

Abstract
Sigma-1 receptor (S1R) radioligands have the potential to detect and monitor various neurological diseases. Herein we report the synthesis, radiofluorination and evaluation of a new S1R ligand 6-(3-fluoropropyl)-3-(2-(azepan-1-yl)ethyl)benzo[d]thiazol-2(3H)-one ([18F]FTC-146, [18F]13). [18F]13 was synthesized by nucleophilic fluorination, affording a product with >99% radiochemical purity (RCP) and specific activity (SA) of 2.6 ± 1.2 Ci/Amol (n = 13) at end of synthesis (EOS). Positron emission tomography (PET) and ex vivo autoradiography studies of [18F]13 in mice showed high uptake of the radioligand in S1R rich regions of the brain. Pre treatment with 1 mg/kg haloperidol (2), non radioactive 13, or BD1047 (18) reduced the binding of [18F]13 in the brain at 60 min by 80%, 82% and 81% respectively, suggesting that [18F]13 accumulation in mouse brain represents specific binding to S1Rs. These results indicate that [18F]13 is a promising candidate radiotracer for further evaluation as a tool for studying S1Rs in living subjects.
Introduction
Initially the sigma receptor (SR) was thought to belong to the opioid class of receptors;1 however, further pharmacological evaluation classified it as a distinct molecular entity, resulting in its recognition as a separate family of receptors.2 There are two known SR subtypes, namely Sigma-1 and sigma-2 (S1R and S2R, respectively).3 At present, S1Rs are the most well characterized of the two subtypes.4
Despite initial controversy and conflicting ideas, recent key discoveries concerning the SR have helped elucidate various biological aspects about this molecular chaperone and its putative functional roles.5-7 Mainly located at the endoplasmic reticulum of cells, S1Rs have been implicated in a host of biochemical processes and pathological conditions including neurodegenerative diseases, psychiatric disorders, pain sensitization, drug addiction, digestive function, regulation of smooth muscle contraction, and ischemia.4, 5, 8-13 S1Rs are also highly expressed in most known human cancers (e.g., breast, lung, colon, ovarian, prostate, brain).9, 14 Agonists for S1Rs influence intracellular and extracellular Ca2+ levels and thus have a broad range of neuromodulatory effects.15, 16 Certain S1R agonists have been shown to regulate endothelial cell proliferation,17 improve cognition,18, 19 provide neuroprotection,20 and act as anti depressant agents,4, 21 while antagonists inhibit cocaine induced seizures,22 and attenuate neuropathic pain,23 highlighting the potential of S1Rs as both a diagnostic and therapeutic target.
There are numerous different compounds that target SRs, many of which fall into the following three classes of compounds; 1) benzomorphans, such as (+)-pentazocine (1, Figure 1) and (+)-N-allylnormetazocine (NANM) that preferentially bind S1Rs (compared to their (-)-enantiomers), 2) endogenous neurosteroids like progesterone (an antagonist of S1Rs) and 3) butyrophenones, such as the antipsychotic agent haloperidol (2, Figure 1) that displays high affinity for both SR subtypes.24, 25 Over the last two decades, several groups have reported the development of high affinity S1R ligands25-33 – and of these, some have been labeled with radioisotopes (Figure 1) for use in positron emission tomography (PET) studies.
Figure 1.

Selected Sigma-1 receptor (S1R) ligands and radioligands.
Examining S1Rs in living subjects with PET is an important step towards understanding the receptor's functional role and involvement in disease. PET radioligands specific for S1Rs could potentially provide a non invasive means of 1) investigating the distribution and function of these sites in living subjects – both in normal and disease states, 2) assessing receptor occupancy, to determine optimal doses of therapeutic drugs, 3) early detection and staging of S1R related disease(s), and 4) monitoring therapeutic response. Some existing S1R radioligands include: [11C]SA4503 (3),34 [18F]FM-SA4503 (4),35 [18F]FPS (5),36 [18F]SFE (6),37, 38 [11C]13 (7),30 and [18F]fluspidine (8)39 (Figure 1). Table 1 within the Supporting Information highlights the affinity and selectivity profiles of these compounds and compares them to 1 and 2. At present, the only radioligand being routinely used in S1R clinical research is 3, however this compound also displays affinity for the emopamil binding protein (EBP, Ki = 1.7 nM)40 and the vesicular acetylcholine transporter (VAChT, Ki = 50 nM)41, and is therefore not entirely specific for S1Rs.11, 42, 43 In order to accurately investigate and characterize the in vivo localization and role of S1Rs in both normal and disease states, a highly selective and specific imaging agent/technique is essential.
With the aim of synthesizing a new, selective fluorinated PET radioligand for studying S1Rs in living subjects, we identified a lead compound from the benzothiazolone class of compounds originally reported by Yous and colleagues in 2005.33 SN56 (9, Figure 1, Supporting Information Table 1) from this class was reported to have high affinity (Ki = 0.56 nM) and apparent high selectivity for the S1R (S2R/S1R > 1000). More recently, a tritiated version of this compound, [3H]9, was assessed in vitro44 and results suggested it may be a favorable alternative to the S1R radioligand [3H]1. We devised a strategy for modifying 9 in a way that would allow incorporation of a fluorine-18 radiolabel without greatly altering the structure of the molecule in the hope of maintaining its high affinity and selectivity for the S1R. Our target molecule, 6-(3-fluoropropyl)-3-(2-(azepan-1-yl)ethyl)benzo[d]thiazol-2(3H)-one 13 (FTC-146) 45 (Scheme 1) contains a fluoropropyl, in place of the propyl group on 9.
Scheme 1.

To the best of our knowledge, no compounds from the benzothiazolone class have been evaluated as PET radioligands for S1Rs. Since 13 has an entirely different scaffold from other known S1R radiotracers, and was born out of a class of highly selective S1R ligands, we believe studies using this compound may generate valuable and novel information about the S1R.
In this paper, we report the synthesis of 13, the radiosynthesis of [18F]13 and the preliminary evaluation of this new S1R radioligand through the use of cellular uptake assays, PET imaging of mice, ex vivo autoradiography and radiometabolite studies.
Results
Synthesis, Log P, and In Vitro Binding of 13
Compound 13 was prepared according to Scheme 1. Compounds 10 and 11 were synthesized using known procedures.44 The fluorinated intermediate 12 was successfully prepared from 11 via halogen exchange using tetra-n-butylammonium fluoride (TBAF) and potassium fluoride. Following this, 12 was alkylated with 2-(hexamethyleneimino)ethylchloride in the presence of potassium carbonate in DMF to afford 13.
The experimental pKa (10.4) of 13 was determined46 to be slightly higher than the calculated pKa value (9.36), and the experimental Log D PBS, pH 7.4 ± SD was measured to be 1.45 ± 0.04.
Using previously described methods,44 13 was subjected to radioligand binding assays, and found to demonstrate high affinity (Ki = 0.0025 nM) and superior selectivity for S1Rs (> 145,000-fold selectivity for S1R compared to S2R) in rat brain homogenates (Supporting Information Table 2). The affinity of 2 was determined in the same assay to serve as a reference standard, and it was found to have a Ki of 3.35 ± 0.79 nM for S1R and a Ki of 80.63 ± 14.08 nM for S2R in rat brain homogenates. In the same assay, 9 displayed a Ki of 1.7 ± 0.1 nM and 627 ± 115 nM in rat brain homogenates for S1R and S2R respectively. All initial binding assays were performed using brain homogenates because the CNS is a primary target for the radiotracer being developed. However, since many reports in the literature utilize liver for SR binding assays, the compounds were also tested under these conditions. Results from rat liver homogenate binding assays were as follows: 13 Ki = 0.96 ± 0.21 nM (S1R) and 467 ± 60 nM (S2R) (Supporting Information Table 2), 2 Ki = 3.3 ± 0.6 nM (S1R) and 57.2 ± 2.4 nM (S2R), 9 Ki = 1.61 ± 0.10 nM (S1R) and 247 ± 14 nM (S2R). In a NovaScreen and in-house profile of 59 targets, 13 displayed >10,000-fold selectivity for S1R compared to all other tested targets (Supporting Information Table 2). At 10,000 nM screening concentration, 13 exhibited <50% displacement of the radioligand, and at 100 nM screening concentration <50% displacement for ten targets, including: α2-adrenoceptors; histamine H2 receptors; muscarinic M2 receptors; peripheral muscarinic receptors; neuronal (α-bungarotoxin insensitive) nicotinic receptors; opioid receptors; norepinephrine transporters; calcium L type channels; sodium, site 2 channels; and acetylcholine esterase. Therefore, even for targets where there was modest displacement at the screening concentrations of 13, this still represented a >10,000-fold preference for S1Rs.
In Vitro Half-life Studies in Mouse and Rat Liver Microsomes
The metabolic stability of 13 was evaluated in mouse and rat liver microsomes. First, 13 was incubated in the presence of an NADPH-generating system at 37°C for 60 min. The reaction was initiated by adding cofactors and quenched at designated time points (0, 5, 10, 15, 30, 45, 60 min) by addition of an equal volume of ice-cold acetonitrile. 13 was found to have a half-life of 4.2 min with a clearance of 0.55 mL/min/g in mouse, and a half-life of 12.6 min with a clearance of 0.18 mL/min/g in rat liver microsomes.
