

SUMMARY
We studied the factors that regulate IL-23 receptor expression and IL-17 production in human tuberculosis infection. Mycobacterium tuberculosis (M. tb)-stimulated CD4+ cells from tuberculosis patients secreted less IL-17 than did CD4+ cells from healthy tuberculin reactors (PPD+). M. tb cultured monocytes from tuberculosis patients and PPD+ donors expressed equal amounts of IL-23p19 mRNA and protein, suggesting that reduced IL-23 production is not responsible for decreased IL-17 production by tuberculosis patients. Freshly isolated and M. tb- stimulated CD4+ cells from tuberculosis patients had reduced IL-23 receptor and phosphorylated STAT3 (pSTAT3) expression, compared to PPD+ donors. STAT3 siRNA reduced IL-23 receptor expression, and IL-17 production by CD4+ cells from PPD+ donors. Tuberculosis patients had increased number of PD-1+ T cells compared to healthy PPD+ individuals. Anti-PD-1 antibody enhanced pSTAT3 and IL-23R expression and IL-17 production by M. tb cultured CD4+ cells of tuberculosis patients. Anti-tuberculosis therapy decreased PD-1 expression, increased IL-17 and IFN-γ production and pSTAT3, IL-23R expression. These findings demonstrate that increased PD-1 expression and decreased pSTAT3 expression reduces IL-23 receptor expression and IL-17 production by CD4+ cells of tuberculosis patients.
Keywords: human, cytokine, IL-17, M. tuberculosis
INTRODUCTION
T cells play a crucial role in protective immunity against Mycobacterium tuberculosis (M. tb) and other intracellular pathogens [1-4]. Recent studies have shown that IL-17, a proinflammatory cytokine that is produced predominantly by memory T cells, contributes to immunity against M. tb [5-7]. IL-17 plays an important role in vaccine-induced protective immune responses against M. tb infection [6]. After vaccination with M. bovis Bacille Calmette Guerin (BCG), IL-17 induces chemokine production that recruits IFN-γ-producing T cells which inhibit bacterial growth after infection with M. tb [6]. IL-1, IL-6, TGF-β and IL-23 produced by antigen presenting cells were shown to induce IL-17 production in various experimental systems [8-10]. Previously, we found that Ag-experienced CD4+ cells are the major source for IL-17, and that IL-23, but not TGF-β or IL-1 or IL-6, contributes to IL-17 production in human M. tb infection [10]. We also found that NKG2D expressed on CD4+ cells interacts with ULBP1 on M. tb-infected mononuclear phagocytes to enhance IL-23 secretion by mononuclear phagocytes, which in turn favors IL-17 production by CD4+CD62L− cells [10].
There are conflicting reports about IL-17 production in tuberculosis patients. In one report, CD4+ cells from tuberculosis patients produced less IL-17 in response to M. tb antigens than CD4+ cells from healthy donors [11], but the underlying mechanisms were not defined. In contrast, other studies have found that tuberculosis patients have increased Th17 responses [12] and that IL-17 production in tuberculosis patients correlates with disease severity [13]. Another study found no difference in IL-17-producing cells between uninfected individuals, persons with latent tuberculosis infection and patients with active tuberculosis [14].
In the current study, we compared M. tb-induced IL-17 production by CD4+ cells of healthy tuberculin reactors (PPD+ donors) with that of tuberculosis patients, and identified the signaling molecules that regulate IL-17 production by CD4+ cells. We found that increased PD-1 expression reduces expression of phosphorylated STAT3 (pSTAT3) leads to decreased IL-23R expression, which in turn decreases IL-17 production by CD4+ cells in tuberculosis patients.
RESULTS
CD4+ cells from tuberculosis patients produce less IL-17 upon M. tb stimulation
CD4+ cells from PPD+ donors produced IL-17 in response to autologous M. tb-stimulated monocytes [10]. In this study, we isolated CD4+ cells and CD14+ cells from PBMC of 10 PPD+ donors and 10 tuberculosis patients and cultured in the presence or absence of γ-irradiated M. tb H37Rv (10 μg/ml) for 96 h. IL-17 levels in M. tb-stimulated culture supernatants from tuberculosis patients were reduced by 85%, compared with those from PPD+ donors (48 + 86 pg/ml versus 314 + 241 pg/ml, p < 0.05, Fig. 1).
Figure 1. IL-17 production by healthy PPD+ donors and tuberculosis patients.
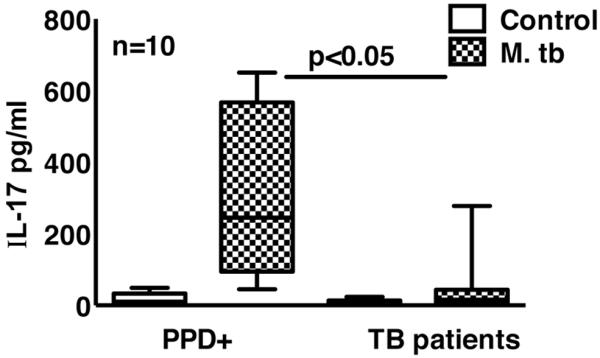
CD4+ cells and CD14+ cells from freshly isolated PBMC of 10 PPD+ individuals and 10 tuberculosis patients were isolated by magnetic beads conjugated to anti-CD14 or anti-CD4 obtained from Miltenyi Biotech. CD4+ cells and CD14+ cells were cultured at a 9:1 ratio, with or without γ-irradiated M. tb H37Rv for 96 h. IL-17 levels in culture supernatants were measured by ELISA. The horizontal line shows the median, the boxes show the 25th and 75th percentile values, and the whiskers show the 5th and 95th percentile values. Each time experiments were performed with the blood obtained from one PPD+ individual and one tuberculosis patient. A total 10 independent experiments were performed. Maximum, minimum and median values, p values and number of donors (n) are shown.
IL-23 production is similar in tuberculosis patients and PPD+ donors
IL-23 produced by M. tb-stimulated monocytes, but not TGF-β, IL-1 or IL-6, contributed to IL-17 production in persons with latent tuberculosis infection [10]. To determine if reduced IL-17 production by CD4+ cells of tuberculosis patients is due to less IL-23 production, CD14+ monocytes from 5 PPD+ individuals and 5 tuberculosis patients were cultured with or without γ-irradiated M. tb H37Rv for 48 h. IL-23 levels in M. tb-stimulated culture supernatants from tuberculosis patients were similar compared to PPD+ donors (21 ± 13 pg/ml versus 18 ± 3 pg/ml, Fig. 2A). When monocytes from 10 PPD+ donors and 9 tuberculosis patients were cultured with γ-irradiated H37Rv, IL-23p19 mRNA expression was also similar (Fig. 2B).
