

Abstract
The nuclear transporter exportin-1 (XPO1) is highly expressed in mantle cell lymphoma (MCL) cells, and is believed to be associated with the pathogenesis of this disease. XPO1-selective inhibitors of nuclear export (SINE) compounds have been shown to induce apoptosis in MCL cells. Given that p53 is a cargo protein of XPO1, we sought to determine the significance of p53 activation through XPO1 inhibition in SINE-induced apoptosis of MCL cells. We investigated the prognostic impact of XPO1 expression in MCL cells using Oncomine analysis. The significance of p53 mutational/functional status on sensitivity to XPO1 inhibition in cell models and primary MCL samples, and the functional role of p53-mediated apoptosis signaling, were also examined. Increased XPO1 expression was associated with poor prognosis in MCL patients. The XPO1 inhibitor KPT-185 induced apoptosis in MCL cells through p53-dependent and -independent mechanisms, and p53 status was a critical determinant of its apoptosis induction. The KPT-185-induced, p53-mediated apoptosis in the MCL cells occurred in a transcription-dependent manner. Exportin-1 appears to influence patient survival in MCL, and the SINE XPO1 antagonist KPT-185 effectively activates p53-mediated transcription and apoptosis, which would provide a novel strategy for the therapy of MCL.
Keywords: Apoptosis, KPT-SINE, mantle cell lymphoma, p53, XPO1
Mantle cell lymphoma (MCL) is an incurable form of B-cell, non-Hodgkin's lymphoma (NHL). Given this grievous outcome, novel, effective treatments are urgently needed for this disease. The main therapeutic challenge in MCL is the implementation of treatment strategies that maximize the efficient induction of lymphoma cell apoptosis without the development of chemoresistant sub-clones.
The nuclear–cytoplasmic transport of proteins and ribonucleic acids is vital to cellular homeostasis in eukaryotic cells. This process is regulated, in part, by the karyopherin-β protein family. The XPO1 protein (exportin-1, also known as CRM1), is among seven exportins. Interestingly, XPO1 is the only exportin that mediates the transport of numerous proteins including tumor suppressor, growth regulatory, and anti-apoptotic proteins as well as several mRNAs and ribosomal proteins that are essential for ribosomal biogenesis.(1–3) Exportin-1 is abnormally highly expressed/upregulated in a variety of solid tumor types,(4–7) as well as hematological malignancies including MCL.(8–10) In fact, the overexpression of XPO1 is positively correlated with poor disease prognosis in some malignancies.(4–7,10,11) Therefore, it has been suggested that alterations in nuclear–cytoplasmic trafficking, and hence the aberrant cytoplasmic localization, of tumor suppressor proteins, cell cycle regulators, and/or pro-apoptotic proteins, as well as the deregulation of ribosomal biogenesis, can cause oncogenesis and the development of resistance to chemotherapeutic agents.(2,3)
More than 90% of MCL patients have extranodal manifestations, including circulating lymphoma cells, bone marrow, and gastrointestinal tract involvement.(12) TP53 mutations occur in 15–20% of the cases of MCL,(13) and wild-type p53 is inactivated by upstream gene amplification of BMI1 (∼10%), homozygous deletion of CDKN2A (INK4a/ARF) (15–20%), the overexpression of human homolog of murine double minute 2 (MDM2) (∼5%), or TP53 gene deletion (25–30%).(14–16) All of these abnormalities essentially lead to the loss of p53 tumor suppressor activity.
The nuclear export of p53 is cooperatively mediated by MDM2 and XPO1.(17) MDM2 activates the nuclear export signal (NES) in p53 through its E3 ubiquitin ligase activity, leading a conformational change in p53 that exposes p53's NES domain. Following ubiquitination, XPO1 recognizes p53's NES and exports the protein from the nucleus to the cytoplasm, where it is unable to execute transcriptional activity to regulate cell fate. As we mentioned previously, XPO1 is highly expressed in MCL cells,(8) which may limit p53-mediated transcriptional activity, and hence the ability of p53 to trigger apoptosis.(18) It has been reported that wild-type p53 is abnormally sequestered in the cytoplasm in certain human tumor cells.(19,20)
Novel small-molecule, drug-like, potent, and covalent XPO1-selective inhibitors of nuclear export (SINE) compounds were recently developed. These compounds selectively bind to the Cys528 of XPO1, thereby inhibiting XPO1 binding to the NES domains of its cargo protein.(21) The SINE KPT-185 has been shown to induce apoptosis in MCL cells.(8) The inhibition of XPO1 is believed to maintain the nuclear localization, and hence function, of p53.(1–3) Furthermore, XPO1 is involved in the nuclear export of numerous proteins including p21, p27, p73, nucleophosmin-1, PP2A, FOXO, β-catenin/APC, topoisomerase II, and IκB.(1) This would suggest that the biological significance of p53 activation in XPO1 inhibition-induced apoptosis in MCL cells is highly unspecified and thus in need of further elucidation. Accordingly, we examined the pathophysiological significance of XPO1's influence on p53 cellular localization and functional activity and its potential as a therapeutic target for enhancing MCL cell apoptosis.
Materials and Methods
Reagents
The selective XPO1 inhibitor KPT-185 was synthesized and provided by Karyopharm (Karyopharm, Natick, MA, USA). The selective small-molecule antagonist of MDM2, Nutlin-3a was purchased from Cayman Chemical Company (Ann Arbor, MI, USA).
