- or third- line treatment.
Teriflunomide
Teriflunomide is an oral agent that demonstrated efficacy in Phase III clinical trials for treatment of RRMS92 and was recently approved in the USA. Teriflunomide is the active metabolite of leflunomide (Fig. 5), a DMT licensed for treatment of RA.93 Teriflunomide inhibits mitochondrial dihydroorotate dehydrogenase (DHODH), an enzyme used for de novo synthesis of pyrimidine nucleotides in proliferating cells. However, teriflunomide does not inhibit the salvage pathway used by resting cells.94
Figure 5.

Chemical structure of leflunomide and teriflunomide.
AEs associated with teriflunomide include lymphopenia, alopecia, elevated liver enzymes, elevation of blood pressure and nausea. Leflunomide and teriflunomide are considered to be teratogenic in humans and are therefore contraindicated in pregnancy.95 Teriflunomide can also penetrate into breast milk.96 As leflunomide treatment of RA is associated with elevated risk of tuberculosis, PPD testing is recommended before commencing teriflunomide treatment in MS patients.96
Teriflunomide undergoes extensive enterohepatic recirculation, leading to chronic exposure of the liver to high concentrations that may result in hepatotoxicity,97 an important safety issue with leflunomide in RA98,99 and teriflunomide in MS.96 As a consequence of its enterohepatic recycling, substantial time is required to achieve steady-state plasma concentrations of teriflunomide. The extended 10-day half-life97 is of potential clinical relevance in case of serious AE or pregnancy, when rapid drug elimination is necessary. In this context, wash-out procedures have been developed involving administration of cholestyramine or activated charcoal to prevent enterohepatic recirculation. Although genetic polymorphisms of p450 isoforms have been associated with AEs to leflunomide,100 cytochrome p450 may have a limited role in teriflunomide metabolism.
Discussion
With introduction of several new MS medications, treatment decisions are becoming more complex. Whereas efficacy remains paramount, choosing new agents necessitates careful consideration of other characteristics, including mechanism(s) of action, duration of effect (i.e. pharmacodynamics) and potential risks. In this article, we have classified DMTs into four categories based upon their ability to (1) inhibit cell trafficking, (2) promote immune cell depletion, (3) influence immune function or (4) inhibit cell replication. While we have provided a framework, it is important to recognize that each category is not mutually exclusive. Agents that reduce lymphocyte proliferation may induce immune modulation and vice versa.63,101 Nevertheless, categorization of agents with similarities can help us anticipate specific side effects of newer agents. In this regard, it is important to recognize that natalizumab and efalizumab, which are SAM inhibitors and therefore block lymphocyte trafficking, are both associated with PML. While newer S1P agonists (e.g. BAF312 and ONO-4641) selectively activate S1P1 receptors on lymphocytes and reduce trafficking, these agents also bind the S1P1 receptors expressed by cells directing atrioventricular conduction and therefore, like fingolimod, can be associated with some level of bradycardia.
Agents specific for one molecular target or immune pathway may have pleiotropic effects. While the intended mechanism of a given DMT may shift immune balance favorably for one disease, it may have paradoxical activity in others. TNF receptor antagonists are widely used in RA, and were considered for MS therapy until their use was associated with increased risk of CNS demyelination. Although T and B cell depletion by alemtuzumab is associated with potent therapeutic effects in MS, its use promotes humoral autoimmunity targeting the thyroid and, more rarely, platelets, kidney or lung. Whether this iatrogenic autoimmunity relates to distinct kinetics of T and B cell reconstitution, or abnormal T cell cytokine secretion is not clear. Prolonged lymphopenia after alemtuzumab treatment may be an important consideration when using other agents sequentially. Specifically, should one wait until there is full reconstitution of both B cells and T cells prior to treatment with another agent? Similarly, if a patient does not respond to fingolimod, one may consider delaying sequential treatment until the fingolimod-associated lymphopenia resolves. Interestingly, prolonged lymphopenia and associated immunosuppression, rather than lack of clinical benefit in MS, probably halted development and use of cladribine. When treating MS with newer agents, we may need to think beyond our next therapy.