Figure 2. IL-23 production by healthy PPD+ donors and tuberculosis patients.

A. CD14+ monocytes from PPD+ individuals and tuberculosis patients were cultured in the presence or absence of γ-irradiated M. tb H37Rv for 48 h. (A) Supernatants were collected and IL-23 levels were measured by ELISA. Five independent experiments were performed. (B) RNA was isolated from control and M. tb cultured monocytes after 48 h and IL-23p19 mRNA expression was measured by real time PCR. The thin lines in all panels show mean values and p values are shown in each panel. Nine independent experiments were performed. Paired and unpaired t tests were performed. Mean values, p values and number of donors (n) in each panel is shown.
IL-23 receptor expression by CD4+ cells is reduced in tuberculosis patients
IL-23 binds to IL-23R on CD4+ cells, leading to initiation of signaling pathways for the production of IL-17. We compared IL-23R expression by CD4+ cells of freshly isolated PBMC obtained from PPD+ donors and tuberculosis patients, using flow cytometry. The mean fluorescence intensity (MFI) was 334 ± 133 for 8 PPD+ donors and 171 ± 67 for 7 tuberculosis patients (p = 0.04, Fig. 3A). Similarly, IL-23R mRNA expression by freshly isolated CD4+ cells of PPD+ individuals was 3 fold higher compared to tuberculosis patients (p = 0.01, Fig. 3B). Next, CD4+ cells from PBMC of PPD+ individuals and tuberculosis patients were cultured with autologous monocytes in medium alone, or with γ-irradiated M. tb H37Rv. After 96 h, surface staining for CD4 and IL-23R was performed. IL-23R expression on gated CD4+ cells was increased in 9 PPD+ donors compared to 7 tuberculosis patients (MFI 332 ± 208 versus 237 ± 172, p = 0.01, Fig. 3C). Further we isolated CD4+ cells in the above cultured cells and measured IL-23R mRNA expression in 7 PPD+ individuals and 9 tuberculosis patients. IL-23R mRNA expression was also reduced in tuberculosis patients, compared to PPD+ donors (0.6 ± 0.5 versus 2.0 ± 0.5 arbitrary units, p = 0.002, Fig. 3E).
Figure 3. IL-23R expression by T cells from healthy PPD+ donors and tuberculosis patients.

A. IL-23R expression by CD4+ cells. PBMC from 8 PPD+ individuals and 7 tuberculosis patients were isolated from the blood and stained with anti-CD4-FITC and anti-IL-23R-APC. CD4+ cells were gated and the mean fluorescence intensity (MFI) of IL-23R was measured by flow cytometry. Seven independent experiments were performed. B. IL-23R mRNA expression by freshly isolated CD4+ cells. CD4+ cells from freshly isolated PBMC of 13 PPD+ individuals and 13 tuberculosis patients were isolated by immunomagnetic selection. RNA was extracted and reverse transcribed to cDNA, and IL-23R mRNA was quantified by real-time PCR, relative to expression of GAPDH. Each point shows the mean of triplicate determinations. Thirteen independent experiments were performed. C. IL-23R expression by M. tb-stimulated CD4+ cells. CD4+ cells and CD14+ cells from freshly isolated PBMC of 9 PPD+ individuals and 7 tuberculosis patients were isolated by magnetic beads conjugated to anti-CD14 or anti-CD4 obtained from Miltenyi Biotech. CD4+ cells and CD14+ cells were cultured at a 9:1 ratio, with or without γ-irradiated M. tb H37Rv. After 96 h, surface staining for CD4 and IL-23R was performed using anti-CD4-FITC and anti-IL-23R-APC. CD4+ cells were gated and the mean fluorescence intensity (MFI) of IL-23R was measured. Seven independent experiments were performed. D. A representative figure for panel C is shown. IL-23R expression by CD4+ cells of medium alone (thin black line), CD4+ cells of γ-irradiated M. tb H37Rv cultured cells (thick black line) and IL-23R isotype antibody staining on CD4+ cells of γ-irradiated M. tb H37Rv cultured cells (thick light line). E. IL-23R mRNA expression by M. tb-stimulated CD4+ cells. CD4+ cells from PBMC of 7 PPD+ individuals and 9 tuberculosis patients were cultured as in panel C. After 96 h, RNA was extracted and reverse transcribed to cDNA, and IL-23R mRNA was quantified by real-time PCR, relative to expression of GAPDH. In all panels, the thin lines show mean values. Seven independent experiments were performed. Paired and unpaired t tests were performed. Mean values, p values and number of donors (n) in each panel is shown.
Expression of pSTAT3, but not SOCS3, is reduced in tuberculosis patients
Because STAT3 and SOCS3 contribute to IL-17 production in various experimental models [15;16], we determined if altered expression of these molecules reduced IL-17 production in tuberculosis patients. PBMC from PPD+ donors and tuberculosis patients were isolated and surface staining for CD4+ cells and intracellular staining for pSTAT3 and SCOS3 was performed. The percentage of CD4+pSTAT3+ and CD4+SCOS3+ cells was determined by flow cytometry. In 10 PPD+ donors the percentage of CD4+pSTAT3+ cells was 7.9 ± 4.9% compared to 3.4 ± 2.3% in 9 tuberculosis patients (Fig. 4A, p = 0.04). The percentage of CD4+SOCS3+ cells was similar in 8 PPD+ individuals and in 8 tuberculosis patients (8.4 ± 5.4% versus 9.7 ± 6.8%, Fig. 4B). We next cultured CD4+ cells from 11 PPD+ donors and 11 tuberculosis patients with autologous monocytes, with or without γ-irradiated M. tb H37Rv. After 96 h, expression of pSTAT3 and SOCS3 were measured by intracellular staining. Similar to findings in fresh cells, the percentage of CD4+pSTAT3+ cells in 11 tuberculosis patients was less compared to 11 PPD+ donors (6.0 ± 3.5% versus 15.9 ± 6.4%, p = 0.04, Fig. 4C). As in fresh cells the percentage of SOCS3+ cells was similar in 9 healthy donors and 9 tuberculosis patients (14.8 ± 7.1% versus 21.8 ± 18.2%, Fig. 4D).
Figure 4. Expression of pSTAT3 and SOCS3 by healthy PPD+ donors and tuberculosis patients.