Cells and cell culture
A total of 16 lymphoid cell lines, including six MCL cell lines, were cultured in RPMI-1640 medium containing 20% heat-inactivated FBS (Table 1). Z-138 and JVM-2 have wild-type p53, whereas MINO, JeKo-1, MAVER-1, and NCEB-1 have defective (i.e., missense mutated or deleted) p53.(22) The Z-138 and JVM-2 cells were transduced with retroviruses encoding either p53-specific shRNA (nucleotides 611–629, Genbank NM000546) or scrambled shRNA and stable shRNA-expressing cells were generated.(23) The cell were harvested in log-phase growth, seeded at a density of 2 × 105 cells/mL and exposed to the indicated compounds.
Table 1.
Effective doses of KPT-185 and Nutlin-3a for inducing 50% killing in lymphoid cells, as measured by annexin V positivity, relative to the cell lines' p53 mutational status
| Cell line | KPT-185, nM | Nutlin-3a, μM | p53 Status |
|---|---|---|---|
| Z-138† | 85 | 1.1 | Wild-type |
| JVM-2† | 641 | 5.9 | Wild-type |
| REH | 371 | 6.1 | Wild-type |
| NALM-6 | 32 | 2.1 | Wild-type |
| RS4;11 | 198 | 2.4 | Wild-type |
| MT-2 | 49 | 5.0 | Wild-type |
| TMD8 | 54 | 1.4 | Wild-type |
| SUP-M2 | 81 | 1.4 | Wild-type |
| MINO† | 1865 | >10 | Mutant (V147G) |
| JeKo-1† | 703 | >10 | Mutant (deletion) |
| MAVER-1† | 391 | >10 | Mutant (D281E) |
| NCEB-1† | 3518 | >10 | Mutant (E221K) |
| Raji | 418 | >10 | Mutant (R213Q, Y234H) |
| Jurkat | 1285 | >10 | Mutant (R196*) |
| ATN-1 | 279 | >10 | Mutant (V272L) |
| SU-DHL-1 | 185 | >10 | Mutant (R273H) |
Mantle cell lymphoma cell lines.
Eμ-TCL1 transgenic mice (kindly provided by Dr. Carlo M. Croce, Ohio State University, Columbus, OH, USA) with different p53 backgrounds (TCL1-Tg:p53WT/WT or TCL1-Tg:p53R172H/R172H) were generated. Eμ-TCL1 transgenic mice spontaneously develop CD19+/CD5+/CD23+/− B-lineage lymphoid malignancies, and have been used as a mouse model for chronic lymphocytic leukemia that typically shows CD19+/CD5+/CD23+ immunophenotype.(24) The immunological profile of MCL cells is defined by CD19+/CD5+/CD23−; p53R172H is equivalent to human p53R175H, one of the hotspot mutations in human cancers.
B-lymphoma cells were collected from affected spleens, cultured in RPMI-1640 medium containing 20% FBS at 2 ×106 cells/mL, and were exposed to the indicated compounds. Heparinized peripheral blood and pleural effusion samples were obtained from MCL patients after informed consent, according to the University of Texas MD Anderson Cancer Center (Houston, TX, USA) guidelines in accordance with the Declaration of Helsinki. Mononuclear cells were purified by density-gradient centrifugation, and non-adherent cells were resuspended at a density of 1 × 106 cells/mL. Cell viability was evaluated by triplicate counts of Trypan blue dye-excluding cells.
Apoptosis analysis
Evaluation of apoptosis by the annexin V–propidium iodide binding assay was carried out as described previously.(11) Apoptosis was quantified as the percentage of annexin V-positive cells, and the percent of drug-specific apoptosis was assessed by the formula: (% test – % control) × 100/(100–% control).
Western blot analysis and co-immunoprecipitation
Western blot analysis was carried out as described previously.(24) The following antibodies were used: mouse monoclonal anti-p53 (DO-1; Santa Cruz Biotechnology, Santa Cruz, CA, USA); rabbit polyclonal anti-XPO1 (Santa Cruz Biotechnology); rabbit polyclonal anti-HSP-90a/b (Santa Cruz Biotechnology); rabbit monoclonal histone H3 (Cell Signaling Technologies, Beverly, MA, USA); rabbit monoclonal GAPDH (Cell Signaling Technologies); and mouse monoclonal anti-β-actin (Sigma Chemical Co., St Louis, MO, USA). Nuclear and cytoplasmic proteins were extracted using a subcellular fractionation kit (ProteoExtract; EMD Millipore, Billerica, MA, USA), according to the manufacturer's protocol. Protein lysates were also subjected to immunoprecipitation using anti-XPO1 and immunoprecipitates were subjected to Western blot analysis with anti-XPO1 or p53. Visualized blots were analyzed by the MultiGauge 3.1 software (Fujifilm, Tokyo, Japan).