MS physicians will need to pay particular attention to metabolic properties when prescribing certain newer agents. In contrast to interferons (natural endogenous proteins) and glatiramer acetate (a polypeptide-based agent), newer oral therapies are synthetic organic molecules, and may be metabolized and excreted differently. Teriflunomide undergoes prolonged hepatobiliary circulation; in certain situations (e.g., pregnancy or AE) it may be necessary to accelerate teriflunomide elimination. Metabolites may be active therapeutically, and also responsible for adverse effects. DMF is rapidly metabolized to MMF, considered the predominant bioactive form responsible for Nrf2 activation. As methanol is produced in metabolism of DMF to MMF, methanol or other DMF metabolites could possibly contribute to its adverse effects.
With introduction of new agents that utilize different MOAs, one can envisage combining MS medications that may act in an additive or synergistic manner.102 Although this is a worthy goal, there are practical concerns. First, to establish that two effective drugs are more efficacious together than either one alone may require enrolling large numbers of patients. Second, as the price of many MS agents increases, it may be unreasonable to consider the added cost in combination. In general, one should be cautious combining pharmacological agents as their metabolism may interfere with one another, and further, paradoxical effects can occur. In this regard, clinical trials have suggested that widely-used cholesterol-lowering statins may interfere with the efficacy of interferon-β,103,104 and it is postulated that this potential antagonistic effect relates to their opposing activity on the pro-inflammatory signaling molecule, STAT1.105
Surrogate markers that associate with risk of adverse effects, or response, to DMTs are particularly helpful in clinical practice. As JCV Ab+ patients have increased risk of PML during natalizumab treatment, anti-JCV seropositivity has become an important biomarker for stratification of this risk. Serum IL-21 levels could be considered to estimate risk of thyroid autoimmunity in alemtuzumab-treated patients. Stratification may include gene polymorphisms. For example, ABC-transporter gene polymorphisms have been associated with response to mitoxantrone.106
In stark contrast to the excitement surrounding our increasing repertoire of treatments for RRMS, the paucity of useful agents for progressive MS is sobering. Thus far, our successes primarily target the peripheral inflammation characterizing RRMS, but not the CNS-resident inflammatory and neurodegenerative processes of progressive MS. Hopefully, this therapeutic gap will be breached through better understanding of MS progression, refining our clinical and imaging metrics of MS progression, and testing of established and novel agents with potential anti-oxidative and neuroprotective MOAs.
While no drug to date ‘cures’ MS, it is clear that major advances have been made in therapeutics of RRMS. However, several current drugs have serious, sometimes life-threatening toxicities. Although the understanding of mechanisms underlying DMT toxicities is incomplete, it is important to develop this knowledge to minimize risk to patients, and to ensure future therapies have the most advantageous risk-benefit profiles. Recognizing the individual classifications of DMTs described here may be beneficial when considering use of such agents sequentially, or eventually in combination.
Search strategy and selection criteria
References for this review were identified through searches of PubMed with the following key words: Drug name: chemical and brand name; mode of action, specific side effects, major metabolites and clinical trials. Search was conducted August 14, 2012. Articles were also identified through searches of the authors' own files. Only papers published in English were reviewed. The final reference list was generated on the basis of originality and relevance to the broad scope of this Review.
Acknowledgments
Role of the funding source: This manuscript was the result of a Working Group Meeting held in Tel Aviv in August 2012 during which a panel of MS experts addressed safety considerations in immune modulation in MS therapy that was supported by Teva Pharmaceuticals, Inc.
Disclosures: Dr. Brück receives research support from Teva Pharmaceutical Industries Ltd., Biogen Idec and Novartis. He is member of scientific advisory boards of Teva Pharmaceutical Industries Ltd., Biogen Idec, Novartis and Genzyme/Sanofi. Dr. Brück serves on speaker's bureaus for Bayer Vital, Biogen Idec, Merck Serono, Teva Pharmaceutical Industries Ltd., Genzyme/Sanofi and Novartis. Dr. Brück serves on editorial boards of Acta Neuropathologica, Neuropathology and Applied Neurobiology and Therapeutic Advances in Neurological Disorders.
Dr. Gold receives research support from Teva Pharmaceutical Industries Ltd., Biogen Idec, Novartis, Merck Serono and ELAN. He is a member of scientific advisory boards of Teva Pharmaceutical, Biogen Idec, Novartis, Baxter and Genzyme/Sanofi. Dr. Gold serves on speaker's bureaus for Bayer Vital, Biogen Idec, Merck Serono, Teva Pharmaceutical, Genzyme/Sanofi, ZLB Behring and Novartis. Dr. Gold serves on editorial boards of American Journal of Pathology, Aktuelle Neurologie and Therapeutic Advances in Neurological Disorders.