Radiochemistry
The strategy for generating [18F]13 involved the preparation of a tosylate precursor 17 and its subsequent radiolabeling with fluorine-18 (Scheme 2). Compound 11 was reacted with benzoic acid to yield 14, which was then alkylated with 2-(hexamethyleneimino)ethylchloride. Hydrolysis of the intermediate 15 afforded the corresponding alcohol 16. The tosylate precursor was then prepared by reacting the alcohol with p toluenesulfonyl chloride in the presence of triethylamine. [18F]13 was successfully synthesized via nucleophilic substitution using an automated GE TRACERlab FXFN radiosynthesis module. Fluorine-18 (half-life = 109.8 min) radiolabeling was accomplished by reaction of tosylate precursor (17) with cyclotron- produced 18F-fluoride as an 18F-labeled Kryptofix-222/K+/[18F]F− complex in DMSO at 150 °C for 15 min. Semi-preparative reverse-phase high performance liquid chromatography (HPLC) of the crude reaction mixture afforded [18F]13 in 3.7 ± 1.9% yield (n=13) at end of bombardment (EOB) (and 2.5 ± 1.2% yield at EOS), >99% radiochemical purity (RCP), with a specific activity (SA) of 3.9 ± 1.9 Ci/Amol (EOB) (and SA of 2.6 ± 1.2 Ci/Amol at EOS) in a total synthesis time of 75 min. The formulated version of [18F]13 in saline/ethanol (9:1, total 10 mL) was shown to be stable for at least 5.5 hours via analytical reverse-phase HPLC.
Scheme 2.

Radiosynthesis of [18F]13
Cell Uptake Studies
Uptake of [18F]13 in Chinese hamster ovarian (CHO) cells was compared to the uptake of the known S1R ligand [3H]1. Control CHO cells (transfected with a vector not containing the S1R gene - to serve as a negative control) and CHO cells transfected with a vector containing S1R cDNA (to serve as a positive control for S1R expression in cells) were used for the uptake assays. Cells were exposed to [18F]13 or [3H]1 for 30 and 120 min (triplicate for each time point). The incubated cells were subsequently washed, lysed and counted for radioactivity. All collected data were normalized for amount of protein present in each well. Data for both uptake assays (Figure 2) showed there was a small increase in uptake for both radioligands between 30 and 120 min in control CHO cells. This increase was more pronounced in CHO cells transfected with S1R cDNA, and numerically higher at both 30 and 120 min compared with negative control CHO cells. The uptake of [18F]13 in cells transfected with S1R cDNA was 4-fold higher than uptake in control CHO cells at 120 min (Figure 2).
Figure 2.
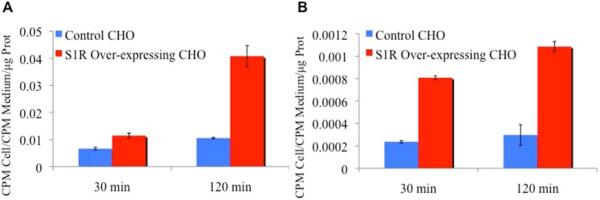
Cell uptake results for A) [18F]13 and B) [3H]1. Uptake of [18F]13 and [3H]1 was determined in control CHO cells and CHO cells transfected with Sigma-1 receptor (S1R) cDNA following incubation for either 30 or 120 min. Results are expressed as counts per minute (CPM) recorded in a sample from a particular well/CPM recorded in medium/amount of protein (μg) present in a sample from that well.
Western Blot
Western blot analysis was performed using Image J (image processing and analysis software in java) and showed that the level of S1R expression in the CHO cells transfected with the S1R cDNA was approximately 4.3 times greater than that found in the control CHO cells that had been transfected with an empty vector (Figure 3).
Figure 3.
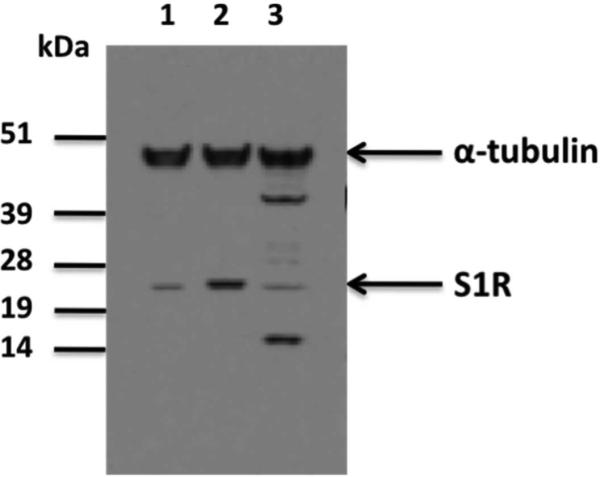
Western blot analysis of Sigma-1 receptor (S1R) expression in control CHO cells, CHO cells transfected with S1R cDNA, and a positive control cell line (JAR cells) known to contain S1R protein. Cell lysates (50 μg of protein) were subjected to gel electrophoresis followed by immunoblot analysis with S1R specific antibody S 18 (400:1). Lane 1: control CHO cells (transfected with empty S1R vector); lane 2: CHO cells over-expressing S1R (transfected with vector containing S1R cDNA); and lane 3: positive control cell lysate for S1R as supplied by Santa Cruz Biotech (JAR cells). Blot was also stained for α-tubulin as a protein loading control.
PET Imaging in Mice
The in vivo kinetics of [18F]13 in normal mice were assessed using small animal PET. Dynamic brain PET scanning was commenced one minute prior to administration of [18F]13 and terminated 60 min later. Graphs depicting uptake of [18F]13 in whole mouse brain as a function of time for baseline and blocking studies are displayed in Figure 4. The baseline time activity curve (TAC) (Figure 4) demonstrated that [18F]13 entered the brain rapidly, peaked within the first few minutes and then gradually decreased over the remaining time of the scan. Pre treatment with 2 (1 mg/kg), 13 (1 mg/kg), or N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino)-ethylamine (BD1047, 18, 1 mg/kg) 10 min prior to radioligand administration reduced the binding of [18F]13 in the brain at 60 min by 80%, 82%, and 81% respectively (Figure 4). PET images provided visual evidence that [18F]13 rapidly crossed the blood brain barrier (BBB) and appeared to accumulate in known S1R rich regions (Figure 5, sagittal brain PET image). There also seemed to be some accumulation in the snout area and spine over the course of the study. Since the spatial resolution of PET and partial volume effects did not allow for accurate determination of radioligand localization in specific brain regions, ex vivo autoradiography was performed (see Figure 5 and below description).
Figure 4.

Time activity curves (TACs) from mouse positron emission tomography (PET) studies. TACs represent accumulation of [18F]13 in whole mouse brain as a function of time for baseline (n=3), pre block with 2 (n=3), pre block with 13 (n=3), and pre block with 18 (n=3). Baseline studies involved iv administration of [18F]13 (95-125 μCi), whereas blocking studies involved pre-treatment of mice with either 2 (1 mg/kg), 13 (1 mg/kg) or 18 (1 mg/kg) 10 min prior to iv administration of [18F]13 (95-125 μCi).
Figure 5.
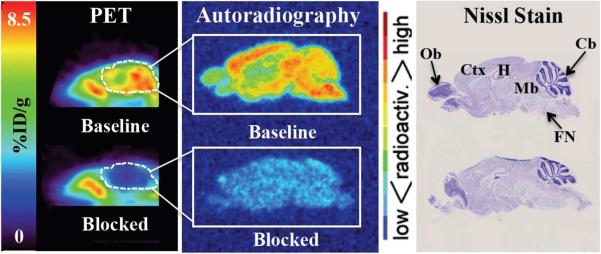
Sagittal mouse PET images and ex vivo autoradiography of sagittal brain sections (12 μm) obtained 60 min post administration of [18F]13. Sections used for autoradiography were stained with nissl for anatomical correlation (right). Baseline study involved injection of radiotracer only ([18F]13, 200 μCi), whereas the blocked study involved pre-treatment with known Sigma-1 receptor ligand 18 (1 mg/kg) 10 min prior to radiotracer administration. PET images (5 min static scans) were acquired just prior to perfusing mice and harvesting brain tissue for autoradiography. White dotted lines indicate location of mouse brain in sagittal PET images. Cb = Cerebellum, Ctx = Cortex, FN = Facial Nucleus, H = Hippocampus, Mb = Midbrain, Ob = Olfactory Bulb.
Ex Vivo Autoradiography
The distribution of [18F]13 in the mouse brain was further evaluated by ex vivo autoradiography at 60 min post injection of tracer. In brief, mice were perfused with saline, brains were snap frozen, sectioned (12 μm), and incubated with phosphor imaging film overnight. Figure 5 shows the autoradiography results for selected 12 ⌈m sagittal brain sections from both the baseline and blocking study (the latter of which was pre-treated with 18 (1 mg/kg) 10 min prior to injection of [18F]13). From these images it can be seen that [18F]13 mainly accumulated in the midbrain, facial nucleus, cortex and hippocampus. There was also some accumulation in the cerebellum, albeit lower than that observed in the midbrain and cortex. The corpus callosum appeared to be devoid of [18F]13 binding. Autoradiography results from the blocking study depicted a dramatic reduction in 18F]13 uptake in the mouse brain. A similar pattern of radioligand distribution can be seen when comparing the sagittal brain PET images with the autoradiography results for both the baseline and blocking study (Figure 5).