Gene expression analysis
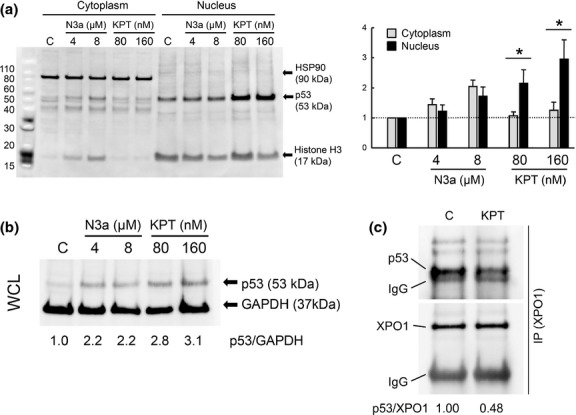
The mRNA expression levels were quantified using TaqMan gene expression assays (TNFRSF10B (DR5), Hs00366272_m1; FAS, Hs00163653_m1; BBC3 (PUMA), Hs00248075_m1; TP53INP1, Hs00264502_m1; GAPDH, Hs99999905_m1; Applied Biosystems, Foster City, CA, USA) on a 7900HT Fast Real-Time PCR System.(25) Relative quantification between different samples was determined according to the 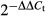 method with a relative quantification (RQ)min/RQmax confidence set at 95%, using SDS RQ Manager 1.2.1 software (Applied Biosystems). The error bars display the calculated maximum (RQmax) and minimum (RQmin) expression levels that represent standard error of the mean (SEM) expression level (RQ value). Collectively, the upper and lower limits define the region of expression within which the true expression level value is likely to occur.
method with a relative quantification (RQ)min/RQmax confidence set at 95%, using SDS RQ Manager 1.2.1 software (Applied Biosystems). The error bars display the calculated maximum (RQmax) and minimum (RQmin) expression levels that represent standard error of the mean (SEM) expression level (RQ value). Collectively, the upper and lower limits define the region of expression within which the true expression level value is likely to occur.
Mutation analysis
Mutation analysis of TP53 was carried out as previously described.(11)
Statistical analyses
The statistical analyses were carried out using the two-sided Student's t-test, the non-parametric Mann–Whitney U-test, and the Pearson correlation coefficient as appropriate. P < 0.05 was considered statistically significant. Where indicated, the mean values of triplicate samples are expressed ± standard deviation (SD).
Results
Overexpression of XPO1 associated with poor disease prognosis in MCL patients
The mRNA expression levels in MCL patient samples were determined using Oncomine data (Compendia Bioscience, Ann Arbor, MI, USA). Our gene expression analyses showed an increase in XPO1 mRNA expression in the MCL samples (n = 8) versus the normal B-cell controls (n = 5) (P < 0.001; GSE2350). In fact, higher XPO1 expression at diagnosis was associated with a poorer prognosis in MCL patients (i.e., a median overall survival of 3.2 years in the low expression XPO1 cases vs 1.9 years in the high expression XPO1 cases, P = 0.033) (Fig. 1a). Patients who survived for 5 years or more with MCL had lower levels of XPO1 mRNA (P = 0.004) (Fig. 1b). In contrast to XPO1, the differential expression of MDM2 did not show clinical significance in MCL patients. The MDM2 expression levels were not statistically significantly higher in the MCL samples (n = 8) compared to normal B-cell controls (n = 5) (P = 0.27; GSE2350), and the levels of MDM2 were not associated with overall disease survival of these patients (P = 0.12 at 5 years, n = 63). These results suggest that XPO1 overexpression has a negative prognostic impact on MCL overall disease survival, and, as such, XPO1 is potentially a therapeutic target in this disease.
Figure 1.
Exportin-1 (XPO1) expression has prognostic impact on mantle cell lymphoma (MCL). (a) Kaplan–Meier plots on prognostic relevance of XPO1 mRNA expression on overall survival for MCL patients. (b) XPO1 mRNA expression levels in patients with different survival status (alive or dead) at 5 years. Higher XPO1 expression was associated with poorer prognosis in MCL patients.
Mutational status of p53 affects lymphoid cell sensitivity to KPT-185
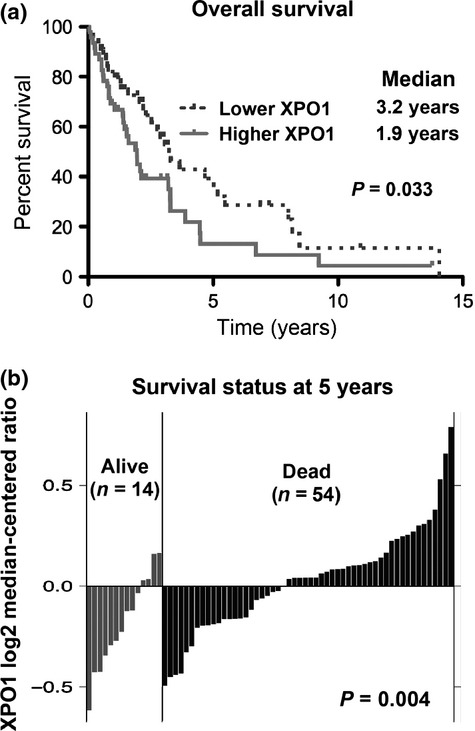
Fifteen to 20% of MCL cases have deleterious p53 mutations,(13) and it has been recently reported that KPT-185 induces apoptosis in MCL cells.(8) Thus, we investigated the possibility that the p53 mutational status of MCL cells could affect their sensitivity to XPO1 inhibition by KPT-185. We determined ED50 values for phosphatidylserine externalization (i.e., the effective dose-induced apoptotic cell killing in ∼50% of the sample population as measured by annexin V positivity) in 16 lymphoid cell lines with known p53 mutational status. As shown in Table 1, the p53 mutational status was generally associated with a decrease in sensitivity to KPT-185.