A. Expression of pSTAT3 by freshly isolated cells. PBMC from 10 healthy tuberculin reactors and 9 tuberculosis patients were isolated and surface staining for CD4+ cells and intracellular staining for pSTAT3 was performed. The percentage of CD4+pSTAT3+ cells was determined by flow cytometry. Nine independent experiments were performed. B. Expression of SOCS3 by freshly isolated cells. PBMC from 8 healthy PPD+ donors and 8 tuberculosis patients were isolated and surface staining for CD4+ cells and intracellular staining for SOCS3 was performed. The percentage of CD4+SOCS3+ cells was determined by flow cytometry. Eight independent experiments were performed. C. Expression of pSTAT3 by γ-irradiated M. tbH37Rv cultured cells. CD4+ and CD14+ cells from the PBMC of 11 PPD+ individuals and 11 tuberculosis patients were isolated by immunomagnetic selection, and cultured at a 9:1 ratio, with or without γ-irradiated M. tb H37Rv. After 96 h, surface staining for CD4+ cells and intracellular staining for pSTAT3 was performed. The percentage of CD4+pSTAT3+ cells was determined by flow cytometry. Eleven independent experiments were performed. D. Expression of SOCS3 by γ-irradiated M. tb H37Rv cultured cells. CD4+ and CD14+ cells from the PBMCs of 9 PPD+ individuals and 9 tuberculosis patients were isolated by immunomagnetic selection, and cultured at a 9:1 ratio, with or without γ-irradiated M. tb H37Rv. After 96 h, surface staining for CD4+ cells and intracellular staining for SOCS3 was performed. The percentage of CD4+SOCS3+ cells was determined by flow cytometry. Nine independent experiments were performed. E. A representative figure for panels C is shown. pSTAT3 expression by CD4+ cells of medium alone, CD4+ cells of γ-irradiated M. tb H37Rv cultured cells and pSTAT3 isotype antibody staining on CD4+ cells of γ-irradiated M. tb H37Rv cultured cells is shown. In all panels, the numbers of subjects are shown, and thin lines show the mean values. Paired and unpaired t tests were performed. Mean values, p values and number of donors (n) in each panel is shown.
STAT3 regulates IL-23R expression and IL-17 production
In the above experiments, expression of pSTAT3 and IL-23R was decreased in tuberculosis patients. We next asked whether STAT3 regulates IL-23R expression and IL-17 production by using STAT3 siRNA. Inhibition of STAT3 was confirmed by real-time PCR analysis (approximately 70% inhibition). In 9 healthy PPD+ donors, STAT3 siRNA inhibited M. tb-induced IL-17 production by CD4+ cells from 549 ± 416 pg/ml to 100 ± 97 pg/ml (p = 0.03, Fig. 5A). Similarly, in 5 healthy PPD+ donors, STAT3 siRNA inhibited IL-23R mRNA expression by M. tb induced CD4+ cells by about 5 folds compared to control siRNA (p = 0.05, Fig. 5B). These findings indicate that STAT3 increases IL-23R expression, which in turn induces IL-17 production by CD4+ cells in response to M. tb.
Figure 5. Effect of STAT3 siRNA on M. tb induced IL-17 production and IL-23R expression by CD4+ cells.
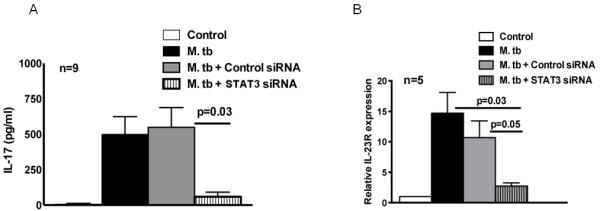
CD4+ cells and CD14+ cells from freshly isolated PBMC of PPD+ individuals were isolated by magnetic beads conjugated to anti-CD14 or anti-CD4 obtained from Miltenyi Biotech. Some CD4+ cells were transfected with STAT3 siRNA or scrambled siRNA. After overnight, CD4+ cells and CD14+ cells were cultured at a 9:1 ratio, with or without γ-irradiated M. tb H37Rv for 96 h. A. IL-17 production in culture supernatants was measured by ELISA. Nine independent experiments were performed. B. The percentages of IL-23R+CD4+ cells were measured by flow cytometry. Five independent experiments were performed. Paired and unpaired t tests were performed. Mean values ± SD, p values and number of donors (n) in each panel are shown.
PD-1 contributes to reduced STAT3, and IL-23R expression and IL-17 production
PD-1 knockout mice are extraordinarily susceptible to M. tb and produce high levels of IL-17 during infection, compared to wild type mice [17;18]. To determine the role of PD-1 in IL-17 production by tuberculosis patients, first we measured PD-1 expression by freshly isolated CD3+ cells. In 5 PPD+ individuals, the percentages of CD3+PD-1+ cells were 9.3 ± 6.6, compared to 27 ± 13 in 7 tuberculosis patients (p < 0.03, Fig. 6A). Next we cultured CD4+ cells and autologous monocytes of tuberculosis patients with γ-irradiated M. tb H37Rv, with anti-PD-1 neutralizing Ab (10 μg/ml) or isotype Ab (10 μg/ml). After 96 h, IL-23R expression was measured by flow cytometry. The MFI of IL-23R-expressing CD4+ cells in M. tb stimulated and isotype Ab cultured cells was 140 ± 95, and this was increased marginally to 161 ± 97 with the addition of anti-PD-1neutralizing Ab (p = 0.05, Fig. 6B). Similarly, anti-PD-1 antibody neutralization increased the percentage of CD4+pSTAT3+ cells from 4.1 + 2.5 (isotype Ab cultured cells) to 6.1 ± 2.4 (p = 0.02, Fig. 6C). In 5 tuberculosis patients, we also measured IL-17 levels in the above culture supernatants. IL-17 levels in unstimulated culture supernatants were 93 ± 119 pg/ml and this was not significantly increased upon culturing with M. tb and isotype antibody (210 ± 260 pg/ml). In contrast, neutralization of PD-1 increased IL-17 levels to 418 ± 355 pg/ml (p = 0.04, Fig. 6D).
Figure 6. Effect of PD-1 on pSTAT3, IL-23R expression and IL-17 production by tuberculosis patients.

A. PD-1 expression by freshly isolated CD3+ cells. Freshly isolated PBMC from 5 PPD+ individuals and 7 tuberculosis patients were stained for CD3+ and PD-1+ cells. Five independent experiments were performed. CD4+ cells and autologous monocytes from PBMCs of tuberculosis patients were cultured with γ-irradiated M. tb H37Rv, in the presence of PD-1 antibody (10 μg/ml) or isotype antibody. After 96 h, B. Surface staining for CD4+ cells and IL-23R was performed. Five independent experiments were performed. C. CD4+pSTAT3+ cells were measured by flow cytometry. Five independent experiments were performed. D. IL-17 production in culture supernatants was measured by ELISA. For panels B, C and D the horizontal line shows the median, the boxes show the 25th and 75th percentile values, and the whiskers show the 5th and 95th percentile values. Five independent experiments were performed. Paired and unpaired t tests were performed. Mean values, p values and number of donors (n) is shown in panel A. Maximum, minimum and median values, p values and number of donors (n) are shown in panels B, C and D.