Ex Vivo Determination of Radiometabolites: Mouse Plasma, Liver and Brain
The percentage of intact [18F]13 in mouse plasma, liver and brain (at 30 and 60 min post administration of tracer) was assessed via HPLC. Figure 6 shows that the percentage of intact [18F]13 in liver was lower than that observed in plasma (34% at 30 min and 27% at 60 min, compared to 60% and 50% for plasma), while the percentage of intact tracer in the brain remained 100% throughout the duration of our studies.
Figure 6.

Graph depicting the percentage of intact radiotracer ([18F]13) in mouse blood, liver, and brain at 30 and 60 min post injection.
Discussion
Since S1Rs are believed to be intimately associated with numerous psychiatric conditions and neurodegenerative diseases, radioligands specific for S1Rs have the potential to serve as novel diagnostic tools, and may be useful in assessing treatment effectiveness. The present study describes the synthesis and radiolabeling of a new S1R PET radioligand from the benzothiazolone class of compounds, together with its preliminary in vitro and in vivo characterization using cell uptake studies, metabolic stability tests, PET imaging of mice, and ex vivo autoradiography.
Compound 13 was successfully synthesized (Scheme 1) and found to have an experimental Log D of 1.45 ± 0.04. We also investigated the in vitro binding properties of 13 and it was found to display high affinity (Ki = 0.0025 nM, Supporting Information Table 2) and superior selectivity for S1Rs (> 145,000-fold selectivity for S1Rs compared to S2Rs) in rat brain homogenate when compared to its parent 9. In vitro binding of 13 was also evaluated in liver homogenates and found to be Ki = 0.96 ± 0.21 nM (S1R) and 467 ± 60 nM (S2R). The affinity of 2 for S1R and S1R was determined in the same binding assays to serve as a standard reference. The Ki of 2 in rat brain for S1R and S2R was found to be 3.35 ± 0.79 nM and 80.63 ± 14.08 nM, respectively, which was comparable to affinities reported for this compound in the literature,49-51 thus validating our assay conditions and technique. Our findings concerning the in vitro binding data for 13 demonstrated that the small structural modification made to 9 in order to form 13 led to an improvement in affinity and selectivity for S1Rs. In fact, both the affinity and selectivity (S1R vs. S2R) of 13 are higher than values reported for most other known S1R ligands (Supporting Information Table 1). The large increase in S1R affinity found in rat brain assay when comparing 13 with its parent 9 is unlikely to be explained through classical structure activity relationships. One possible explanation for the large increase in affinity could be that in addition to 13 binding S1Rs, it may interact with one or more proteins when engaged, which could affect the affinity at the S1R site. This is because S1Rs are ligand-gated chaperones that participate in protein protein interactions to modulate activity at various cellular targets, such as ion channels, receptors, and signaling pathways. The reason for the difference in affinity of 13 in the rat brain and rat liver assay is unknown, but thought to be related to differences in the assay environment, most likely related to lipid composition and/or variations in protein partners that may be present in liver versus brain.
Radiosynthesis of [18F]13 was achieved by nucleophilic aliphatic [18F]fluorination of compound 17 (Scheme 2). Since heating the reaction (precursor concentration 2 mg/mL) at 150 °C for 15 min afforded high purity product in sufficient yields (2-5%, 1-5 mCi/mL) for preliminary in vitro and in vivo investigations, no further optimizations were pursued at this stage.
It is typical to differentiate between S1R and S2Rs using benzomorphan-type opiates such as the well-known selective S1R radioligand [3H]1. For this reason we selected [3H]1 as the “gold standard” S1R ligand to compare with [18F]13 in cell uptake studies. Results obtained from cell uptake studies demonstrated a 4-fold higher uptake of [18F]13 in CHO cells transfected with S1R cDNA compared to control CHO cells at 120 min (Figure 2). This was comparable to the 3.6-fold greater uptake of [3H]1 in transfected CHO cells versus uptake in control cells at 120 min, indicating that [18F]13 behaves similarly to [3H]1, and that it might be a more sensitive marker of S1R levels. Western blot results (Figure 3) verified the level of [18F]13 uptake in cell assays (at 120 min) correlated to the level of S1R protein levels and therefore highlight its potential as a radioligand for accurately identifying and visualizing S1Rs.
Following these encouraging in vitro cell uptake results, the in vivo kinetics and binding of this radiofluorinated ligand were evaluated in living, normal mice using small animal PET. Baseline TACs (Figure 4) showed that [18F]13 rapidly crossed the BBB, reached a maximum uptake of ~17%ID/g within the first few minutes, and then slowly washed out of the brain throughout the remainder of the scan to a level of 6%ID/g at 60 min. Pre-treating mice with 2 (1 mg/kg) or 13 (1 mg/kg) 10 min prior to radioligand administration led to a marked reduction of [18F]13 binding in the brain (80% or 82% reduction respectively, at 60 min) (Figure 4). Comparable results have been reported for other S1R-related rodent blocking studies with 2.35, 52 Since 2 also displays affinity for dopamine receptor type 2 (D2), S2R, and VAChT, we also performed blocking studies with 18 (a known S1R selective antagonist, structurally unrelated to 13). Results from these blocking studies demonstrated 81% reduction in [18F]13 uptake in the brain at 60 min, indicating that [18F]13 accumulation in mouse brain likely represents specific S1R binding. Ex vivo autoradiography (Figure 5) showed that [18F]13 mainly accumulated in midbrain (especially facial nucleus), cortical regions, hippocampus, and to a lesser extent in the cerebellum and thalamus, which are all known to contain S1Rs.34 Our autoradiography results were similar to that seen in other S1R ex vivo autoradiography studies.28, 39 PET images and autoradiography results from the blocking studies with 18 (1 mg/kg) demonstrate notable reduction of [18F]13 accumulation in mouse brain (Figure 5). The PET signal present outside the brain after blocking with 18 could be due to peripheral defluorination of [18F]13 leading to uptake of [18F]fluoride ion in jaw bone and spine. Importantly, ex vivo radiometabolite analysis revealed that [18F]13 in brain remained intact throughout the duration of our studies (Figure 6), meaning that the PET signal observed in the brain was entirely due to intact [18F]13.
Although [18F]13 is yet to be evaluated alongside other fluorinated S1R radioligands, its initial kinetics (i.e. rapid uptake in mouse brain within the first few minutes) appear similar to that reported for [18F]4 and [18F]8 in normal mice.39 On the other hand, the binding profile of [18F]13 at later time points is quite different from the reported uptake levels for other known S1R radioligands at corresponding times. For example, [18F]13 reached its maximum uptake in mouse brain within the first few minutes of imaging, and gradually washed out to 65% of its maximum at 60 min post injection. Whereas, [18F]4 reached its maximum uptake in the brain at 30 min post injection and did not experience significant washout over the remainder of the study (120 min post injection). Biodistribution studies with [18F]8 demonstrated that it reached maximum uptake in the mouse brain at 30 min post injection and then washed out to a level 81% of its maximum at 60 min post injection. Future studies involving the use of available S1R tracers in the same animal (and/or animal model of disease) are needed to properly understand the relative advantages/disadvantages of each tracer.
Since there was some observed bone uptake in our mouse PET studies we had reason to suspect possible defluorination of [18F]13. Further investigation of the metabolic profile of 13 revealed the major metabolites were hydroxylation (oxidation) products, and that a minor metabolite (<10%, data not shown) of a defluorinated product was apparent at all time points. The molecular formula suggests a radical defluorination mechanism, although this needs to be confirmed through more detailed studies. Importantly, these results are from in vitro measurements and do not indicate the actual in vivo metabolic profile of this compound, and cannot predict the metabolism of [18F]13 in different species.
Bone uptake has also been reported in studies using other S1R tracers35, 39 and has been postulated to be due to high levels of S1Rs in highly proliferative tissues (e.g., bone marrow). Furthermore, Mavlyutov and colleagues showed that S1Rs are expressed in the mouse spinal cord.53 We are currently evaluating [18F]13 in rats and non-human primates and preliminary results in rats show less bone uptake. Both rat and monkey data will be reported in a later publication. Future studies will involve further characterization of [18F]13, especially concerning its selectivity and specificity for S1Rs. Binding to EBP and VAChT will be determined, along with PET imaging and ex vivo autoradiography in S1R knockout mice. Other studies will involve comparative analysis of [18F]13 with other known S1R radioligands in the same animal models of disease (e.g., Alzheimer's disease, neuropathic pain) to determine which is most suitable for clinical applications.
In conclusion, we have successfully prepared a new benzothiazolone 18F-labeled S1R ligand, [18F]13, that demonstrates promise as a specific PET radiotracer for visualizing S1Rs in living subjects, and therefore warrants further investigation.
Experimental Section
Chemistry
Unless otherwise stated, reagents and starting materials were obtained from commercial suppliers and were used without purification. Pre-coated silica gel GF Uniplates (Analtech) were used for thin-layer chromatography (TLC). Column chromatography was performed on silica gel 60 (Sorbent Technologies). 1H and 13C NMR spectra were obtained on a Bruker APX400 at 400 and 100 MHz, respectively. The high-resolution mass spectra (HRMS) were recorded on a Waters Micromass Q-Tof Micro mass spectrometer with a lock spray source. The mass spectra (MS) were recorded on a WATERS ACQUITY Ultra Performance LC with ZQ detector in ESI mode.
The purity of final compounds (>97%) was verified by HPLC on a Waters 2695 system equipped with a Waters X-Bridge C18 column and a Waters 996 UV detector set at 254 nm. The following conditions were used: a flow rate of 1 mL/min, a gradient run from 50% eluent A (H2O), 40% eluent B (acetonitrile), 10% eluent C (H2O/triethylamine, 98.2:0.2) to 0% eluent A, 90% eluent B, 10% eluent C for 12 min. Chemical names were generated using ChemDraw Ultra (CambridgeSoft, version 10.0).