As a positive control for wild-type p53-mediated apoptosis induction, we used the selective MDM2 inhibitor Nutlin-3a.(26,27) As shown in Table 1, Nutlin-3a, like KPT-185, had decreased efficacy in the p53-mutated MCL cells. The mutant p53 cell lines were significantly less sensitive to KPT-185 than those with wild-type p53 (P < 0.01, Fig 2a). Next, we correlated the extent of apoptosis induced by KPT-185 with that induced by Nutlin-3a, specifically in eight of the p53 wild-type cell lines, as a potential cellular outcome of wild-type p53 activation. The extent of apoptosis induced by KPT-185 significantly, positively correlated (i.e., P = 0.04, r = 0.73, Fig. 2b) with that induced by Nutlin-3a, implying that KPT-185 could utilize p53 signaling to induce apoptosis in lymphoid cells.
Figure 2.
Status of p53 signaling determines exportin-1 (XPO1) inhibition-induced apoptosis. (a) ED50 values for annexin V induction (effective dose inducing 50% cell killing as measured by annexin V positivity) of 72-h exposure to KPT-185 were determined in 16 lymphoid cell lines with known p53 mutational status (eight cell lines with wild-type p53 [WT] and eight with mutant p53 [MUT]), and the values were compared between p53 wild-type and mutant cells. Average values were expressed as mean ± SEM. (b) ED50 values for annexin V induction of 72-h exposure to KPT-185 were correlated with those to Nutlin-3a in p53 wild-type cell lines. (c) Levels of p53 determined sensitivity to MDM2/XPO1 inhibition in p53 wild-type MCL cells. Transduced Z-138 and JVM-2 cells (virus encoding either negative control shRNA [shC] or p53-specific shRNA [shp53]) were incubated with the indicated concentrations of KPT-185 or Nutlin-3a for 72 h, and the annexin V-positive fractions were measured by flow cytometry. Results are expressed as mean ± SD of triplicate measurements. Intensity of the p53 immunoblot signals relative to that of β–actin was calculated, and the levels in shC cells were set as 1.0. (d) Mutational status of p53 affects lymphoma cell sensitivity to Nutlin-3a and KPT-185 in an Eμ-TCL1 mouse model. Eμ-TCL1 mice spontaneously develop B-lineage lymphomas. Cells were collected from affected spleens of TCL1-Tg:p53WT/WT mice (WT; n = 9) and TCL1-Tg:p53R172H/R172H mice (MUT; n = 6), and they were exposed to KPT-185 (100 nM or 400 nM) or Nutlin-3a (10 μM) for 72 h. The annexin V-positive fractions were measured by flow cytometry. Average values were expressed as mean ± SEM.
We wanted to determine if wild-type p53 expression levels could determine susceptibility of the MCL cells to XPO1 inhibition. To this end, p53 wild-type Z-138 and JVM-2 MCL cells were transduced with lentivirus encoding either negative control shRNA or p53-specific shRNA, and stable shRNA-expressing cells were generated. The p53-specific shRNA reduced p53 levels by 80–90%, and these p53 knockdown cells were also significantly less sensitive to both Nutlin-3a- and KPT-185-induced apoptosis (i.e., P < 0.05 in drug-specific annexin V induction at all concentrations examined; Fig. 2c), suggesting that both the MDM2 inhibitor Nutlin-3a and the XPO1 inhibitor KPT-185 activate p53-mediated signaling to induce apoptosis in MCL cells.
To further define the observed wild-type p53-dependent apoptosis induced by KPT-185, we used isogenic B-lymphoma cells from Eμ-TCL1 transgenic mice with different p53 backgrounds. As expected, the lymphoma cells derived from the TCL1-Tg:p53R172H/R172H mice were markedly less sensitive to Nutlin-3a than those derived from the TCL1-Tg:p53WT/WT mice (Fig. 2d). The presence of p53R172H also limited the apoptotic activity of KPT-185. However, higher concentrations of KPT-185 (400 nM) were able to induce some extent of apoptosis in lymphoma cells from the TCL1-Tg:p53R172H/R172H mice, indicating a p53-independent effect of KPT-185.(28) Taken together, data suggest that the functional status of p53 is a determinant of XPO1 inhibition-induced apoptosis in lymphoma cells.