Anti-tuberculosis therapy decreases PD-1 expression and increases IL-17 production
We determined the effect of anti-tuberculosis therapy on PD-1, pSTAT3, IL-23R expression and IL-17 and IFN-γ production. We first measured PD-1 expression by CD3+ cells. In 11 untreated patients, the percentages of CD3+PD-1+ cells were 18.3 ± 5.4, compared to 6 ± 2 in 10 treated (2 years after anti-tuberculosis therapy, p < 0.0001, Fig. 7A). Next, we isolated CD4+ cells and CD14+ cells from PBMC of 5 untreated tuberculosis patients and 5 treated (2 years after anti-tuberculosis therapy) and cultured in the presence or absence of γ-irradiated M. tb H37Rv for 96 h. IL-17 levels in M. tb-stimulated culture supernatants of treated tuberculosis patients were 22 fold higher compared with those from untreated patients (189 ± 75 pg/ml versus 8 ± 7 pg/ml, p = 0.006, Fig. 7B). Similarly, IFN-γ levels in M. tb-stimulated culture supernatants of treated tuberculosis patients were 3.5 fold higher compared with those from untreated patients (409 ± 87 pg/ml versus 125 ± 45 pg/ml, p = 0.0002, Fig. 7C). We also determined pSTAT3 expression and IL-23R expression by CD4+ of the above cultured cells. The percentage of pSTAT3 expressing CD4+ cells of treated tuberculosis patients was 12 ± 2.5 % compared to 8.5 ± 2 % (p = 0.02, Fig. 7D) in untreated tuberculosis patients. Similar to the above findings the mean florescence intensity of IL-23R expressing CD4+ cells of treated patients was 320 ± 67 compared to 167 ± 61 in untreated patients (p = 0.01, Fig. 7E).
Figure 7. Effect of anti-tuberculosis therapy on IL-17 and IFN-γ production, PD-1, pSTAT3, and IL-23R expression by tuberculosis patients.

A. PD-1 expression by freshly isolated CD3+ cells. Freshly isolated PBMC from 11 untreated tuberculosis patients and 10 treated tuberculosis patients (2 years after anti-tuberculosis therapy) were stained for CD3+ and PD-1+ cells. Ten independent experiments were performed. CD4+ cells and CD14+ cells from PBMC of 5 untreated tuberculosis patients and 5 treated tuberculosis patients (2 years after anti-tuberculosis therapy) were cultured in the presence or absence of γ-irradiated M. tb H37Rv for 96 h. B. IL-17 levels in culture supernatants were measured by ELISA. Five independent experiments were performed. C. IFN-γ levels in culture supernatants were measured by ELISA. Seven independent experiments were performed. D. pSTAT3 expression by CD4+ of the above cultured cells was determined by flow cytometry. Five independent experiments were performed. E. IL-23R expression by CD4+ of the above cultured cells was determined by flow cytometry. Five independent experiments were performed. Paired and unpaired t tests were performed. Mean values for panel A, p values and number of donors (n) in each panel are shown.
DISCUSSION
IL-17 contributes to BCG vaccine-induced immunity to M. tb in mouse models [5-7]. In humans with latent tuberculosis infection, we previously found that antigen-experienced CD4+CD62L− cells are the major source for IL-17, and that NKG2D expressed on T cells interacts with ULBP-1 on M. tb-stimulated monocytes to induce IL-23, which in turn enhances IL-17 production [10]. In this report, we found that in response to M. tb, CD4+ cells from tuberculosis patients produce less IL-17 compared to healthy PPD+ individuals. Reduced IL-17 production by CD4+ cells of tuberculosis patients was not due to decreased IL-23 secretion by monocytes, but was associated with decreased expression of transcription factor, STAT3 and IL-23R by CD4+ cells. STAT3 siRNA inhibited IL-23R expression and IL-17 production by CD4+ cells of healthy PPD+ individuals. The percentage of PD-1+ T cells were significantly higher in tuberculosis patients PBMC compared to healthy PPD+ individuals PBMCs. Neutralization of PD-1, significantly increased pSTAT3, and IL-23R expression and IL-17 production by CD4+ T cells of tuberculosis patients in response to M. tb. Anti-tuberculosis therapy decreased PD-1 expression and increased IL-17 production. Our study provides the first evidence that defective IL-17 production in tuberculosis patients is due to reduced IL-23R expression, which is in turn regulated by pSTAT3 and PD-1.
IL-17 is an inflammatory cytokine produced by CD4+, CD8+ and γδ T cells, that can cause autoimmunity, enhance transplant graft rejection, and contribute to anti-tumor immunity and resistance to several intracellular pathogens, including M. tb [5-7;19-23]. The overall effects of IL-17 may favor or harm the host, depending on the pathogen and stage of infection. In HIV infection, disease progression is associated with loss of Th17 cells [24] and IL-17-secreting innate lymphoid cells are essential for host defense against certain fungi [25;26]. In contrast, IL-17 aggravates chronic hepatitis B infection [27], increases pathology in human schistosomiasis [28] and worsens inflammation in patients with Dengue virus infection [29]. In mouse models, IL-17 also increases lung inflammaton in Mycoplasmapneumoniae and Chlamydophilapneumoniae infection, and exacerbates pathology in Pseudomonasaeruginosa infection [30-32].
Studies in murine models have clearly shown that IL-17contributes to protective immunity against challenge with M. tb after BCG vaccination, given either subcutaneously or through mucosal routes [32-35]. However, the role of IL-17 during primary M. tb infection is less clear. IL-17 production precedes the development of IFN-γ-producing cells [6;36], and BCG-infected IL-17 knockout mice have reduced granuloma formation and decreased DTH responses to M. tb antigens, suggesting that IL-17 contributes to induction of optimal Th1 responses [21;36]. However, bacterial burdens are not consistently higher in IL-17 knockout mice that are infected with M. tb or M. bovis BCG [6;36;37].
We found that M. tb-induced IL-17 production by CD4+ cells of tuberculosis patients was significantly reduced, compared to that in PPD+ donors, similar to most published studies [11;38-40]. One report found that IL-17 production was increased in tuberculosis patients, but this may have been because control subjects were BCG-vaccinated without latent tuberculosis infection [13], and IL-17 production in response to M. tb antigens may be lower in BCG-vaccinated persons than in those with latent tuberculosis infection.