The UPLC/MS/MS mass spectrophotometer consisted of a Waters Micromass Quattro Micro™ triple-quadrupole system (Manchester, UK). The system was controlled by MassLynx software version 4.0. Ionization was performed in the positive electrospray mode. The MS/MS parameters for the analysis were as follows: capillary voltage 4.95 kV, cone voltage 31 V, extractor voltage 5V, RF lens voltage 0.5V. The source and desolvation temperatures were 110 °C and 400 °C, respectively, and the desolvation and cone gas flows were 252 and 76 L/hr, respectively. The selected mass-to-charge (m/z) ratio transition of 13 ion [M+H]+ used in the single ion recording (SIR) was m/z 337.03 The dwell time was set at 500 ms. Calculated pKa and Log P were determined using PALLAS 3.1.2.4 Software from CompuDrug Chemistry, Ltd (Sedona, AZ USA).
For the reported radiochemistry, Semi-preparative HPLC separations were performed on Dionex 680 pump with KANUR UV detector K-2001 (for purification of [18F]13). Analytical HPLC was performed on Lab Alliance with Model 500 UV detector. Radioactivity in HPLC eluates was detected with a model 105S single-channel radiation detector (Carroll & Ramsey Associates). The UPLC system, consisted of Water's Acquity UPLC (Milford, MA, USA) equipped with a binary solvent manager, vacuum degasser, thermostatted column compartment, and an autosampler. Chromatographic separations were performed on a Waters Acquity UPLC™ BEH C18 column (1.7 μm, 2.1 × 50 mm). For the metabolite studies, an isocratic method was developed using a mobile phase containing 0.1% formic acid in water: 0.1% formic acid in methanol (50:50, v/v). For the metabolite separation, a linear gradient method was developed with a mobile phase containing 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The linear gradient elution program was as follows: 0-80% B over 6 min, followed by an isocratic hold at 80% B for another 4 min. At 10 min, B was returned to 0% in 2 min and the column was equilibrated for 3 min before the next injection. The total run time for each injection was 15 min. The flow rate was 0.2 mL/min. The column temperature was maintained at 25 °C and the injection volume was 10 μL. For metabolite studies an Agilent 1200 HPLC system with autosampler and Gabi radioactivity detector (Raytest) was used.
Synthetic procedures for 6-(3-chloropropanoyl)benzo[d]thiazol-2(3H)-one (10) and 6-(3-chloropropyl)benzo[d]thiazol-2(3H)-one (11) are described in the Supporting Information.
6-(3-fluoropropyl)benzo[d]thiazol-2(3H)-one (12)
A mixture of 11 (0.30 g, 1.32 mmol), KF (0.23 g, 3.95 mmol) and TBAF (1M in THF, 3.95 mL, 3.95 mmol) in THF (10 mL) was heated at reflux for 4 h. After completion of the reaction, the reaction mixture was partitioned between ethyl acetate and water, and the organic layer was washed with brine and dried. The solvent was removed in vacuo, and the residue was purified by chromatography on a silica gel column using petroleum ether/ether (8:2) as the eluent to give 0.010 g (36%) of 6-(3-fluoropropyl)benzo[d]thiazol-2(3H)-one as a white solid. 1H NMR (CDCl3): δ 10.33 (br s, 1H), 7.23 (s, 1H), 7.10 (s, 2H), 4.45 (dt, J = 47.2, 5.8 Hz, 2H), 2.76 (t, J = 7.6 Hz, 2H), 2.00 (dquint, J = 25.2, 6.8 Hz, 2H). 13C NMR (CDCl3): δ 173.26, 136.39, 133.69, 126.83, 124.09, 122.13, 111.79, 82.77 (d, J = 164.2 Hz), 32.13 (d, J = 19.7 Hz), 31.01 (d, J = 5.2 Hz). MS (ESI ) m/z 210 (M+-1).
3-(2-(azepan-1-yl)ethyl)-6-(3-fluoropropyl)benzo[d]thiazol-2(3H)-one hydrochloride (13)
K2CO3 (0.180 g, 1.30 mmol) and 2-(hexamethyleneimino)ethylchloride hydrochloride (0.080 g, 0.40 mmol) were added, under mechanical stirring, to a solution of 12 (0.090 g, 0.43 mmol) in anhydrous DMF (2 mL). The reaction mixture was heated at 55 °C for 2 h. After cooling, the mixture was poured into 10 mL of water, extracted with ethyl acetate (3 × 20 mL), washed with saturated aqueous NaCl and dried. The solvent was removed in vacuo, and the residue was purified by chromatography on a silica gel column using methylene chloride/methanol (9.5:0.5) as the eluent. 3-(2-(azepan-1-yl)ethyl)-6-(3-fluoropropyl)benzo[d]thiazol-2(3H)-one was isolated as a hydrochloride salt (white solid, 0.120 g, 80%) by addition of HCl/dioxane. 1H NMR (D2O): δ 7.34 (br s, 1H), 7.26-7.24 (m, 1H), 7.16-7.14 (m, 1H), 4.46 (dt, J = 47.2, 4.5 Hz, 1H), 4.28 (t, J = 4.8 Hz, 2H), 3.49-3.37 (m, 6H), 2.70-2.66 (m, 2H), 1.97-1.66 (m, 11H). 13C NMR (D2O): δ 173.02 (C=O), 137.92 (Cq), 133.68 (Cq), 127.31 (CHar), 122.67 (CHar), 122.09 (Cq), 110.90 (CHar), 84.33 (d, J = 157.6 Hz, CH2), 55.23 (CH2), 53.46 (CH2), 37.47 (CH2), 31.34 (d, J = 18.8 Hz, CH2 ), 30.30 (d, J = 5.5 Hz, CH2), 25.61 (CH2), 23.37 (CH2). HRMS (ESI+) calculated for C18H26N2OFS [M+H]+ 337.1750, found 337.1764.
3-(2-oxo-2,3-dihydrobenzo[d]thiazol-6-yl)propyl benzoate (14)
K2CO3 (5.31 g, 38.4 mmol) and benzoic acid (9.38 g, 76.8 mmol) were added, under mechanical stirring, to a solution of 11 (3.51 g, 15.4 mmol) in anhydrous DMF (250 mL). The reaction mixture was heated at 110 °C for 6 h. After cooling, the mixture was poured into 100 mL of a 2.5 N HCl solution in water, extracted with ethyl acetate (3 × 70 mL), and the organic phase was washed with brine. The solvent was dried over Na2SO4 and evaporated in vacuo and the residue was purified via chromatography on a silica gel column using a gradient of petroleum ether/ethyl ether (4:6 to 6:4) as the eluent. The product was then recrystallized in toluene to give 2.97 g (62%) of 3-(2-oxo-2,3-dihydrobenzo[d]thiazol-6-yl)propyl benzoate as a white solid. 1H NMR (DMSO-d6): δ 11.70 (br s, 1H), 7.91 (d, J = 7.6 Hz, 2H), 7.63 (t, J = 7.5 Hz, 1H), 7.49 (t, J = 7.6 Hz, 2H), 7.41 (s, 1H), 7.12 (d, J = 8.1 Hz, 1H), 7.02 (d, J = 8.1 Hz, 1H), 4.25 (t, J = 6.3 Hz, 2H), 2.71 (t, J = 7.4 Hz, 2H), 2.03-1.97 (m, 2H). 13C NMR (DMSO-d6): δ 170.00, 165.70, 135.77, 134.42, 133.22, 129.76, 129.10, 128.64, 126.58, 123.44, 122.18, 111.34, 64.02, 31.34, 29.93. MS (ESI−) m/z 312 (M+-1, 100), 190 (52) .
3-(3-(2-(azepan-1-yl)ethyl)-2-oxo-2,3-dihydrobenzo[d]thiazol-6-yl)propyl benzoate (15)
K2CO3 (0.75 g, 5.47 mmol) and 2-(hexamethyleneimino)ethylchloride hydrochloride (0.47 g, 2.37 mmol) were added, under mechanical stirring, to a solution of 14 (0.57 g, 1.82 mmol) in anhydrous DMF (10 mL). The reaction mixture was heated at 65 °C for 2 h. After cooling, the mixture was poured into 80 mL of water, extracted with ethyl acetate (3 × 60 mL), and the combined organic layers were washed with brine and dried. The solvent was removed in vacuo, and the residue was chromatographed on a silica gel column using diethyl ether as the eluent to give 0.72 g (90%) of 3-(-3-(2-(azepan-1-yl)ethyl)-2-oxo-2,3-dihydrobenzo[d]thiazol-6-yl)propyl benzoate as a colorless oil. A sample was prepared as a hydrochloride salt for analysis. 1H NMR (DMSO-d6): δ 11.29 (br s, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.66-7.57 (m, 3H), 7.50 (t, J = 7.6 Hz, 2H), 7.28 (d, J = 8.0 Hz, 1H), 4.43-4.40 (m, 2H), 4.27 (t, J = 6.0 Hz, 2H), 3.44-3.18 (m, 6H), 2.77 (t, J = 7.2 Hz, 2H), 2.06-1.56 (m, 10H). 13C NMR (DMSO-d6): δ 168.74 (CO), 165.52 (CO), 136.68 (Cq), 134.23 (Cq), 133.07 (CHar), 129.59 (Cq), 128.92 (CHar), 128.50 (CHar), 126.78 (CHar), 122.45 (CHar), 121.44 (Cq), 111.36 (CHar), 63.83 (CH2), 53.62 (CH2), 52.05 (CH2), 37.02 (CH2), 31.10 (CH2), 29.72 (CH2), 25.58 (CH2), 22.88 (CH ). HRMS (ESI+) calculated for C25H31N2O3S [M+H]+ 439.2055, found 439.2056.