Dr. Lund receives research support from Teva Pharmaceutical Industries Ltd., Genzyme/Sanofi and Novartis and has served as a consultant and received honoraria from Teva Pharmaceuticals Inc.
Dr. Oreja-Guevara has received honoraria as consultant in scientific advisory boards by Biogen Idec, Bayer-Schering, Merck Serono, Teva and Novartis and has also participated in clinical trials and other research projects promoted by Biogen Idec, GSK, Teva and Novartis.
Dr. Prat receives research grant support from the CIHR, The MS Society of Canada, the FRQ-S, Teva Pharmaceuticals, Inc. and ELAN Corporation. He has served as a consultant and received honoraria from ELAN, Biogen Idec, EMD Serono, Genzyme/Sanofi, Novartis and Teva Pharmaceuticals.
Mr. Spencer has received travel compensation from Teva Pharmaceuticals.
Dr. Steinman receives research grant support from the NIH (RO1 NS55997, R21AI0955055), the NMSS, and The Guthy Jackson Charitable Foundation, He has served as a consultant and received honoraria from Biogen Idec, Genzyme, MedImmune, Novartis, Questcor, Roche, Sanofi, and Teva Pharmaceuticals, Inc.
Dr. Tintoré has received compensation for consulting services and speaking from Bayer Schering, Merck Serono, Biogen Idec, Teva, Sanofi-Aventis, Genzyme and Novartis.
Dr. Vollmer receives research grant support from the NIH, Teva Neuroscience, Biogen Idec, Ono Pharmaceuticals, Genzyme, and Janssen Research & Development. He has served as a consultant and has received honoraria from Biogen Idec, Teva Neuroscience, EMD Serono, Genzyme, Novartis, Questcor, Roche, Elan Pharmaceuticals, Bristol-Myers Squibb, Acord Pharmaceuticals, Ono, Accelerated Cure Project, Eli Lilly, Medical Logix, MSDx, Esai Pharmaceuticals, Schering-Plough Biopharma, Xenoport, Daiichi Sanyko.
Dr. Weiner serves as a consultant to Teva, and has ancillary grants from Teva. He has served on an Advisory Board for Novartis and is the USC Principal Investigator for an alemtuzumab study sponsored by Genzyme/Sanofi. He is also a member of the Safety Board for ACTH-Solu-medrol study and was appointed by the NIH and NMSS as Chair of the Data Safety Monitoring Committee for the Phase 2 Estriol trial.
Dr. Ziemssen has received speaker honoraria from Biogen Idec, Genzyme, GSK, Sanofi-Aventis, Merck Serono, MSD, Novartis, Teva, and Bayer Healthcare. He serves as a consultant for Biogen Idec, Genzyme, Teva, Novartis, and Bayer HealthCare.
Dr. Weber received or receives research support from Teva Pharmaceutical Industries Ltd. and Roche. He participated or participates in scientific advisory boards of Teva Pharmaceutical Industries Ltd. and Roche. He has served as a consultant and received honoraria from Genzyme, Novartis, Roche, and Teva Pharmaceuticals, Inc. Dr. Weber serves on the editorial board of PLoS One.
Dr. Zamvil receives research grant support from the NIH (RO1 AI073737 and RO1 NS063008), the NMSS (RG 4124), The Guthy Jackson Charitable Foundation, The Maisin Foundation, Biogen Idec, Inc., Teva Pharmaceuticals, Inc., Five Prime, Inc. and Boehringer-Ingelheim, Inc. He has served as a consultant and received honoraria from Biogen Idec, EMD Serono, Genzyme, Novartis, Questcor, Roche, and Teva Pharmaceuticals, Inc., and has served or serves on the editorial boards of The Journal of Clinical Investigation, The Journal of Immunology, Neurotherapeutics, and The Journal of Neurological Sciences.
Contributor Information
Wolfgang Brück, Pr, Department of Neuropathology, University Medical Centre, Göttingen, Germany.
Gold Ralf, Pr, Research Department of Neuroscience, Neurologische Universitätsklinik, St Josef Hospital Faculty of Medicine, Bochum, Germany.
Brett T. Lund, Dr, Department of Neurology, Keck School of Medicine of University of Southern California, USA.
Oreja-Guevara Celia, Dr, Department of Neurology, Multiple Sclerosis Unit, Hospital Universitario San Carlos, Madrid, Spain.