KPT-185 increases nuclear p53 levels and activates p53-mediated transcription in MCL cells
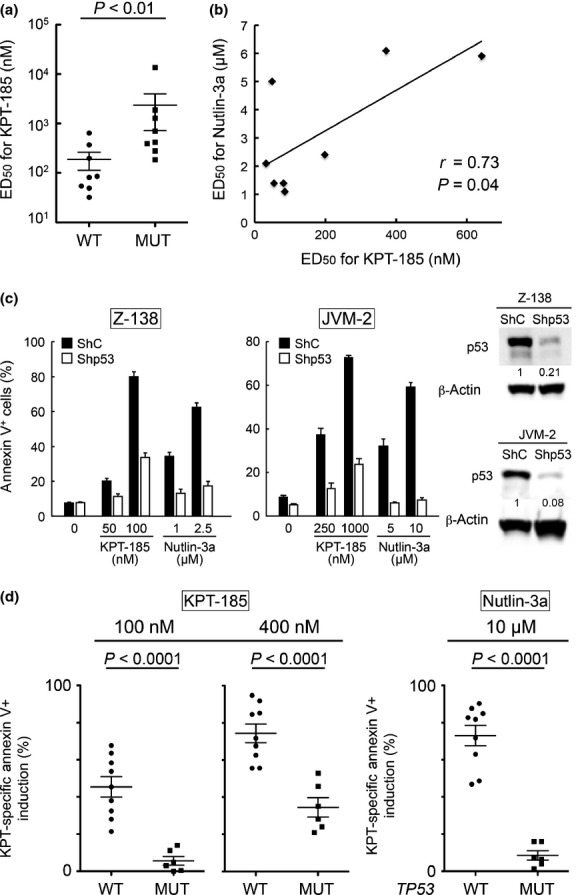
Next, we investigated the possible mechanistic differences between Nutlin-3a- and KPT-185-induced apoptosis. The primary function of p53 is as a transcription factor to induce p53 targets and apoptosis, which occurs in the nucleus, although in some circumstances cytoplasmic p53 may trigger apoptosis in a transcription-independent manner.(18,29) To determine localization of induced p53, Z-138 MCL cells were treated with Nutlin-3a or KPT-185 and were subjected to subcellular fractionation, and the levels of p53 in the nuclear and cytoplasmic fractions were determined by immunoblotting. In contrast to Nutlin-3a, which increased p53 levels both in the cytoplasm and the nucleus, KPT-185 accumulated p53 only in the nucleus (Fig. 3a,b). KPT-185 treatment resulted in a reduced interaction between XPO1 and p53 (Fig. 3c), indicating that KPT-185 disrupts XPO1–p53 interaction.(30) These results suggest that Nutlin-3a treatment causes an increase in the total levels of cellular p53, whereas KPT-185 treatment induces nuclear p53 by disrupting the XPO1–p53 interaction.
Figure 3.
Exportin-1 (XPO1) inhibitor KPT-185 increases nuclear p53 levels. (a) Z-138 mantle cell lymphoma cells were treated with indicated concentrations of Nutlin-3a (N3a) or KPT-185 (KPT) for 14 h and subjected to subcellular fractionation. The levels of p53 in the cytoplasmic and nuclear fractions were determined by immunoblotting. The relative purity of the cytoplasmic and nuclear fractions was respectively determined by sequential probing for the cytoplasmic marker HSP90 and the nuclear marker histone H3. The relative intensity of p53 compared to HSP90 (cytoplasm) or histone H3 (nucleus) was calculated, and the value in untreated cells (C) was set as 1. Results are expressed as mean ± SD of triplicate experiments. *Significance at P < 0.05. (b) p53 expression in whole cell lysate (WCL) of Z-138 cells, which were treated with indicated concentrations of Nutlin-3a or KPT-185 for 14 h. The relative intensity of p53 compared to GAPDH was calculated, and the value in untreated cells (C) was set as 1. Results are representative of three independent experiments. (c) KPT-185 disrupts CRM1–p53 interaction. Z-138 cells were treated for 1 h with 160 nM KPT-185. Exportin-1 was immunoprecipitated (IP) from total cell lysates and its association with p53 was determined by Western blot analysis. The relative p53 expression levels were normalized against XPO1 in which untreated Z-138 cells (C) served as 1. IgG, heavy chain IgG.
To determine the activation potential for p53-mediated transcription in MCL, Z-138 and JVM-2 cells were treated with KPT-185 or Nutlin-3a at, or twice, their ED50 concentration (Table 1), and the transcriptional activation of p53-regulated target genes were assessed. As shown in Figure 4, only KPT-185 induced the expression of p53 target genes in Z-138 cells, indicating different effector pathways for p53-induced apoptosis in these cells. In JVM-2 cells, both KPT-185 and Nutlin-3a activated p53 target genes. The data suggest that KPT-185 primarily activates p53-mediated transcription to induce apoptosis. To elucidate this, Z-138 cells expressing p53-specific or scrambled shRNA were exposed to KPT-185 at 2 × ED50 concentration, and KPT-induced DR5 and FAS transcripts compared to untreated cells were determined. KPT-185 induced significantly less DR5 and FAS in p53 knockdown Z-138 cells than Z-138 cells expressing scrambled shRNA (3.20 ± 0.50 vs 1.75 ± 0.32-fold increase in DR5, P = 0.01; and 3.40 ± 0.19 vs 0.63 ± 0.12-fold increase in FAS, P < 0.0001). However, although MDM2 contributes to p53 nuclear export, Nutlin-3a may not always activate p53-mediated transcription, probably in a cell-specific manner. Cytoplasmic retention of Nutlin-induced p53 has been reported in neoplastic cell lines and patient cells.(31–34) Neither KPT-185 nor Nutin-3a altered p53 mRNA levels (data not shown), suggesting that they have little effect on p53 synthesis.
Figure 4.