3-(2-(azepan-1-yl)ethyl)-6-(3-hydroxypropyl)benzo[d]thiazol-2(3H)-one (16)
To a solution of 16 (0.67 g, 1.53 mmol) in methanol (10 mL) was added a solution of sodium hydroxide (0.15 g, 3.75 mmol) in water (10 mL). The mixture was heated at 90 °C for 1 h, concentrated in vacuo, poured into 1N HCl (20 mL) and extracted with ethyl acetate (10 mL). The pH of the aqueous layer was adjusted to 10 with potassium carbonate and the mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine, dried and evaporated. The residue was chromatographed on a silica gel column using methylene chloride/methanol (9.7:0.3) as the eluent to give 0.47 g (92%) of 3-(2-(azepan-1-yl)ethyl)-6-(3-hydroxypropyl)benzo[d]thiazol-2(3H)-one as a white solid. A sample was prepared as a hydrochloride salt for analysis. 1H NMR (DMSO-d6): δ 11.35 (br s, 1H), 7.46 (d, J = 1.2 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.50 (dd, J = 8.0, 1.2 Hz, 1H), 4.31 (t, J = 6.8 Hz, 2H), 3.80 (br s, 2H), 3.53 (s, 1H), 3.39-3.29 (m, 6H), 2.60 (t, J = 7.6 Hz, 2H), 1.79 (br s, 4H), 1.68 (quint, J = 8.0 Hz, 2H), 1.58 (br s, 4H). 13C NMR (DMSO-d6): δ 170.04 (CO), 138.34 (Cq), 134.45 (Cq), 127.41 (CHar), 122.92 (CHar), 121.91 (Cq), 111.58 (CHar), 60.26 (CH2), 54.60 (CH2), 53.07 (CH2), 37.70 (CH2), 34.50 (CH2), 31.47 (CH2), 25.99 (CH2), 23.58 (CH2). HRMS (ESI+) calculated for C28H27N2O2S [M+H]+ 335.1793, found 335.1786.
3-(3-(2-(azepan-1-yl)ethyl)-2-oxo-2,3-dihydrobenzo[d]thiazol-6-yl)propyl 4-methyl benzenesulfonate (17)
A solution of p-toluenesulfonyl chloride (0.22 g, 1.26 mmol) in methylene chloride (10 mL) was slowly added to a solution of 16 (0.38 g, 1.15 mmol) and triethylamine (0.34 mL, 2.42 mmol) in methylene chloride (20 mL). The mixture was stirred for 3 days at room temperature and the solvent was evaporated. The residue was purified by chromatography on a silica gel column using a gradient of methylene chloride/methanol (10:0 to 9.7:0.3) as the eluent to give 0.50 g (89%) of 3-(-3-(2-(azepan-1-yl)ethyl)-2-oxo-2,3-dihydrobenzo[d]thiazol-6-yl)propyl 4-methyl benzenesulfonate as a pale yellow oil. 1H NMR (DMSO-d6): δ 7.78 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.30 (s, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 4.00-3.93 (m, 4H), 2.71 (t, J = 6.8 Hz, 2H), 2.59-2.55 (m, 6H), 2.41 (s, 3H), 1.88-1.85 (m, 2H), 1.45 (br s, 8H). 13C NMR (DMSO-d6): δ 168.36, 144.68, 135.24, 135.11, 132.32, 129.98, 127.41, 126.45, 122.10, 121.25, 111.11, 69.77, 54.84, 54.21, 40.64, 30.13, 29.80, 27.93, 26.27, 20.95. MS (ESI+) m/z 489 (M++1, 100), 335 (4).
Determination of pKA for 13
The pKa of 13 was determined using the potentiometric titration method.46 A solution of 0.01 M sodium hydroxide was prepared and the pH measured as 11.9. Similarly, 0.01 M hydrochloric acid solution was prepared and the pH measured as 2.07. To 50 mL of a 1 mM 13 solution, 0.1 mL volumes of sodium hydroxide were added and pH recorded (Mettler Toledo SevenEasyTM pH meter S20) until the pH of the solution became constant. To the same sample, 0.1 mL portions of hydrochloric acid were added and pH recorded until it became constant. A titration curve was then plotted as pH versus volume of base/acid added. The intersection point of these two curves was noted as the pKa value of 10.4.
Determination of Partition coefficient (Log D) for 13
Using the Stir flask method,47 n-Octanol and water/PBS, pH 7.4 (equal quantity) were added to a glass vial (25 mL). The contents were sealed and stirred continuously for 24 h at 25 °C to achieve mutual saturation of the phases. Water/PBS, pH 7.4 phase was brought into a vessel together with a Teflon-coated magnetic stirring bar. The n-octanol phase containing the known quantity of test substance was poured very carefully on top of the aqueous phase in order to avoid emulsion formation as far as possible. The vessel was not shaken; instead the system was stirred for an extended period of time (at least 36 h) allowing equilibrium to be reached. The contents were allowed to separate upon standing, and then centrifuged. An aliquot of aqueous layer was taken and diluted (1000 times) for quantitative analysis by UPLC/MS/MS.
In Vitro Radioligand Binding Assays
Competition binding assays were performed as previously described.54,56 Briefly, radioligands were used to tag the targeted sites under standard conditions, with S2R binding performed in the presence of 300 nM 1 to mask radioligand binding to S1Rs. In addition, SR binding was performed for non radioactive 13, 2, and 9 in rat liver homogenates using a 96-well platform.57 Non radioactive 13 was initially evaluated at non SR sites using a screening concentration of 10,000 nM. If <50% displacement of the radioligand was observed, then the results were reported as Ki >10,000 nM. If >50% displacement was observed, for assays run by NovaScreen/Caliper Life Sciences (Hanover, MD), a second screening concentration of 100 nM was examined, and Ki values were estimated based on radioligand displacement of the two screening concentrations of 13. If the estimated Ki yielded a selectivity ratio relative to S1R of less than 100-fold, then full competition curves were run; all targets with moderate affinity for non-SR sites from the NovaScreen assays greatly exceeded the 100-fold selectivity criterion and it was not necessary to run full competition assays. A panel of seven non-SR sites that have been historically problematic for earlier sigma ligands (three monoamine transporters as well as opioid, NMDA, dopamine D2, and serotonin 5-HT2 receptors) were performed in-house and full competition assays were conducted if there was >30% displacement of the radioligand at the 10,000 nM screening concentration. For the in-house assays, at least 10 concentrations of 13 were tested to generate IC50 values, which were converted to Ki values using the Cheng-Prufsoff equation.
In Vitro Half-life Studies in Mouse and Rat Liver Microsomes
Non-radioactive 13 was incubated in the presence of an NADPH-generating system at 37 °C for 60 min in test tubes. The basic incubation mixture consisted of 5 mM substrate, 1 mg/mL microsomal protein, 3 mM MgCl2, 1 mM NADP, 5 mM glucose-6-phosphate, 1 IU/mL glucose-6-phosphate dehydrogenase, 100 mM Tris HCl buffer (pH 7.4) in a final volume of 1 mL. The reaction was initiated by adding cofactors and quenched at designated time points (0, 5, 10, 15, 30, 45, 60 min) by addition of an equal volume of ice-cold acetontrile. The mixture was centrifuged at 3000 rpm for 10 min, and the supernatant was analyzed by UPLC/MS/MS.
Radiosynthesis of [18F]13
No carrier added-aqueous [18F]fluoride ion was produced on a PETtrace cyclotron (GE Healthcare, Sweden) by irradiation of a 1.6 mL water target using a 16 MeV proton beam on 95% enriched [18O]H2O by the [18O(p,n)18F] nuclear reaction. [18F]Fluoride in [18O]H2O was transferred to a GE TRACERlab FXFN synthesizer and passed through an anion exchange resin (QMA cartridge in carbonate form, prepared by washing with 1 mL EtOH and 1 mL of water) under vacuum. Trapped [18F]fluoride ions were then eluted from the QMA cartridge and transferred to the reactor using an eluent solution containing 3.5 mg of K2CO3 and 15 mg of Kryptofix-222 (K222: 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo [8.8.8] hexacosan) in acetronitrile (0.9 mL) and water (0.1 mL) mixture. The solution was then evaporated at 65 °C under helium flow and vacuum, followed by heating at 88 °C under vacuum. Tosylate precursor 17, 3-(2-Oxo-3-(2-(azepan-1-yl)ethyl)-2,3-dihydrobenzo[d]thiazol-6-yl)propyl 4-methylbenzenesulfonate (2 mg) was dissolved in DMSO (1 mL) and added to the dry Kryptofix-222/K+[18F]F− complex. The mixture was allowed to react at 150 °C for 15 min. Upon completion, the reaction mixture was diluted with sterile water (8 mL) and passed through a C18 Sep-Pak cartridge. The C18 trapped-radiolabelled-product was then eluted from the C18 Sep-Pak with acetonitrile (1.5 mL) and sterile water (1.5 mL). The resulting crude mixture was then injected onto two serial HPLC Phenomenex Gemini C 18, 5 μm (10 × 250 mm) Semi-preparative reversed phase column. Using a mobile phase of H2O (0.1% TEA) : acetonitrile (0.1% TEA), (pH = 8) : (20/80, v:v), and with a flow rate of 5.0 mL/min, the retention time (tR) of [18F]13 was 13 min. Tosylate precursor and decomposed hydroxyl derivative were eluted at 17.5 min and 10 min respectively. The radioactive fraction corresponding to [18F]13 was collected in a round bottom flask containing sterile water (15 mL) and then passed through a C18 Sep-Pak. A further 10 mL of sterile water was passed through the C18 Sep-Pak. The trapped, purified radiolabelled product was eluted from the C18 Sep-Pak using ethanol (1 mL) and saline (9 mL). The formulated solution was then filtered through a sterile 13 mm Millipore GV 0.22 μm filter into a sterile pyrogen free evacuated 30 mL vial. Solutions in saline containing no more than 10% ethanol by volume were used for the studies described in this article.