The IL-23R is essential for differentiation and maintenance of IL-17-producing effector T cells [41]. IL-23R gene polymorphisms are associated with several autoimmune diseases, including Crohn’s disease and psoriasis [34], and genetic variants in the IL-23R promoter that increase receptor mRNA expression are associated with an increased risk of hepatocellular carcinoma [42]. However, limited information is available about the role of IL-23R in infections due to intracellular pathogens. The IL-23R is important for expansion of γδ T cells and CD4-CD8-αβ T cells during the murine immune response to Listeria infection [43]. Although IL-23 is not required for early control of tuberculosis, it is essential for long term containment of infection through expression of CXCL13, which directs trafficking of T cells from blood vessels to the site of infection [44;45]. Previously we found that IL-23, but not IL-6 or TGF-β, is required for T cells from persons with M. tb infection to produce IL-17 in response to mycobacterial antigens [10]. In the current study, we found that monocytes from PPD+ individuals and tuberculosis patients produce equal amounts of IL-23 in response to γ-irradiated M. tb H37Rv, indicating that reduced IL-23 expression did not account for decreased IL-17 production by T cells from tuberculosis patients. However, CD4+ cells from tuberculosis patients had reduced expression of IL-23R mRNA and protein compared to findings in PPD+ donors. We found that in response to M. tb, CD4+ cells of treated tuberculosis patients (2 years after anti-tuberculosis therapy) produce more IL-17, express higher percentage of pSTAT3+ cells, increased IL-23R expression compared to untreated tuberculosis patients suggesting active tuberculosis inhibits IL-17 production, pSTAT3 and IL-23R expression.
Limited information is available regarding the signaling molecules that regulate IL-23R expression. CD5 costimulation induces stable Th17 development by promoting IL-23R expression and sustained STAT3 activation [46] and a specific microRNA, Let-7f, inhibits IL-23R expression in human CD4+ memory T cells [47]. STAT3 and SOCS3 regulate IL-17 production in several experimental systems, and transfection of STAT3 constructs into murine T cells increases IL-23R expression and production of IL-17 [15;16]. There are conflicting reports about the role of SOCS3 in Th17 cytokine production. Some investigators found that TGF-β inhibits SOCS3 to enhance Th17 development [48] and overexpression of SOCS3 in T-lymphocytes impairs IL-17 production [49;50]. On the other hand, one study showed that increased SOCS3 expression in CD4+ cells increased IL-17 production [51]. In the current study, we found that tuberculosis patients have decreased expression of STAT3 but no changes in SOCS3 expression. STAT3 siRNA significantly reduced IL-23R expression and IL-17 production by CD4+ cells of PPD+ donors. Our results provide the first evidence that pSTAT3 regulates IL-23R expression and IL-17 production in human disease due to a widespread intracellular pathogen.
Programmed death 1 (PD-1) is a type I membrane protein also known as CD279. PD-1 and its ligands, programmed death ligand 1 (PDL-1) and programmed death ligand 2 (PDL-2), negatively regulate T cell proliferation and cytokine production [52;53]. PD-1 knockout mice are extraordinarily susceptible to M. tb and produce high levels of IL-17 during infection, compared to wild type mice [17;18]. There is a limited information available on PD-1 mediated regulation of Th17 responses. PD-1 negatively regulates anti-mycobacterial responses by suppressing innate immune cells which help to differentiate IL-17-producing cells [54] and IL-27-primed T cells restrict differentiation of Th17 cells [55]. In the current study, we found that increased PD-1 expression reduces pSTAT3 and IL-23R expression and IL-17 production by CD4+ cells in tuberculosis patients and anti-tuberculosis therapy (2 years after anti-tuberculosis therapy) decreased PD-1 expression. We have not determined how PD-1 regulates pSTAT3 and IL-23R expression and IL-17 production by CD4+ cells. Previously we found that tuberculosis patients have increased number of T-regulatory cells compared to healthy PPD+ individuals [56] and PD-1 control expansion of M. tb-induced Foxp3+ Treg cells [57]. Foxp3 can inhibit Th17 differentiation by antagonizing RORγt function [58] and it is not known whether Foxp3 can regulate pSTAT3 and IL-23R expression. Alternatively, PD-1 can regulate IL-23R expression, which results in decreased pSTAT3 and IL-17 production. Further work is needed to determine if PD-1 dependent increase in Foxp3 expression in CD4+ cells of tuberculosis patients can inhibit pSTAT3 and IL-23R expression and IL-17 production by CD4+ cells or PD-1 regulates IL-23R expression, which results in decreased pSTAT3 and IL-17 production.
In summary, we found that reduced IL-17 production by CD4+ cells of tuberculosis patients is due to increased PD-1 expression. PD-1 inhibits expression of pSTAT3, which in turn lowers T-cell expression of IL-23R. Because IL-17 is important for vaccine-induced protection against tuberculosis, understanding the mechanisms that control production of this cytokine may allow development of immunotherapeutic strategies to enhance IL-17 production in response to anti-tuberculosis vaccines. Parallel approaches may also be useful to upregulate protective Th17 responses against fungi and other pathogens.
Materials and Methods
Patient population
Blood was obtained from 55 PPD+ donors, 16 healthy donors (2 years after anti-tuberculosis therapy) and 50 human immunodeficiency virus (HIV)-seronegative patients with culture-proven pulmonary tuberculosis who had received anti-tuberculosis therapy for <1 week. Acid-fast stains of sputum were positive for all patients. All studies were approved by the Institutional Review Boards of the Blue Peter Public Health Research Center, Hyderabad, AP, India and the University Of Texas Health Science Center at Tyler. Informed consent was obtained from all participants.
Antibodies and other reagents
For flow cytometry, we used FITC anti-CD4, APC anti-CD3 (BD Biosciences), FITC anti-PD-1 (Biolegend), APC anti-IL-23R, anti-pSTAT3 (R&D), anti-SOCS3, IgG PE (all from Abcam). For neutralization, we used mAb to PD-1 and isotype Abs (R&D). γ-irradiated M. tb H37Rv was obtained from Colorado State University, Fort Collins, CO.
Isolation of monocytes and CD4+ cells
PBMC were isolated by centrifugation over Ficoll-Paque (Sigma). Monocytes and CD4+ cells were isolated by magnetic beads conjugated to anti-CD14 or anti-CD4 (all from Miltenyi Biotech), respectively. Cell purity was >95%, as measured by flow cytometry.