Alexandre Prat, Dr, Center of Excellence in Neuromics, CHUM-Université de Montreal, Montréal, Canada.
Lawrence Steinman, Pr, Department of Neurology, Stanford University School of Medicine, Stanford, USA.
Tintoré Mar, Dr, Centre d'Esclerosi Múltiple de Catalunya (CEM-Cat), Unitat de Neuroimmunologia Clínica, Servei de Neurologia, Hospital Vall d'Hebron, Barcelona, Spain.
Timothy Vollmer, Pr, Department of Neurology, University of Colorado Denver School of Medicine, Aurora, USA.
Martin S. Weber, Pr, Department of Neuropathology, Department of Neurology, University Medical Centre, Göttingen, Germany.
Leslie P. Weiner, Pr, Department of Neurology, Keck School of Medicine of University of Southern California, USA.
Tjalf Ziemssen, Pr, MS Center Dresden, Neuroimmunological Lab, Center of Clinical Neuroscience, Department of Neurology, University Clinic Carl Gustavus Carus, Dresden University of Technology, Dresden, Germany.
Scott S. Zamvil, Pr, Department of Neurology, University of California San Francisco School of Medicine, San Francisco, USA.
References
- 1.Koch-Henriksen N, Sorensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol. 2010;9(5):520–532. doi: 10.1016/S1474-4422(10)70064-8. [DOI] [PubMed] [Google Scholar]
- 2.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 3.Bruck W, Stadelmann C. Inflammation and degeneration in multiple sclerosis. Neurol Sci. 2003;24 Suppl 5:S265–267. doi: 10.1007/s10072-003-0170-7. [DOI] [PubMed] [Google Scholar]
- 4.Prod'homme T, Zamvil SS. Bench to bedside: tempering antigen-presenting cells in multiple sclerosis. Nat Med. 2008;14(6):614–615. doi: 10.1038/nm0608-614. [DOI] [PubMed] [Google Scholar]
- 5.Weber MS, Prod'homme T, Youssef S, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med. 2007;13(8):935–943. doi: 10.1038/nm1620. [DOI] [PubMed] [Google Scholar]
- 6.Kieseier BC, Stuve O. A critical appraisal of treatment decisions in multiple sclerosis--old versus new. Nat Rev Neurol. 2011;7(5):255–262. doi: 10.1038/nrneurol.2011.41. [DOI] [PubMed] [Google Scholar]
- 7.Lexchin J. New Drugs and Safety: What Happened to New Active Substances Approved in Canada Between 1995 and 2010? Arch Intern Med. 2012:1–2. doi: 10.1001/archinternmed.2012.4444. [DOI] [PubMed] [Google Scholar]
- 8.Rawlins MD, Thomson JW. Mechanisms of adverse drug reactions. In: Davies DM, editor. Textbook of adverse drug reactions. 4th. Oxford; New York: Oxford University Press; 1991. [Google Scholar]
- 9.Wingerchuk DM. Multiple sclerosis disease-modifying therapies: adverse effect surveillance and management. Expert Rev Neurother. 2006;6(3):333–346. doi: 10.1586/14737175.6.3.333. [DOI] [PubMed] [Google Scholar]
- 10.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 11.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 12.Sandborn WJ, Yednock TA. Novel approaches to treating inflammatory bowel disease: targeting alpha-4 integrin. Am J Gastroenterol. 2003;98(11):2372–2382. doi: 10.1111/j.1572-0241.2003.08703.x. [DOI] [PubMed] [Google Scholar]
- 13.Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology. 2008;71(17):1350–1354. doi: 10.1212/01.wnl.0000327671.91357.96. [DOI] [PubMed] [Google Scholar]
- 14.Stuve O, Marra CM, Jerome KR, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol. 2006;59(5):743–747. doi: 10.1002/ana.20858. [DOI] [PubMed] [Google Scholar]
- 15.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353(4):375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- 16.Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 17.Monaco MC, Major EO. The link between VLA-4 and JC virus reactivation. Expert Rev Clin Immunol. 2012;8(1):63–72. doi: 10.1586/eci.11.85. [DOI] [PubMed] [Google Scholar]
- 18.Gorelik L, Lerner M, Bixler S, et al. Anti-JC virus antibodies: implications for PML risk stratification. Ann Neurol. 2010;68(3):295–303. doi: 10.1002/ana.22128. [DOI] [PubMed] [Google Scholar]
- 19.Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med. 2010;61:35–47. doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- 20.TYSABRI. Cambridge, MA: Biogen Idec, Inc.; 2012. package insert. [Google Scholar]
- 21.Lenhard T, Biller A, Mueller W, Metz I, Schonberger J, Wildemann B. Immune reconstitution inflammatory syndrome after withdrawal of natalizumab? Neurology. 2010;75(9):831–833. doi: 10.1212/WNL.0b013e3181f07362. [DOI] [PubMed] [Google Scholar]