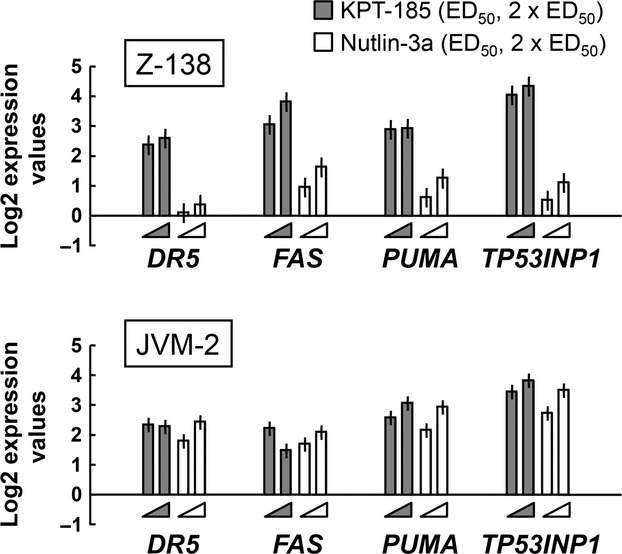
Exportin-1 inhibitor KPT-185 induces p53-regulated apoptotic genes. Z-138 and JVM-2 mantle cell lymphoma cells were treated for 12 h with KPT-185 or Nutlin-3a at their ED50 or 2 × ED50 concentrations, and transcripts were quantitated by real-time PCR. Data shown are from one of three independent experiments with similar results.
KPT-185 induces apoptosis in primary MCL cells
We investigated whether KPT-185 could induce apoptosis in MCL cells from five primary samples (three with wild-type p53 and two with mutant p53, as determined by annexin V induction). The primary MCL cells were cultured in medium in the presence or absence of KPT-185 (100 or 400 nM) or 2.5 μM Nutlin-3a. The p53 mutant samples were resistant to Nutlin-3a (Fig. 5), which was consistent with previously reported findings.(26,27) The two p53-mutant MCL samples were less sensitive to 100 nM KPT-185 exposure than p53 wild-type MCL samples. However, when the concentration was increased to 1 μM, the KPT-185 exposure induced apoptosis in 71% of the p53 mutant cells from patient 1. Of note, serum concentrations of >1 μM KPT-330, a SINE XPO1 antagonist related to KPT-185 and currently in phase I clinical studies, are achievable at well-tolerated doses in humans.(35,36) Data suggest that p53-mediated and transcription-dependent apoptosis is the primary signaling of SINE-induced apoptosis and that KPT-185 may induce p53-independent apoptosis at higher concentrations.
Figure 5.
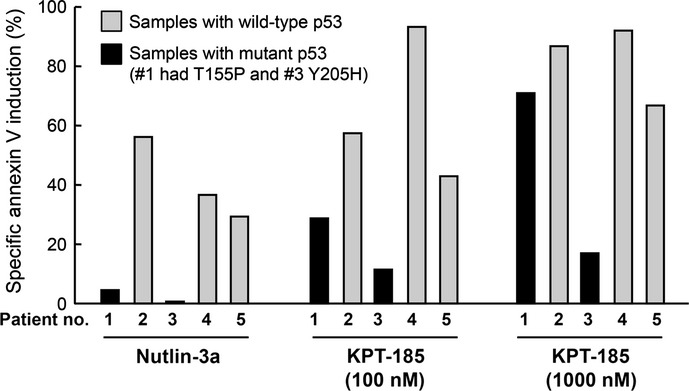
Exportin-1 inhibitor KPT-185 induces apoptosis in primary mantle cell lymphoma cells. Primary cells from five patients were incubated for 72 h with Nutlin-3a (2.5 μM) or KPT-185 (100 or 1000 nM), and the annexin V-positive fractions were measured by flow cytometry. Three samples had wild-type p53 and two had mutant p53 (one had T155P and another Y205H).
Absence of acquired TP53 mutations in KPT-adapted MCL cells
Nutlin-3a has been shown to induce or select TP53 mutations in p53 wild-type cancer cells, resulting in the development of subclones that are resistant to p53-mediated apoptosis.(35,36) As KPT-185 appeared to activate p53-dependent apoptotic signaling in MCL cells, we investigated if long-term KPT-185 exposure induces or selects p53 mutant clones. Both Z-138 and JVM-2 cells were continuously exposed for 2 months to KPT-185 starting at ED50. The JVM-2 cells adapted to grow in the presence of KPT-185 in two independent experiments, as determined by more than 90% viability (Trypan blue-negative cell percentage) at 8 weeks of exposure to ×1.5 ED50. Acquired TP53 mutations were not detected in the SINE-adapted cells, suggesting that the mechanism of resistance is independent of TP53 mutations.
Discussion
We found that p53 is a determinant of apoptosis induction by the SINE XPO1 inhibitor KPT-185 in MCL. XPO1 carries ∼230 XPO1 cargo proteins,(1) and many of them are responsible for MCL cell survival, proliferation, and cell death. Our data highlight the importance of p53 in the cellular survival network mediated by XPO1 cargos.