Culture of Monocytes
Freshly isolated CD14+ cells were cultured in 12-well plates at 2 ×106 cells/well in Roswell Park Memorial Institute (RPMI 1640) containing penicillin/streptomycin (Sigma) and 10% heat-inactivated human serum, with or without γ-irradiated M. tb H37Rv (10 μg/ml) at 37°C in a humidified 5% CO2 atmosphere. In some experiments, after 48 h, cell-free culture supernatants were collected, aliquoted and stored at −70°C until cytokine concentrations were measured. For other experiments, RNA was extracted to perform real time PCR analysis to measure expression of IL-23p19 mRNA.
Culture of CD4+ cells and monocytes
Freshly isolated CD4+ cells were cultured in 12-well plates at 2 ×106 cells/well in RPMI 1640 containing penicillin/streptomycin and 10% heat-inactivated human serum, with or without 2×105 autologous monocytes/well. CD4+ cells and monocytes were cultured in the presence or absence of γ-irradiated M. tbH37Rv (10 μg/ml) at 37°C in a humidified 5% CO2 atmosphere. In some experiments, neutralization mAb to PD-1 or isotype Abs (10 μg/ml each) were added to the cultures. After 96 h, cell-free culture supernatants were collected, aliquoted and stored at −70°C until cytokine concentrations were measured. In some experiments cells were stained to detect pSTAT3 and SOCS3, and for other experiments, RNA was extracted for real time PCR analysis to determine the expression of IL-23R.
CD4+ cell transfection
CD4+ cells isolated as outlined above, were transfected with small interfering RNA (siRNA) for STAT3 or scrambled siRNA (Santa Cruz Biotechnology), using the Amaxa nucleofector (Amaxa Biosystems). Briefly, 2 × 106 freshly isolated cells were resuspended in 100 μl of the Amaxa transfection solution and transfected with 30 pmol of siRNA, using the manufacturer’s protocol U-01. After overnight incubation, cells were washed, transferred to 0.6 ml RPMI 1640 complete medium with or without 2 × 105 autologous monocytes/well. CD4+ cells and monocytes were cultured, with or without γ-irradiated M. tb H37Rv (10 μg/ml) at 37°C in a humidified 5% CO2 atmosphere. After 4 days, some cells were stained for IL-23R expression and analyzed by flow cytometry. RNA was extracted to measure IL-23R expression by real time PCR. Cell-free culture supernatants were collected, aliquoted and stored at −70°C until IL-17 and IFN-γ concentrations were measured.
Real-time PCR for quantification of IL-23p19 and IL-23R mRNA
Total RNA was extracted from cultured cells, using TRIzol reagent (Invitrogen). Total RNA was reverse transcribed, using the Clone AMV First-Strand cDNA synthesis kit (Life Technologies). The forward and reverse primers for IL-23p19 were 5′-GAGCAGCAACCCTGAGTCCCTA-3′ and 5′-CAAATTTCCCTTCCCATCTAATAA-3′, respectively. For IL-23R, the forward and reverse primers were 5′-CATGACTTGCACCTGGAATG-3′ and 5′-GCTTGGACCCAAACCAAGTA-3′, respectively. Primers for GAPDH were, forward, 5′-GCCATCAATGACCCCTTCATT-3′ and reverse, 5′-TTGACGGTGCCATGGAATTT-3′. Real-time PCR was performed using the Quantitect SYBR Green PCR kit (Qiagen) on a spectrofluorometric thermal cycler (Mini Opticon, Bio-Rad). PCR were performed in triplicate as follows: 95°C for 10 min, and 45 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. All samples were normalized to the amount of GAPDH transcript present in each sample.
Detection of intracellular pSTAT3 and SOCS3
To measure intracellular pSTAT3 and SOCS3 in freshly isolated PBMC and cultured CD4+ cells, we used the intracellular staining kits from BD Biosciences. For surface staining, FITC anti-CD4 was added. After washing with PBS and 2% FCS, cells were fixed in Cytofix/Cytoperm and washed twice in 1x permeabilization/wash solution. Primary anti-pSTAT3 or anti-SOCS3 were then added to cells re-suspended in staining buffer. After incubation at 4°C for 40 min, cells were washed in PBS with 2% FCS, and the secondary antibody was added and incubated for 30 min on ice. Cells were washed and analyzed by flow cytometry.
Measurement of IL-23, IFN-γ and IL-17 concentrations
IL-23, IFN-γ and IL-17 levels were measured by ELISA (eBioscience). For measurement of IFN-γ and IL-17, supernatants of cultured CD4+ and CD14+ cells were collected after 96 h of culture. Supernatants from cultured CD14+ monocytes were collected after 48 h for measurement of IL-23. Supernatants were stored at −70°C until cytokine concentrations were measured.
Statistical analysis
Results are shown as the mean ± SD. For data that were normally distributed, comparisons between groups were performed by a paired or unpaired t test, as appropriate. For data that were not normally distributed, the non-parametric Mann-Whitney-U-test was performed.
Acknowledgments
Funding: This work was supported by grants from the National Institutes of Health (AI085135 and AI094692 to R.V), the Cain Foundation for Infectious Disease Research, the Center for Pulmonary and Infectious Disease Control, Department of Biotechnology, GoI, India (BT/01/COE/07/02 to VLV) and a Potts Memorial Foundation grant to RD. Anuradha Bandaru received a post-doctoral fellowship from Council of Scientific and Industrial Research, GoI, India (09/0980(001)09/EMR-1).
Footnotes
Conflict of interest: The authors have no financial or commercial conflict of interest.