- 22.Ramos-Cejudo J, Oreja-Guevara C, Stark Aroeira L, Rodriguez de Antonio L, Chamorro B, Diez-Tejedor E. Treatment with natalizumab in relapsing-remitting multiple sclerosis patients induces changes in inflammatory mechanism. J Clin Immunol. 2011;31(4):623–631. doi: 10.1007/s10875-011-9522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rigau V, Mania A, Befort P, et al. Lethal multiple sclerosis relapse after natalizumab withdrawal. Neurology. 2012;79(22):2214–2216. doi: 10.1212/WNL.0b013e318275979d. [DOI] [PubMed] [Google Scholar]
- 24.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 25.Kappos L, Radue EW, O'Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 26.Cohen JA, Chun J. Mechanisms of fingolimod's efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69(5):759–777. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]
- 27.Foster CA, Howard LM, Schweitzer A, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323(2):469–475. doi: 10.1124/jpet.107.127183. [DOI] [PubMed] [Google Scholar]
- 28.Ratchford JN, Costello K, Reich DS, Calabresi PA. Varicella-zoster virus encephalitis and vasculopathy in a patient treated with fingolimod. Neurology. 2012;79(19):2002–2004. doi: 10.1212/WNL.0b013e3182735d00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MS Study Evaluating Safety and Efficacy of Two Doses of Fingolimod Versus Copaxone. [ NCT01633112] http://clinicaltrials.gov/ct2/show/NCT01633112.
- 30.GILENYA. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2012. package insert. [Google Scholar]
- 31.Johnson TA, Shames I, Keezer M, et al. Reconstitution of circulating lymphocyte counts in FTY720-treated MS patients. Clin Immunol. 2010;137(1):15–20. doi: 10.1016/j.clim.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Brinkmann V, Baumruker T. Pulmonary and vascular pharmacology of sphingosine 1-phosphate. Curr Opin Pharmacol. 2006;6(3):244–250. doi: 10.1016/j.coph.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Olsson T, Boster A, Fernandez O, et al. Efficacy and safety of ponesimod, an oral, selective sphingosine 1-phosphate receptor-1 modulator, in patients with relapsing-remitting multiple sclerosis: results from a phase IIb, randomised, double-blind, placebo-controlled trial. Mult Scler J. 2012;18(S4):49–50. [Google Scholar]
- 34.Stuve O, Selmaj K, Li D, et al. BAF312, a Selective Sphingosine-1-Phosphate Receptor Modulator Improves MRI and Clinical Outcomes in Relapsing-Remitting Multiple Sclerosis (RRMS) (S30.001) Neurology. 2012;78(Meeting Abstracts 1):S30.001. [Google Scholar]
- 35.Vollmer T, Selmaj K, Bar-Or A, Zipp F. A Double-Blind, Placebo-Controlled, Phase 2, 26-Week DreaMS Trial of a Selective S1P Receptor Agonist ONO-4641 in patients with Relapsing-Remitting Multiple Sclerosis. Neurology. 2012;79(11):e87–e91. [Google Scholar]
- 36.Investigators CT, Coles AJ, Compston DA, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359(17):1786–1801. doi: 10.1056/NEJMoa0802670. [DOI] [PubMed] [Google Scholar]
- 37.Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–1828. doi: 10.1016/S0140-6736(12)61769-3. [DOI] [PubMed] [Google Scholar]
- 38.Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829–1839. doi: 10.1016/S0140-6736(12)61768-1. [DOI] [PubMed] [Google Scholar]
- 39.Coles AJ, Wing M, Smith S, et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet. 1999;354(9191):1691–1695. doi: 10.1016/S0140-6736(99)02429-0. [DOI] [PubMed] [Google Scholar]
- 40.Coles A, Brinar V, Arnold D, et al. Efficacy and Safety Results from Comparison of Alemtuzumab and Rebif(R) Efficacy in Multiple Sclerosis I (CARE-MS I): A Phase 3 Study in Relapsing-Remitting Treatment-Naive Patients (S01.006) Neurology. 2012;78(Meeting Abstracts 1):S01.006. [Google Scholar]
- 41.Cossburn M, Pace AA, Jones J, et al. Autoimmune disease after alemtuzumab treatment for multiple sclerosis in a multicenter cohort. Neurology. 2011;77(6):573–579. doi: 10.1212/WNL.0b013e318228bec5. [DOI] [PubMed] [Google Scholar]
- 42.Jones JL, Phuah CL, Cox AL, et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H) J Clin Invest. 2009;119(7):2052–2061. doi: 10.1172/JCI37878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 44.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378(9805):1779–1787. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 45.Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 46.Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010;67(4):452–461. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- 47.Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010;68(3):369–383. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roche and Biogen Idec decide to suspend Ocrelizumab treatment - Rheumatoid Arthritis development programme on hold. http://www.roche.com/media/media_releases/med-cor-2010-03-08.htm.