Quality control of [18F]13
For determination of specific activity and radiochemical and chemical purity, an aliquot of the final solution of known volume and radioactivity was injected onto an analytical reversed phase HPLC column (Phenomenex Gemini C18 5Am (4.6 × 250 mm). A mobile phase of H2O (0.1% TEA) : acetonitrile (0.1% TEA) : (20:80; v:v) at a flow rate of 1.0 mL/min was used to elute [18F]13 with a retention time (tR) of 8.33 min. The area of the UV absorbance peak measured at 254 nm corresponding to the carrier product was measured (integrated) on the HPLC chromatogram and compared to a standard curve relating mass to UV absorbance.
Cell Uptake Studies using Transfected Cells
CHO cells were grown in Ham's F-12 medium. For uptake studies CHO cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and either pcDNA (empty vector, negative control) or S1R gene (OPRS1, accession number NM_005866.2, OriGene, Rockville, MD, USA) following directions from manufacturer. The cells were harvested and 2 × 105 cells were seeded per well in 24 well plates. Twenty-four hours later, CHO cells were transfected with either 0.8 μg pcDNA (empty vector, control), or 0.8 μg Sigma-1 DNA. Media was refreshed 12 hours later. Twenty-four hours after initial transfection, Ham's F-12 medium was prepared containing enough [18F]13 for 2 μCi per well. After 30 and 120 min uptake, medium from each of the triplicate wells was aspirated and cells were washed twice with cold PBS (500 μL). Following this, cells were lyzed with 1 N NaOH (500 μL). A portion of each lysate (250 μL) was transferred to a glass tube and activity was measured with a Cobra II γ counter (Packard-Perkin Elmer, Waltham, MA, USA). Protein content from each well was measured by Bradford assay. The same protocol was followed for [3H]1 (NEN Life Science Products, Boston MA), except the activity was measured with a liquid scintillation counter (Beckman Coulter LS 6500, Brea, CA, USA) following addition of scintillation fluid.
Western Blot
Cell lysates from 2 × 106 cells were prepared by scraping cells in ice-cold harvesting buffer (RIPA Lysis Buffer, Santa Cruz Biotechnology). The lysates were centrifuged (14,000 rpm, 20 min at 4 °C) and supernatants were collected. The protein concentration of the supernatant was determined by Bradford assay. Equal amounts of protein (50 μg) were loaded onto 10% SDS-polyacrylamide mini-gels. After gel electrophoresis, proteins were transferred to a nitrocellulose membrane and blocked at room temperature using 5% non-fat milk blocking buffer (15 mL 1X TBS, 0.01% Tween 20 and 0.75 g milk powder). Following this, the membrane was incubated overnight at 4 °C with goat polyclonal anti-S1R ((S-18): sc-22948, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) primary antibody. The primary antibody was diluted 1:400 in 5% non fat milk blocking buffer. After washing three times with TBST (TBS with 0.01% Tween 20), bovine anti-goat-IgG horseradish peroxidase-conjugated antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) diluted 1:5000 in TBST, was added and incubated for 1 h at room temperature. After washing three times with TBST, S1R protein was visualized using ECL reagent (Pierce, Rockford, IL, USA) and images were obtained using film. Blot was also stained for alpha tubulin as a protein loading control. Image J (image processing and analysis software in java) was used for western blot analysis.
Animals
All experimental procedures involving animals were performed under humane conditions following approval from the Stanford University or University of Mississippi animal research internal review board. Animals had access to food and H2O ad libitum and were kept under a 12 h light/dark cycle.
Small-Animal PET Imaging in mice
All PET imaging was performed on a microPET R4 model scanner (Siemens) fitted with a computer-controlled bed, 10.8 cm transaxial and 8 cm axial field of view (FOV), no septa and operated exclusively in 3-dimensional list mode. PET images were reconstructed with 2-dimensional OSEM (Ordered Subsets Expectation Maximization) and analyzed using AMIDE (A Medical Image Data Examiner) software.58 Normal Balb C mice (20-30 g) were anaesthetized using isoflurane gas (3% for induction and 1-3% for maintenance). Acquisition of the PET data in list mode was commenced just prior to iv administration of [18F]13 (95-125 μCi in 100 μL 0.9% saline) via the tail vein, and was continued for a period of 60 min. Blocking studies involved pre treatment of mice with either 2 (1 mg/kg), 13 (1 mg/kg), or 18 (1 mg/kg) 10 min prior to tracer administration.
Ex Vivo Autoradiography
Sagittal brain sections were obtained 60 min following iv injection of 200-250 μCi [18F]13 for both baseline and blocking studies. Baseline study involved injection of radiotracer only, whereas blocking study involved pre treatment with Sigma-1 specific ligand 18 (1 mg/kg) 10 min prior to radiotracer administration. Each mouse underwent a 5 min static brain PET scan just prior to perfusion. After perfusing each mouse with saline (10 mL), the brain was removed, snap-frozen in isopentane (in liquid nitrogen), and embedded in optimal cutting temperature (O.C.T.) compound (Tissue-Tek, Sakura, USA). Subsequently, 12μm-thick sagittal brain sections were cut using a cryostat microtome HM500 (Microm, Germany). The sections were mounted on microscope slides (Fisherbrand Superfrost™ Plus Microscope Slides), air-dried for 10 min, and then exposed to 18F-sensitive storage phosphor screens (Perkin Elmer, USA) for 12 hours at 4 °C. The image plates were analyzed using a Typhoon 9410 Variable Mode Imager (Amersham Biosciences, USA) and image data were visualized and processed by Image J (image processing and analysis software in java). Anatomy of brain sections was confirmed by Nissl staining (cresyl violet acetate, Sigma Aldrich).
Ex Vivo Determination of Radiometabolites: Mouse Plasma, Liver and Brain
At 30 and 60 min post iv injection of 630-690 μCi [18F]13, blood, brain, and liver from Balb C mice (n=2 per time point) were obtained. Blood samples were collected via cardiac puncture and were immediately centrifuged at 1,800 g for 4 min at room temperature to separate plasma. Each plasma sample (100 μL) was transferred to a tube containing acetonitrile (300 μL) and mixed thoroughly. Water (40 μL) was then added to each tube and after mixing was centrifuged at 9,400 g for 4 min. The resulting supernatants were collected and an aliquot of each supernatant (100 μL) was analyzed via the same HPLC method used for quality control of [18F]13 (fitted with a highly sensitive positron detector for radioactivity). The percent ratio of intact [18F]13 (tR = 8.33 min) to the total radioactivity (decay corrected to the time of dose administration) on the HPLC chromatogram was calculated as % = (peak area for [18F]13/total peak area)*100. Brain and liver samples were homogenized in 500 μL ice-cold acetonitrile and protein precipitates were sedimented by centrifugation (9,400g, 4 min). The resulting supernatants were analyzed by HPLC as described above. Samples (100 μL) from each supernatant were measured in gamma counter to assess the extraction efficiency into supernatant liquids. Pelleted cells were washed twice with 1 mL EtOH and then counted. The activity in the supernatants were compared to that in the corresponding pellets to afford the percentage of the tracer bound to serum proteins. No attempt was made to enhance the efficiency of [18F]fluoride ion extraction by adding carrier fluoride ion prior to extractions.
Supplementary Material
Acknowledgements
This project was supported in part by the NCI ICMIC P50 CA114747 (SSG), and NIDA DA023205 (CRM). We appreciate the technical assistance of Jamaluddin Shaikh (West Virginia University, Morgantown, WV) during some of the binding assays.