References
- 1.Flynn JL, Chan J. Immunology of tuberculosis. Annu.Rev.Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 2.Flynn JL, Goldstein MM, Triebold KJ, Koller B, Bloom BR. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc.Natl.Acad.Sci.U.S.A. 1992;89:12013–12017. doi: 10.1073/pnas.89.24.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rojas RE, Balaji KN, Subramanian A, Boom WH. Regulation of human CD4(+) alphabeta T-cell-receptor-positive (TCR(+)) and gammadelta TCR(+) T-cell responses to Mycobacterium tuberculosis by interleukin-10 and transforming growth factor beta. Infect.Immun. 1999;67:6461–6472. doi: 10.1128/iai.67.12.6461-6472.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orme IM, Collins FM. Protection against Mycobacterium tuberculosis infection by adoptive immunotherapy. Requirement for T cell-deficient recipients. J.Exp.Med. 1983;158:74–83. doi: 10.1084/jem.158.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scriba TJ, Kalsdorf B, Abrahams DA, Isaacs F, Hofmeister J, Black G, Hassan HY, Wilkinson RJ, Walzl G, Gelderbloem SJ, Mahomed H, Hussey GD, Hanekom WA. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J.Immunol. 2008;180:1962–1970. doi: 10.4049/jimmunol.180.3.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat.Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 7.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J.Immunol. 2006;177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 8.O’Garra A, Stockinger B, Veldhoen M. Differentiation of human T(H)-17 cells does require TGF-beta! Nat.Immunol. 2008;9:588–590. doi: 10.1038/ni0608-588. [DOI] [PubMed] [Google Scholar]
- 9.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Paidipally P, Periasamy S, Barnes PF, Dhiman R, Indramohan M, Griffith DE, Cosman D, Vankayalapati R. NKG2D-dependent IL-17 production by human T cells in response to an intracellular pathogen. J.Immunol. 2009;183:1940–1945. doi: 10.4049/jimmunol.0803578. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Li J, Tian J, Zhu B, Zhang Y, Yang K, Ling Y, Hu Y. IL-17 and IFN-gamma production in peripheral blood following BCG vaccination and Mycobacterium tuberculosis infection in human. Eur.Rev.Med.Pharmacol.Sci. 2012;16:2029–2036. [PubMed] [Google Scholar]
- 12.Marin ND, Paris SC, Rojas M, Garcia LF. Reduced frequency of memory T cells and increased Th17 responses in patients with active tuberculosis. Clin.Vaccine Immunol. 2012;19:1667–1676. doi: 10.1128/CVI.00390-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jurado JO, Pasquinelli V, Alvarez IB, Pena D, Rovetta AI, Tateosian NL, Romeo HE, Musella RM, Palmero D, Chuluyan HE, Garcia VE. IL-17 and IFN-gamma expression in lymphocytes from patients with active tuberculosis correlates with the severity of the disease. J.Leukoc.Biol. 2012;91:991–1002. doi: 10.1189/jlb.1211619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marin ND, Paris SC, Rojas M, Garcia LF. Functional profile of CD4+ and CD8+ T cells in latently infected individuals and patients with active TB. Tuberculosis.(Edinb.) 2013 doi: 10.1016/j.tube.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J.Biol.Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 16.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat.Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 17.Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J.Immunol. 2011;186:1598–1607. doi: 10.4049/jimmunol.1003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazar-Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, Porcelli SA, Almo SC, Nathenson SG, Jacobs WR., Jr. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc.Natl.Acad.Sci.U.S.A. 2010;107:13402–13407. doi: 10.1073/pnas.1007394107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J.Exp.Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antonysamy MA, Fanslow WC, Fu F, Li W, Qian S, Troutt AB, Thomson AW. Evidence for a role of IL-17 in organ allograft rejection: IL-17 promotes the functional differentiation of dendritic cell progenitors. J.Immunol. 1999;162:577–584. [PubMed] [Google Scholar]
- 21.Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, Oshiro K, Okamoto Y, Watanabe H, Kawakami K, Roark C, Born WK, O’Brien R, Ikuta K, Ishikawa H, Nakae S, Iwakura Y, Ohta T, Matsuzaki G. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver. J.Immunol. 2008;181:3456–3463. doi: 10.4049/jimmunol.181.5.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuzaki G, Umemura M. Interleukin-17 as an effector molecule of innate and acquired immunity against infections. Microbiol.Immunol. 2007;51:1139–1147. doi: 10.1111/j.1348-0421.2007.tb04008.x. [DOI] [PubMed] [Google Scholar]
- 23.Peng MY, Wang ZH, Yao CY, Jiang LN, Jin QL, Wang J, Li BQ. Interleukin 17-producing gamma delta T cells increased in patients with active pulmonary tuberculosis. Cell Mol.Immunol. 2008;5:203–208. doi: 10.1038/cmi.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh A, Vajpayee M, Ali SA, Mojumdar K, Chauhan NK, Singh R. HIV-1 diseases progression associated with loss of Th17 cells in subtype ‘C’ infection. Cytokine. 2012;60:55–63. doi: 10.1016/j.cyto.2012.06.288. [DOI] [PubMed] [Google Scholar]
- 25.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M, Gaffen SL. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J.Exp.Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng SC, van, d. V, Smeekens S, Joosten LA, Van der Meer JW, Kullberg BJ, Netea MG. Candida albicans dampens host defense by downregulating IL-17 production. J.Immunol. 2010;185:2450–2457. doi: 10.4049/jimmunol.1000756. [DOI] [PubMed] [Google Scholar]
- 27.Yang B, Wang Y, Zhao C, Yan W, Che H, Shen C, Zhao M. Increased Th17 cells and interleukin-17 contribute to immune activation and disease aggravation in patients with chronic hepatitis B virus infection. Immunol.Lett. 2013;149:41–49. doi: 10.1016/j.imlet.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Mbow M, Larkin BM, Meurs L, Wammes LJ, de Jong SE, Labuda LA, Camara M, Smits HH, Polman K, Dieye TN, Mboup S, Stadecker MJ, Yazdanbakhsh M. T-helper 17 cells are associated with pathology in human schistosomiasis. J.Infect.Dis. 2013;207:186–195. doi: 10.1093/infdis/jis654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain A, Pandey N, Garg RK, Kumar R. IL-17 Level in Patients with Dengue Virus Infection & its Association with Severity of Illness. J.Clin.Immunol. 2012 doi: 10.1007/s10875-012-9855-0. [DOI] [PubMed] [Google Scholar]
- 30.Kurai D, Nakagaki K, Wada H, Saraya T, Kamiya S, Fujioka Y, Nakata K, Takizawa H, Goto H. Mycoplasma pneumoniae Extract Induces an IL-17-Associated Inflammatory Reaction in Murine Lung: Implication for Mycoplasmal Pneumonia. Inflammation. 2012 doi: 10.1007/s10753-012-9545-3. [DOI] [PubMed] [Google Scholar]