- 49.Palazzo E, Yahia SA. Progressive multifocal leukoencephalopathy in autoimmune diseases. Joint Bone Spine. 2012;79(4):351–355. doi: 10.1016/j.jbspin.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Rituxan. South San Francisco, CA: Genentech, Inc.; 2012. package insert. [Google Scholar]
- 51.Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 52.Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 53.Litjens NH, Burggraaf J, van Strijen E, et al. Pharmacokinetics of oral fumarates in healthy subjects. Br J Clin Pharmacol. 2004;58(4):429–432. doi: 10.1111/j.1365-2125.2004.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghoreschi K, Bruck J, Kellerer C, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208(11):2291–2303. doi: 10.1084/jem.20100977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134(Pt 3):678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284(20):13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rostami-Yazdi M, Mrowietz U. Fumaric acid esters. Clin Dermatol. 2008;26(5):522–526. doi: 10.1016/j.clindermatol.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Rostami-Yazdi M, Clement B, Schmidt TJ, Schinor D, Mrowietz U. Detection of metabolites of fumaric acid esters in human urine: implications for their mode of action. J Invest Dermatol. 2009;129(1):231–234. doi: 10.1038/jid.2008.197. [DOI] [PubMed] [Google Scholar]
- 59.Frycak P, Zdrahal Z, Ulrichova J, Wiegrebe W, Lemr K. Evidence of covalent interaction of fumaric acid esters with sulfhydryl groups in peptides. J Mass Spectrom. 2005;40(10):1309–1318. doi: 10.1002/jms.910. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt TJ, Ak M, Mrowietz U. Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine--preparation of S-substituted thiosuccinic acid esters. Bioorg Med Chem. 2007;15(1):333–342. doi: 10.1016/j.bmc.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 61.Albrecht P, Bouchachia I, Goebels N, et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J Neuroinflammation. 2012;9:163. doi: 10.1186/1742-2094-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Litjens NH, Rademaker M, Ravensbergen B, Thio HB, van Dissel JT, Nibbering PH. Effects of monomethylfumarate on dendritic cell differentiation. Br J Dermatol. 2006;154(2):211–217. doi: 10.1111/j.1365-2133.2005.07002.x. [DOI] [PubMed] [Google Scholar]
- 63.Lehmann JC, Listopad JJ, Rentzsch CU, et al. Dimethylfumarate induces immunosuppression via glutathione depletion and subsequent induction of heme oxygenase 1. J Invest Dermatol. 2007;127(4):835–845. doi: 10.1038/sj.jid.5700686. [DOI] [PubMed] [Google Scholar]
- 64.Tecfidera. Vol Cambridge, MA: Biogen Idec; 2013. package insert. [Google Scholar]
- 65.Phillips JT, Fox RJ, Selmaj K, et al. Long-term safety and tolerability of oral BG-12 (dimethyl fumarate) in relapsing-remitting multiple sclerosis: interim results from ENDORSE. Mult Scler J. 2012;18(S4):517–518. [Google Scholar]
- 66.Sweetser MT, Dawson KT, Bozic C. Manufacturer's response to case reports of PML. N Engl J Med. 2013;368(17):1659–1660. doi: 10.1056/NEJMc1300283. [DOI] [PubMed] [Google Scholar]
- 67.Ermis U, Weis J, Schulz JB. PML in a patient treated with fumaric acid. N Engl J Med. 2013;368(17):1657–1658. doi: 10.1056/NEJMc1211805. [DOI] [PubMed] [Google Scholar]
- 68.van Oosten BW, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med. 2013;368(17):1658–1659. doi: 10.1056/NEJMc1215357. [DOI] [PubMed] [Google Scholar]
- 69.Litjens NH, van Strijen E, van Gulpen C, et al. In vitro pharmacokinetics of anti-psoriatic fumaric acid esters. BMC Pharmacol. 2004;4:22. doi: 10.1186/1471-2210-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barceloux DG, Bond GR, Krenzelok EP, Cooper H, Vale JA. American Academy of Clinical Toxicology Ad Hoc Committee on the Treatment Guidelines for Methanol P American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. J Toxicol Clin Toxicol. 2002;40(4):415–446. doi: 10.1081/clt-120006745. [DOI] [PubMed] [Google Scholar]