Abbreviations
- AMIDE
a medical image data examiner
- BBB
blood brain barrier
- Cb
cerebellum
- cDNA
complimentary deoxyribonucleic acid
- CHO
chinese hamster ovarian
- CPM
counts per minute
- Ctx
cortex
- DCM
dichloromethane
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EBP
emopamil binding protein
- ECL
enhanced chemiluminescence
- EOB
end of bombardment
- EOS
end of synthesis
- ESI
electrospray ionization
- FN
facial nucleus
- FOV
field of view
- H
hippocampus
- HPLC
high performance liquid chromatography
- HRMS
high-resolution mass spectra
- ID/g
injected dose per gram
- iv
intravenous
- Kryptofix 222
4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo [8.8.8] hexacosan
- LC
liquid chromatography
- Mb
midbrain
- MS
mass spectra
- m/z
mass-to-charge ratio
- NADPH
nicotinamide adenine dinucleotide phosphate
- NANM
(+)-N-allylnormetazocine
- NMR
nuclear magnetic resonance
- Ob
olfactory bulb
- O.C.T.
optimal cutting temperature
- OSEM
ordered subsets expectation maximization
- PBS
phosphate buffered saline
- PET
positron emission tomography
- QMA
quaternary methylammonium
- RCP
radiochemical purity
- S1Rs
sigma-1 receptors
- SA
specific activity
- SR
sigma receptor
- S2R
sigma-2 receptor
- TACs
time activity curves
- TBAF
tetra-n-butylammonium fluoride
- TBS
tris buffered saline
- TEA
triethylamine
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- tR
retention time
- UPLC
ultra performance liquid chromatography
- UV
ultraviolet
- VAChT
vesicular acetylacholine transporter
Footnotes
Supporting Information Available: Synthetic procedures for compounds 10 and 11 are detailed, along with tables outlining affinity and selectivity of some S1R ligands, and on/off-target binding data for compound 13. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- 2.Martin WR. A steric theory of opioid agonists, antagonists, agonist-antagonists, and partial agonists. NIDA Res Monogr. 1984;49:16–23. [PubMed] [Google Scholar]
- 3.Hellewell SB, Bruce A, Feinstein G, Orringer J, Williams W, Bowen WD. Rat liver and kidney contain high densities of sigma 1 and sigma 2 receptors: characterization by ligand binding and photoaffinity labeling. Eur J Pharmacol. 1994;268:9–18. doi: 10.1016/0922-4106(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 4.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guitart X, Codony X, Monroy X. Sigma receptors: biology and therapeutic potential. Psychopharmacology. 2004;174:301–319. doi: 10.1007/s00213-004-1920-9. [DOI] [PubMed] [Google Scholar]
- 6.Walker JM, Bowen WD, Walker FO, Matsumoto RR, De Costa B, Rice KC. Sigma receptors: biology and function. Pharmacol Rev. 1990;42:355–402. [PubMed] [Google Scholar]
- 7.Hayashi T, Su TP. Cholesterol at the endoplasmic reticulum: roles of the sigma-1 receptor chaperone and implications thereof in human diseases. Subcell Biochem. 2010;51:381–398. doi: 10.1007/978-90-481-8622-8_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su TP. Delineating biochemical and functional properties of sigma receptors: emerging concepts. Crit Rev Neurobiol. 1993;7:187–203. [PubMed] [Google Scholar]
- 9.Vilner BJ, John CS, Bowen WD. Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 1995;55:408–413. [PubMed] [Google Scholar]
- 10.Diaz JL, Zamanillo D, Corbera J, Baeyens JM, Maldonado R, Pericas MA, Vela JM, Torrens A. Selective sigma-1 receptor antagonists: emerging target for the treatment of neuropathic pain. Cent Nerv Syst Agents Med Chem. 2009;9:172–183. doi: 10.2174/1871524910909030172. [DOI] [PubMed] [Google Scholar]
- 11.Toyohara J, Sakata M, Ishiwata K. Imaging of sigma-1 receptors in the human brain using PET and [11C]SA4503. Cent Nerv Syst Agents Med Chem. 2009;9:190–196. doi: 10.2174/1871524910909030190. [DOI] [PubMed] [Google Scholar]
- 12.Narayanan S, Mesangeau C, Poupaert JH, McCurdy CR. Sigma receptors and cocaine abuse. Curr Top Med Chem. 2011;11:1128–1150. doi: 10.2174/156802611795371323. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa M, Hashimoto K. The role of sigma-1 receptors in the pathophysiology of neuropsychiatric diseases. Journal of Receptor, Ligand and Channel Research. 2009;2010:25–36. [Google Scholar]
- 14.Wang B, Rouzier R, Albarracin CT, Sahin A, Wagner P, Yang Y, Smith TL, Meric-Bernstam F, Marcelo Aldaz C, Hortobagyi GN, Pusztai L. Expression of sigma 1 receptor in human breast cancer. Breast Cancer Res Treat. 2004;87:205–214. doi: 10.1007/s10549-004-6590-0. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez GM, Werling LL. Release of [3H]dopamine from guinea pig striatal slices is modulated by sigma1 receptor agonists. Naunyn Schmiedebergs Arch Pharmacol. 1997;356:455–461. doi: 10.1007/pl00005076. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi T, Matsuno K, Nakata K, Mita S. Enhancement of acetylcholine release by SA4503, a novel sigma 1 receptor agonist, in the rat brain. J Pharmacol Exp Ther. 1996;279:106–113. [PubMed] [Google Scholar]
- 17.Collier TL, Waterhouse RN, Kassiou M. Imaging sigma receptors: applications in drug development. Curr Pharm Des. 2007;13:51–72. doi: 10.2174/138161207779313740. [DOI] [PubMed] [Google Scholar]
- 18.Maurice T. Improving Alzheimer's Disease-Related Cognitive Deficits with sigma-1 Receptor Agonists. Drug News Perspect. 2002;15:617–625. doi: 10.1358/dnp.2002.15.10.740241. [DOI] [PubMed] [Google Scholar]
- 19.Senda T, Matsuno K, Kobayashi T, Nakazawa M, Nakata K, Mita S. Ameliorative effect of SA4503, a novel cognitive enhancer, on the basal forebrain lesion-induced impairment of the spatial learning performance in rats. Pharmacol Biochem Behav. 1998;59:129–134. doi: 10.1016/s0091-3057(97)00385-7. [DOI] [PubMed] [Google Scholar]
- 20.Harukuni I, Bhardwaj A, Shaivitz AB, DeVries AC, London ED, Hurn PD, Traystman RJ, Kirsch JR, Faraci FM. Sigma-1 receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine affords neuroprotection from focal ischemia with prolonged reperfusion. Stroke. 2000;31:976–982. doi: 10.1161/01.str.31.4.976. [DOI] [PubMed] [Google Scholar]
- 21.Volz HP, Stoll KD. Clinical trials with sigma ligands. Pharmacopsychiatry. 2004;37(Suppl 3):S214–220. doi: 10.1055/s-2004-832680. [DOI] [PubMed] [Google Scholar]
- 22.Xu YT, Kaushal N, Shaikh J, Wilson LL, Mesangeau C, McCurdy CR, Matsumoto RR. A novel substituted piperazine, CM156, attenuates the stimulant and toxic effects of cocaine in mice. J Pharmacol Exp Ther. 333:491–500. doi: 10.1124/jpet.109.161398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romero L, Zamanillo D, Nadal X, Sanchez-Arroyos R, Rivera-Arconada I, Dordal A, Montero A, Muro A, Bura A, Segales C, Laloya M, Hernandez E, Portillo-Salido E, Escriche M, Codony X, Encina G, Burgueno J, Merlos M, Baeyens J, Giraldo J, Lopez-Garcia J, Maldonado R, Plata-Salaman C, Vela J. Pharmacological properties of S1RA, a new sigma-1 receptor antagonist that inhibits neuropathic pain and activity-induced spinal sensitization. Br J Pharmacol. 2012 doi: 10.1111/j.1476-5381.2012.01942.x. DOI: 10.1111/j.1476-5381.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio JM, Rothman RB, Su TP, Tam SW, Taylor DP. A proposal for the classification of sigma binding sites. Trends Pharmacol Sci. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- 25.Ucar H, Cacciaguerra S, Spampinato S, Van derpoorten K, Isa M, Kanyonyo M, Poupaert JH. 2(3H)-benzoxazolone and 2(3H)-benzothiazolone derivatives: novel, potent and selective sigma1 receptor ligands. Eur J Pharmacol. 1997;335:267–273. doi: 10.1016/s0014-2999(97)01248-x. [DOI] [PubMed] [Google Scholar]
- 26.Berardi F, Ferorelli S, Abate C, Pedone MP, Colabufo NA, Contino M, Perrone R. Methyl substitution on the piperidine ring of N-[ω-(6-methoxynaphthalen-1-yl)alkyl] derivatives as a probe for selective binding and activity at the sigma-1 receptor. J Med Chem. 2005;48:8237–8244. doi: 10.1021/jm050654o. [DOI] [PubMed] [Google Scholar]
- 27.Hudkins RL, Mailman RB, DeHaven-Hudkins DL. RLH-033, a novel, potent and selective ligand for the sigma 1 recognition site. Eur J Pharmacol. 1994;271:235–236. doi: 10.1016/0014-2999(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 28.Maestrup EG, Fischer S, Wiese C, Schepmann D, Hiller A, Deuther-Conrad W, Steinbach J, Wunsch B, Brust P. Evaluation of spirocyclic 3-(3-fluoropropyl)-2-benzofurans as sigma 1 receptor ligands for neuroimaging with positron emission tomography. J Med Chem. 2009;52:6062–6072. doi: 10.1021/jm900909e. [DOI] [PubMed] [Google Scholar]