- 31.Mosolygo T, Korcsik J, Balogh EP, Faludi I, Virok DP, Endresz V, Burian K. Chlamydophila pneumoniae re-infection triggers the production of IL-17A and IL-17E, important regulators of airway inflammation. Inflamm.Res. 2013 doi: 10.1007/s00011-013-0596-1. [DOI] [PubMed] [Google Scholar]
- 32.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, deSauvage F, Cooper AM. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J.Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 33.Wozniak TM, Ryan AA, Britton WJ. Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. J.Immunol. 2006;177:8684–8692. doi: 10.4049/jimmunol.177.12.8684. [DOI] [PubMed] [Google Scholar]
- 34.Yannam GR, Gutti T, Poluektova LY. IL-23 in infections, inflammation, autoimmunity and cancer: possible role in HIV-1 and AIDS. J.Neuroimmune.Pharmacol. 2012;7:95–112. doi: 10.1007/s11481-011-9315-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gopal R, Rangel-Moreno J, Slight S, Lin Y, Nawar HF, Fallert Junecko BA, Reinhart TA, Kolls J, Randall TD, Connell TD, Khader SA. Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal.Immunol. 2013 doi: 10.1038/mi.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K, Suda T, Sudo K, Nakae S, Iwakura Y, Matsuzaki G. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J.Immunol. 2007;178:3786–3796. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- 37.Okamoto YY, Umemura M, Yahagi A, O’Brien RL, Ikuta K, Kishihara K, Hara H, Nakae S, Iwakura Y, Matsuzaki G. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J.Immunol. 2010;184:4414–4422. doi: 10.4049/jimmunol.0903332. [DOI] [PubMed] [Google Scholar]
- 38.Cowan J, Pandey S, Filion LG, Angel JB, Kumar A, Cameron DW. Comparison of interferon-gamma-, interleukin (IL)-17- and IL-22-expressing CD4 T cells, IL-22-expressing granulocytes and proinflammatory cytokines during latent and active tuberculosis infection. Clin.Exp.Immunol. 2012;167:317–329. doi: 10.1111/j.1365-2249.2011.04520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Zhang M, Liao M, Graner MW, Wu C, Yang Q, Liu H, Zhou B. Reduced Th17 response in patients with tuberculosis correlates with IL-6R expression on CD4+ T Cells. Am.J.Respir.Crit Care Med. 2010;181:734–742. doi: 10.1164/rccm.200909-1463OC. [DOI] [PubMed] [Google Scholar]
- 40.Kumar NP, Anuradha R, Suresh R, Ganesh R, Shankar J, Kumaraswami V, Nutman TB, Babu S. Suppressed type 1, type 2, and type 17 cytokine responses in active tuberculosis in children. Clin.Vaccine Immunol. 2011;18:1856–1864. doi: 10.1128/CVI.05366-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat.Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y, Liu Y, Pan S, Liu L, Liu J, Zhai X, Shen H, Hu Z. IL-23R polymorphisms, HBV infection, and risk of hepatocellular carcinoma in a high-risk Chinese population. J.Gastroenterol. 2013;48:125–131. doi: 10.1007/s00535-012-0620-1. [DOI] [PubMed] [Google Scholar]
- 43.Riol-Blanco L, Lazarevic V, Awasthi A, Mitsdoerffer M, Wilson BS, Croxford A, Waisman A, Kuchroo VK, Glimcher LH, Oukka M. IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J.Immunol. 2010;184:1710–1720. doi: 10.4049/jimmunol.0902796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khader SA, Cooper AM. IL-23 and IL-17 in tuberculosis. Cytokine. 2008;41:79–83. doi: 10.1016/j.cyto.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BA, Fountain JJ, Martino C, Pearl JE, Tighe M, Lin YY, Slight S, Kolls JK, Reinhart TA, Randall TD, Cooper AM. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J.Immunol. 2011;187:5402–5407. doi: 10.4049/jimmunol.1101377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Wit J, Souwer Y, van Beelen AJ, de Groot R, Muller FJ, Klaasse BH, Jorritsma T, Kapsenberg ML, de Jong EC, van Ham SM. CD5 costimulation induces stable Th17 development by promoting IL-23R expression and sustained STAT3 activation. Blood. 2011;118:6107–6114. doi: 10.1182/blood-2011-05-352682. [DOI] [PubMed] [Google Scholar]
- 47.Li Z, Wu F, Brant SR, Kwon JH. IL-23 receptor regulation by Let-7f in human CD4+ memory T cells. J.Immunol. 2011;186:6182–6190. doi: 10.4049/jimmunol.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, Reynolds SL, Weaver CT, Roarty K, Serra R, Benveniste EN, Cong Y. TGF-beta promotes Th17 cell development through inhibition of SOCS3. J.Immunol. 2009;183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romain M, Taleb S, Dalloz M, Ponnuswamy P, Esposito B, Perez N, Wang Y, Yoshimura A, Tedgui A, Mallat Z. Overexpression of SOCS3 in T Lymphocytes Leads to Impaired Interleukin-17 Production and Severe Aortic Aneurysm Formation in Mice--Brief Report. Arterioscler.Thromb.Vasc.Biol. 2013;33:581–584. doi: 10.1161/ATVBAHA.112.300516. [DOI] [PubMed] [Google Scholar]
- 50.Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, Esposito B, Perez N, Yasukawa H, Van Snick J, Yoshimura A, Tedgui A, Mallat Z. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J.Exp.Med. 2009;206:2067–2077. doi: 10.1084/jem.20090545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleinsteuber K, Heesch K, Schattling S, Sander-Juelch C, Mock U, Riecken K, Fehse B, Fleischer B, Jacobsen M. SOCS3 promotes interleukin-17 expression of human T cells. Blood. 2012;120:4374–4382. doi: 10.1182/blood-2011-11-392738. [DOI] [PubMed] [Google Scholar]
- 52.Riley JL. PD-1 signaling in primary T cells. Immunol.Rev. 2009;229:114–125. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat.Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 54.Rui Y, Honjo T, Chikuma S. Programmed cell death 1 inhibits inflammatory helper T-cell development through controlling the innate immune response. Proc.Natl.Acad.Sci.U.S.A. 2013;110:16073–16078. doi: 10.1073/pnas.1315828110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirahara K, Ghoreschi K, Yang XP, Takahashi H, Laurence A, Vahedi G, Sciume G, Hall AO, Dupont CD, Francisco LM, Chen Q, Tanaka M, Kanno Y, Sun HW, Sharpe AH, Hunter CA, O’Shea JJ. Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity. 2012;36:1017–1030. doi: 10.1016/j.immuni.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garg A, Barnes PF, Roy S, Quiroga MF, Wu S, Garcia VE, Krutzik SR, Weis SE, Vankayalapati R. Mannose-capped lipoarabinomannan- and prostaglandin E2-dependent expansion of regulatory T cells in human Mycobacterium tuberculosis infection. Eur.J.Immunol. 2008;38:459–469. doi: 10.1002/eji.200737268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Periasamy S, Dhiman R, Barnes PF, Paidipally P, Tvinnereim A, Bandaru A, Valluri VL, Vankayalapati R. Programmed death 1 and cytokine inducible SH2-containing protein dependent expansion of regulatory T cells upon stimulation With Mycobacterium tuberculosis. J.Infect.Dis. 2011;203:1256–1263. doi: 10.1093/infdis/jir011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]