- 71.Kraut JA, Kurtz I. Toxic alcohol ingestions: clinical features, diagnosis, and management. Clin J Am Soc Nephrol. 2008;3(1):208–225. doi: 10.2215/CJN.03220807. [DOI] [PubMed] [Google Scholar]
- 72.Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA. Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem Res Toxicol. 2004;17(1):3–16. doi: 10.1021/tx034170b. [DOI] [PubMed] [Google Scholar]
- 73.Hanson J, Gille A, Offermanns S. Role of HCA(2) (GPR109A) in nicotinic acid and fumaric acid ester-induced effects on the skin. Pharmacol Ther. 2012;136(1):1–7. doi: 10.1016/j.pharmthera.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 74.Reisman SA, Chertow GM, Hebbar S, Vaziri ND, Ward KW, Meyer CJ. Bardoxolone methyl decreases megalin and activates nrf2 in the kidney. J Am Soc Nephrol. 2012;23(10):1663–1673. doi: 10.1681/ASN.2012050457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366(11):1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- 76.Vollmer T, Soelberg Sorensen P, Arnold DL Group obotBS. A placebo-controlled and active comparator phase III trial (BRAVO) for relapsing-remitting multiple sclerosis. Mult Scler J. 2011;17(10 suppl):S507–508. [Google Scholar]
- 77.Noseworthy JH, Wolinsky JS, Lublin FD, et al. Linomide in relapsing and secondary progressive MS: part I: trial design and clinical results. North American Linomide Investigators. Neurology. 2000;54(9):1726–1733. doi: 10.1212/wnl.54.9.1726. [DOI] [PubMed] [Google Scholar]
- 78.Jonsson S, Andersson G, Fex T, et al. Synthesis and biological evaluation of new 1,2-dihydro-4-hydroxy-2-oxo-3-quinolinecarboxamides for treatment of autoimmune disorders: structure-activity relationship. J Med Chem. 2004;47(8):2075–2088. doi: 10.1021/jm031044w. [DOI] [PubMed] [Google Scholar]
- 79.Schulze-Topphoff U, Shetty A, Varrin-Doyer M, et al. Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PLoS One. 2012;7(3):e33797. doi: 10.1371/journal.pone.0033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mishra MK, Wang J, Silva C, Mack M, Yong VW. Kinetics of proinflammatory monocytes in a model of multiple sclerosis and its perturbation by laquinimod. Am J Pathol. 2012;181(2):642–651. doi: 10.1016/j.ajpath.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 81.Bruck W, Pfortner R, Pham T, et al. Reduced astrocytic NF-kappaB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol. 2012;124(3):411–424. doi: 10.1007/s00401-012-1009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ruffini F, Rossi S, Bergamaschi A, et al. Laquinimod prevents inflammation-induced synaptic alterations occurring in experimental autoimmune encephalomyelitis. Mult Scler. 2012 doi: 10.1177/1352458512469698. [DOI] [PubMed] [Google Scholar]
- 83.Thone J, Ellrichmann G, Seubert S, et al. Modulation of autoimmune demyelination by laquinimod via induction of brain-derived neurotrophic factor. Am J Pathol. 2012;180(1):267–274. doi: 10.1016/j.ajpath.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 84.Rose JW, Burns JB, Bjorklund J, Klein J, Watt HE, Carlson NG. Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results. Neurology. 2007;69(8):785–789. doi: 10.1212/01.wnl.0000267662.41734.1f. [DOI] [PubMed] [Google Scholar]
- 85.Bielekova B, Richert N, Howard T, et al. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon beta. Proc Natl Acad Sci U S A. 2004;101(23):8705–8708. doi: 10.1073/pnas.0402653101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wynn D, Kaufman M, Montalban X, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010;9(4):381–390. doi: 10.1016/S1474-4422(10)70033-8. [DOI] [PubMed] [Google Scholar]
- 87.Gold R, Giovannoni G, Selmaj K, et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013 doi: 10.1016/S0140-6736(12)62190-4. [DOI] [PubMed] [Google Scholar]