The nuclear export of p53 is cooperatively mediated by MDM2 and XPO1, and it is anticipated that their inhibition would lead to nuclear p53 retention and transcriptional activation of p53 target genes. However, our data suggest that MDM2 inhibition may not sufficiently accumulate p53 into the nucleus and activate transcription-dependent p53 signaling in MCL. This is probably due to the E3 ubiquitin ligase activity of MDM2. MDM2 is the major ubiquitin ligase of p53, which promotes p53 proteasomal degradation both in the nucleus and the cytoplasm. Therefore, MDM2 inhibition may increase p53 levels in the cytoplasm where p53 is unable to execute transcriptional activity.(31–34) Although cytoplasmic p53 can mediate transcription-independent apoptosis is some circumstances,(33,34) the quantitative contribution to p53-mediated apoptosis still remains unclear. In contrast, KPT-185 treatment activated transcription-dependent p53 signaling toward apoptosis in MCL cells. As increased XPO1 expression predicted poor survival in MCL patients, and MDM2 inhibition might not fully activate p53-mediated transcription, p53 activation by targeting XPO1 might offer a novel and plausible therapeutic strategy for MCL that retains wild-type p53. This hypothesis may be extended to solid cancers in which XPO1 overexpression is associated with poor prognosis.(4–7) Interestingly, preliminary data in patients with advanced, relapsed/refractory MCL and other NHLs treated in the ongoing phase I study (clinicaltrials.gov NCT01607892) of the related SINE compound selinexor (KPT-330), show that XPO1 inhibition can induce responses even in heavily pretreated NHL patients.(37)
Clinical concern about p53 activators would be the selection or induction of p53 mutant subclones during therapy, which has been reported in in vitro cultures with Nutlin-3a.(37,38) Although p53 status was a determinant of apoptotic response to KPT-185 in MCL, KPT-185 showed significant p53-independent apoptotic activity at higher concentrations (prominent at 400–1000 nM). A phase I pharmacokinetic and pharmacodynamic study of KPT-330 in patients with advanced refractory solid tumors showed that after consecutive doses of 30 mg/m2 daily, peak plasma concentrations were above 1000 nM,(36) and similar data have been reported in the ongoing study in advanced NHL.(35) Together, these data suggest that SINE may show both p53-dependent and -independent apoptotic activities in MCL patients at clinically relevant concentrations. This is comparable to the in vitro concentration that caused cytotoxicity of KPT-185 in multiple myeloma cells,(10) as well as the MCL cells examined here. Importantly, studies have shown that in addition to p53, SINEs block nuclear factor-κB and c-MYC signaling pathways that are crucial components of MCL pathogenesis.(8,10,39,40) Targeting multiple key arms of the apoptotic regulatory machinery by XPO1 inhibition in MCL, including nuclear factor-κB and c-MYC in addition to p53, may not only efficiently eradicate lymphoma cells but also block emergence of p53 mutant clones. These hypotheses are consistent with our findings on SINE activity in p53 mutant MCL.
In conclusion, our work suggests that SINEs may provide a novel therapeutic concept for MCL and may be only partially dependent on p53 apoptotic signaling. Induction of p53 would potently induce cell death in MCL, including those with high levels of XPO1 (i.e., poor prognostic MCL) and/or mutant p53 and high levels of XPO1, at clinically relevant concentrations. The multifaceted activities of XPO1 inhibitors may not only enhance p53-mediated MCL cell death but also delay or prevent the selection of p53 mutant subclones during therapy.
Acknowledgments
The authors would like to thank Connie Larsson, Department of Leukemia, The University of Texas MD Anderson Cancer Center (Houston, TX, USA) for valuable technical help. The authors acknowledge elucidating discussions and editing by Dr. Numsen Hail Jr, Molecular Hematology and Therapy, Department of Leukemia, The University of Texas MD Anderson Cancer Center, and Drs. Takero Shindo, Yasushi Kubota, Eisaburo Sueoka, and Naoko Sueoka-Aragane, Department of Medicine, Saga University (Saga, Japan). This study was supported in part by grants from the National Institutes of Health Lymphoma SPORE (CA136411), Leukemia SPORE (CA100632), P01 “The Therapy of AML” (CA55164), Cancer Center Support Grant (CA16672), and the Paul and Mary Haas Chair in Genetics (to M. Andreeff) and Osaka Cancer Research Foundation, Japan Leukemia Research Fund, and a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology in Japan (to K. Kojima). The University of Texas MD Anderson Cancer Center Lymphoma Tissue Bank is supported by the National Institutes of Health Lymphoma SPORE grant P50CA136411 and the Fredrick B. Hagemeister Research Fund.
Disclosure Statement
The research was supported in part by funds/materials provided by Karyopharm Therapeutics (to M. Andreeff). Sharon Shacham and Michael Kauffman are founders and executives of Karyopharm Therapeutics, Inc., where they receive compensation and hold equity positions.