- 29.Matsuno K, Nakazawa M, Okamoto K, Kawashima Y, Mita S. Binding properties of SA4503, a novel and selective sigma 1 receptor agonist. Eur J Pharmacol. 1996;306:271–279. doi: 10.1016/0014-2999(96)00201-4. [DOI] [PubMed] [Google Scholar]
- 30.Moussa IA, Banister SD, Beinat C, Giboureau N, Reynolds AJ, Kassiou M. Design, synthesis, and structure affinity relationships of regioisomeric N-benzyl alkyl ether piperazine derivatives as sigma-1 receptor ligands. J Med Chem. 2010;53:6228–6239. doi: 10.1021/jm100639f. [DOI] [PubMed] [Google Scholar]
- 31.Piergentili A, Amantini C, Del Bello F, Giannella M, Mattioli L, Palmery M, Perfumi M, Pigini M, Santoni G, Tucci P, Zotti M, Quaglia W. Novel highly potent and selective sigma 1 receptor antagonists related to spipethiane. J Med Chem. 2010;53:1261–1269. doi: 10.1021/jm901542q. [DOI] [PubMed] [Google Scholar]
- 32.Quaglia W, Giannella M, Piergentili A, Pigini M, Brasili L, Di Toro R, Rossetti L, Spampinato S, Melchiorre C. 1′-Benzyl-3,4-dihydrospiro[2H-1-benzothiopyran-2,4′-piperidine] (spipethiane), a potent and highly selective sigma 1 ligand. J Med Chem. 1998;41:1557–1560. doi: 10.1021/jm970740r. [DOI] [PubMed] [Google Scholar]
- 33.Yous S, Wallez V, Belloir M, Caignard DH, McCurdy CR, Poupaert JH. Novel 2(3H)-benzothiazolones as highly potent and selective sigma-1 receptor ligands. Med Chem Res. 2005;14:158–168. [Google Scholar]
- 34.Kawamura K, Ishiwata K, Tajima H, Ishii S, Matsuno K, Homma Y, Senda M. In vivo evaluation of [11C]SA4503 as a PET ligand for mapping CNS sigma-1 receptors. Nucl Med Biol. 2000;27:255–261. doi: 10.1016/s0969-8051(00)00081-0. [DOI] [PubMed] [Google Scholar]
- 35.Kawamura K, Tsukada H, Shiba K, Tsuji C, Harada N, Kimura Y, Ishiwata K. Synthesis and evaluation of fluorine-18 labeled SA4503 as a selective sigma-1 receptor ligand for positron emission tomography. Nucl Med Biol. 2007;34:571–577. doi: 10.1016/j.nucmedbio.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Waterhouse RN, Collier TL. In vivo evaluation of [18F]1-(3-fluoropropyl)-4-(4-cyanophenoxymethyl)piperidine: a selective sigma-1 receptor radioligand for PET. Nucl Med Biol. 1997;24:127–134. doi: 10.1016/s0969-8051(96)00184-9. [DOI] [PubMed] [Google Scholar]
- 37.Waterhouse RN, Zhao J, Stabin MG, Ng H, Schindler-Horvat J, Chang RC, Mirsalis JC. Preclinical acute toxicity studies and dosimetry estimates of the novel sigma-1 receptor radiotracer, [18F]SFE. Mol Imaging Biol. 2006;8:284–291. doi: 10.1007/s11307-006-0056-1. [DOI] [PubMed] [Google Scholar]
- 38.Waterhouse RN, Chang RC, Zhao J, Carambot PE. In vivo evaluation in rats of [18F]1-(2-fluoroethyl)-4-[(4-cyanophenoxy)methyl]piperidine as a potential radiotracer for PET assessment of CNS sigma-1 receptors. Nucl Med Biol. 2006;33:211–215. doi: 10.1016/j.nucmedbio.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 39.Fischer S, Wiese C, Grosse Maestrup E, Hiller A, Deuther-Conrad W, Scheunemann M, Schepmann D, Steinbach J, Wunsch B, Brust P. Molecular imaging of sigma receptors: synthesis and evaluation of the potent sigma-1 selective radioligand [18F]fluspidine. Eur J Nucl Med Mol Imaging. 2011;38:540–551. doi: 10.1007/s00259-010-1658-z. [DOI] [PubMed] [Google Scholar]
- 40.Berardi F, Ferorelli S, Colabufo NA, Leopoldo M, Perrone R, Tortorella V. A multireceptorial binding reinvestigation on an extended class of sigma ligands: N-[omega-(indan-1-yl and tetralin-1-yl)alkyl] derivatives of 3,3-dimethylpiperidine reveal high affinities towards sigma1 and EBP sites. Bioorg Med Chem. 2001;9:1325–1335. doi: 10.1016/s0968-0896(01)00011-6. [DOI] [PubMed] [Google Scholar]
- 41.Shiba K, Ogawa K, Ishiwata K, Yajima K, Mori H. Synthesis and binding affinities of methylvesamicol analogs for the acetylcholine transporter and sigma receptor. Bioorg Med Chem. 2006;14:2620–2626. doi: 10.1016/j.bmc.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 42.Ishikawa M, Ishiwata K, Ishii K, Kimura Y, Sakata M, Naganawa M, Oda K, Miyatake R, Fujisaki M, Shimizu E, Shirayama Y, Iyo M, Hashimoto K. High occupancy of sigma-1 receptors in the human brain after single oral administration of fluvoxamine: a positron emission tomography study using [11C]SA4503. Biol Psychiatry. 2007;62:878–883. doi: 10.1016/j.biopsych.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Mishina M, Ohyama M, Ishii K, Kitamura S, Kimura Y, Oda K, Kawamura K, Sasaki T, Kobayashi S, Katayama Y, Ishiwata K. Low density of sigma 1 receptors in early Alzheimer's disease. Ann Nucl Med. 2008;22:151–156. doi: 10.1007/s12149-007-0094-z. [DOI] [PubMed] [Google Scholar]
- 44.Fishback JA, Mesangeau C, Poupaert JH, McCurdy CR, Matsumoto RR. Synthesis and characterization of [3H]-SN56, a novel radioligand for the σ1 receptor. Eur J Pharmacol. 2011;653:1–7. doi: 10.1016/j.ejphar.2010.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCurdy CR, Mesangeau C, Chin FT, James ML, Shen B, Gambhir SS. Highly Selective Sigma Receptor Radioligands. 2011 U.S. Patent. 13/151084.
- 46.Poole SK, Patel S, Dehring K, Workman H, Poole CF. Determination of acid dissociation constants by capillary electrophoresis. J Chromatogr A. 2004;1037:445–454. doi: 10.1016/j.chroma.2004.02.087. [DOI] [PubMed] [Google Scholar]
- 47.Danielsson LG, Zhang YH. Methods for determining n-octanol-water partition constants. TrAC. 1996;15:188–196. [Google Scholar]
- 48.Poupaert J, Gbaguidi FA, Kapanda CN, Aichaoui H, McCurdy CR. Is cocaine a social drug? Exploration of the stereo-structure of cocaine's pharmacophore. Med Chem Res. 2012;21:1–6. [Google Scholar]
- 49.McCann DJ, Weissman AD, Su TP. Sigma-1 and sigma-2 sites in rat brain: comparison of regional, ontogenetic, and subcellular patterns. Synapse. 1994;17:182–189. doi: 10.1002/syn.890170307. [DOI] [PubMed] [Google Scholar]
- 50.Matsumoto RR, Pouw B. Correlation between neuroleptic binding to sigma-1 and sigma-2 receptors and acute dystonic reactions. Eur J Pharmacol. 2000;401:155–160. doi: 10.1016/s0014-2999(00)00430-1. [DOI] [PubMed] [Google Scholar]
- 51.Vilner BJ, Bowen WD. Sigma receptor-active neuroleptics are cytotoxic to C6 glioma cells in culture. Eur J Pharmacol. 1993;244:199–201. doi: 10.1016/0922-4106(93)90029-9. [DOI] [PubMed] [Google Scholar]
- 52.Fischer S, Wiese C, Grosse Maestrup E, Hiller A, Deuther-Conrad W, Scheunemann M, Schepmann D, Steinbach J, Wunsch B, Brust P. Molecular imaging of sigma receptors: synthesis and evaluation of the potent sigma-1 selective radioligand [18F]fluspidine. Eur J Nucl Med Mol Imaging. 2010;38:540–551. doi: 10.1007/s00259-010-1658-z. [DOI] [PubMed] [Google Scholar]
- 53.Mavlyutov TA, Epstein ML, Andersen KA, Ziskind-Conhaim L, Ruoho AE. The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience. 2010;167:247–255. doi: 10.1016/j.neuroscience.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsumoto RR, Shaikh J, Wilson LL, Vedam S, Coop A. Attenuation of methamphetamine induced effects through the antagonism of sigma receptors: Evidence from in vivo and in vitro studies. Eur Neuropsychopharmacol. 2008;18:871–881. doi: 10.1016/j.euroneuro.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaushal N, Robson MJ, Vinnakota H, Narayanan S, Avery BA, McCurdy CR, Matsumoto RR. Synthesis and pharmacological evaluation of 6-acetyl-3-(4-(4-(4-fluorophenyl)piperazin-1-yl)butyl)benzo[d]oxazol-2(3H)-one (SN79), a cocaine antagonist, in rodents. AAPS J. 2011;13:336–346. doi: 10.1208/s12248-011-9274-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu YT, Kaushal N, Shaikh J, Wilson LL, Mesangeau C, McCurdy CR, Matsumoto RR. A novel substituted piperazine, CM156, attenuates the stimulant and toxic effects of cocaine in mice. J Pharmacol Exp Ther. 2010;333:491–500. doi: 10.1124/jpet.109.161398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fishback JA. Ph.D. Dissertation. West Virginia University; Morgantown, WV: 2011. New Methodologies for In Vitro Analysis of Binding and Functional Activity of Sigma Receptor Ligands. [Google Scholar]
- 58.Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging. 2003;2:131–137. doi: 10.1162/15353500200303133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.