- 88.Bielekova B, Catalfamo M, Reichert-Scrivner S, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006;103(15):5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- 90.Malonne H, Atassi G. DNA topoisomerase targeting drugs: mechanisms of action and perspectives. Anticancer Drugs. 1997;8(9):811–822. doi: 10.1097/00001813-199710000-00001. [DOI] [PubMed] [Google Scholar]
- 91.Marriott JJ, Miyasaki JM, Gronseth G, O'Connor PW Therapeutics, Technology Assessment Subcommittee of the American Academy of N. Evidence Report: The efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2010;74(18):1463–1470. doi: 10.1212/WNL.0b013e3181dc1ae0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O'Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- 93.Fox RI, Herrmann ML, Frangou CG, et al. Mechanism of action for leflunomide in rheumatoid arthritis. Clin Immunol. 1999;93(3):198–208. doi: 10.1006/clim.1999.4777. [DOI] [PubMed] [Google Scholar]
- 94.Fairbanks LD, Bofill M, Ruckemann K, Simmonds HA. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. Disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J Biol Chem. 1995;270(50):29682–29689. [PubMed] [Google Scholar]
- 95.Fukushima R, Kanamori S, Hirashiba M, et al. Inhibiting the teratogenicity of the immunosuppressant leflunomide in mice by supplementation of exogenous uridine. Toxicol Sci. 2009;108(2):419–426. doi: 10.1093/toxsci/kfp022. [DOI] [PubMed] [Google Scholar]
- 96.Aubagio. Cambridge, MA: Genzyme Corporation; 2012. package insert. [Google Scholar]
- 97.Rozman B. Clinical pharmacokinetics of leflunomide. Clin Pharmacokinet. 2002;41(6):421–430. doi: 10.2165/00003088-200241060-00003. [DOI] [PubMed] [Google Scholar]
- 98.Alcorn N, Saunders S, Madhok R. Benefit-risk assessment of leflunomide: an appraisal of leflunomide in rheumatoid arthritis 10 years after licensing. Drug Saf. 2009;32(12):1123–1134. doi: 10.2165/11316650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 99.U.S. Food and Drug Administration. FDA Drug Safety Communication: New boxed warning for severe liver injury with arthritis drug Arava (leflunomide) Website http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm218679.htm.
- 100.Bohanec Grabar P, Rozman B, Tomsic M, Suput D, Logar D, Dolzan V. Genetic polymorphism of CYP1A2 and the toxicity of leflunomide treatment in rheumatoid arthritis patients. Eur J Clin Pharmacol. 2008;64(9):871–876. doi: 10.1007/s00228-008-0498-2. [DOI] [PubMed] [Google Scholar]
- 101.Claussen MC, Korn T. Immune mechanisms of new therapeutic strategies in MS: teriflunomide. Clin Immunol. 2012;142(1):49–56. doi: 10.1016/j.clim.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 102.Stuve O, Youssef S, Weber MS, et al. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Invest. 2006;116(4):1037–1044. doi: 10.1172/JCI25805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Birnbaum G, Cree B, Altafullah I, Zinser M, Reder AT. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology. 2008;71(18):1390–1395. doi: 10.1212/01.wnl.0000319698.40024.1c. [DOI] [PubMed] [Google Scholar]
- 104.Sorensen PS, Lycke J, Eralinna JP, et al. Simvastatin as add-on therapy to interferon beta-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): a placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011;10(8):691–701. doi: 10.1016/S1474-4422(11)70144-2. [DOI] [PubMed] [Google Scholar]
- 105.Zamvil SS, Steinman L. Combining statins with interferon beta in multiple sclerosis: think twice, it might not be all right. Lancet Neurol. 2011;10(8):672–673. doi: 10.1016/S1474-4422(11)70153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cotte S, von Ahsen N, Kruse N, et al. ABC-transporter gene-polymorphisms are potential pharmacogenetic markers for mitoxantrone response in multiple sclerosis. Brain. 2009;132(Pt 9):2517–2530. doi: 10.1093/brain/awp164. [DOI] [PubMed] [Google Scholar]
- 107.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85(4):241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]