References
- 1.Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell. 2012;23:3673–6. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kau TR, Way JC, Silver PA. Nuclear transport and cancer: from mechanism to intervention. Nat Rev Cancer. 2004;4:106–17. doi: 10.1038/nrc1274. [DOI] [PubMed] [Google Scholar]
- 3.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–32. doi: 10.1016/j.bcp.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao Y, Dong Y, Lin F, et al. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep. 2009;21(1):229–35. [PubMed] [Google Scholar]
- 5.Shen A, Wang Y, Zhao Y, Zou L, Sun L, Cheng C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery. 2009;65(1):153–9. doi: 10.1227/01.NEU.0000348550.47441.4B. [DOI] [PubMed] [Google Scholar]
- 6.Noske A, Weichert W, Niesporek S, et al. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer. 2008;112:1733–43. doi: 10.1002/cncr.23354. [DOI] [PubMed] [Google Scholar]
- 7.van der Watt PJ, Maske CP, Hendricks DT, et al. The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer. 2009;124:1829–40. doi: 10.1002/ijc.24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang K, Wang M, Tamayo AT, et al. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol. 2013;41(1):67–78. doi: 10.1016/j.exphem.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120:4621–34. doi: 10.1182/blood-2012-05-429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tai YT, Landesman Y, Acharya C, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28:155–65. doi: 10.1038/leu.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kojima K, Kornblau SM, Ruvolo V, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121:4166–74. doi: 10.1182/blood-2012-08-447581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jares P, Campo E. Advances in the understanding of mantle cell lymphoma. Br J Haematol. 2008;142:149–65. doi: 10.1111/j.1365-2141.2008.07124.x. [DOI] [PubMed] [Google Scholar]
- 13.Greiner TC, Dasgupta C, Ho VV, et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2006;103:2352–7. doi: 10.1073/pnas.0510441103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubio-Moscardo F, Climent J, Siebert R, et al. Mantle-cell lymphoma genotypes identified with CGH to BAC microarrays define a leukemic subgroup of disease and predict patient outcome. Blood. 2005;105:4445–54. doi: 10.1182/blood-2004-10-3907. [DOI] [PubMed] [Google Scholar]
- 15.Halldórsdóttir AM, Lundin A, Murray F, et al. Impact of TP53 mutation and 17p deletion in mantle cell lymphoma. Leukemia. 2011;25:1904–8. doi: 10.1038/leu.2011.162. [DOI] [PubMed] [Google Scholar]
- 16.Hernández L, Beà S, Pinyol M, et al. CDK4 and MDM2 gene alterations mainly occur in highly proliferative and aggressive mantle cell lymphomas with wild-type INK4a/ARF locus. Cancer Res. 2005;65:2199–206. doi: 10.1158/0008-5472.CAN-04-1526. [DOI] [PubMed] [Google Scholar]
- 17.Boyd MT, Vlatkovic N, Rubbi CP. The nucleolus directly regulates p53 export and degradation. J Cell Biol. 2011;194:689–703. doi: 10.1083/jcb.201105143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Brate A, Giannakakou P. The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist Updat. 2003;6:313–22. doi: 10.1016/j.drup.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Moll UM, LaQuaglia M, Bénard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci U S A. 1995;92:4407–11. doi: 10.1073/pnas.92.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013;27(1):66–74. doi: 10.1038/leu.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 23.Verhaegen M, Bauer JA, Martín de la Vega C, et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006;66:11348–59. doi: 10.1158/0008-5472.CAN-06-1748. [DOI] [PubMed] [Google Scholar]
- 24.Bichi R, Shinton SA, Martin ES, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A. 2002;99:6955–60. doi: 10.1073/pnas.102181599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kojima K, Duvvuri S, Ruvolo V, Samaniego F, Younes A, Andreeff M. Decreased sensitivity of 17p-deleted chronic lymphocytic leukemia cells to a small molecule BCL-2 antagonist ABT-737. Cancer. 2012;118:1023–31. doi: 10.1002/cncr.26360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 27.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azmi AS, Aboukameel A, Bao B, et al. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology. 2013;144:447–56. doi: 10.1053/j.gastro.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–30. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azmi AS, Al-Katib A, Aboukameel A, et al. Selective inhibitors of nuclear export for the treatment of non-Hodgkin's lymphomas. Haematologica. 2013;98:1098–106. doi: 10.3324/haematol.2012.074781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elison JR, Cobrinik D, Claros N, Abramson DH, Lee TC. Small molecule inhibition of HDM2 leads to p53-mediated cell death in retinoblastoma cells. Arch Ophthalmol. 2006;124:1269–75. doi: 10.1001/archopht.124.9.1269. [DOI] [PubMed] [Google Scholar]
- 32.Kojima K, Konopleva M, Samudio IJ, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106:3150–9. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaseva AV, Marchenko ND, Moll UM. The transcription-independent mitochondrial p53 program is a major contributor to nutlin-induced apoptosis in tumor cells. Cell Cycle. 2009;8:1711–9. doi: 10.4161/cc.8.11.8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kojima K, Konopleva M, McQueen T, O'Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108:993–1000. doi: 10.1182/blood-2005-12-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuruvilla J, Gutierrez M, Shah BD, et al. Preliminary Evidence Of Anti Tumor Activity Of Selinexor (KPT-330) In a Phase I Trial Ofa First-In-Class Oral Selective Inhibitor Of Nuclear Export (SINE) In Patients (pts) With Relapsed/Refractory Non Hodgkin's Lymphoma (NHL) and Chronic Lymphocytic Leukemia (CLL) [abstract] Blood. 2013;122 Abstract 90. [Google Scholar]
- 36.Razak ARA, Soerensen MM, Mahipal A, et al. First-in-class, first-in-human phase I trial of KPT-330, a selective inhibitor of nuclear export (SINE) in patients (pts) with advanced solid tumors [abstract] J Clin Oncol. 2013;31 Abstract 2505. [Google Scholar]
- 37.Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene. 2011;30:4678–86. doi: 10.1038/onc.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michaelis M, Rothweiler F, Barth S, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011;2:e243. doi: 10.1038/cddis.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaturvedi NK, Rajule RN, Shukla A, et al. Novel treatment for mantle cell lymphoma including therapy-resistant tumor by NF-κB and mTOR dual-targeting approach. Mol Cancer Ther. 2013;12:2006–17. doi: 10.1158/1535-7163.MCT-13-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oberley MJ, Rajguru SA, Zhang C, et al. Immunohistochemical evaluation of MYC expression in mantle cell lymphoma. Histopathology. 2013;63:499–508. doi: 10.1111/his.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]