

Abstract
We investigated the protective effects of melatonin and its metabolites: 6-hydroxymelatonin (6-OHM), N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), N-acetylserotonin (NAS), and 5-methoxytryptamine (5-MT) in human keratinocytes against a range of doses (25, 50, and 75 mJ/cm2) of ultraviolet B (UVB) radiation. There was significant reduction in the generation of ROS (50–60%) when UVB-exposed keratinocytes were treated with melatonin or its derivatives. Similarly melatonin and its metabolites reduced the nitrite and hydrogen peroxide levels that were induced by UVB as early as 30 min after the exposure. Moreover, melatonin and its metabolites enhanced levels of reduced glutathione in keratinocytes within 1 h after UVB exposure in comparison to control cells. Using proliferation assay, we observed a dose-dependent increase in viability of UVB-irradiated keratinocytes that were treated with melatonin or its derivatives after 48 h. Using the dot-blot technique and immunofluorescent staining we also observed that melatonin and its metabolites enhanced the DNA repair capacity of UVB-induced 6-4PP or CPD generation in human keratinocytes. Additional evidence for induction of DNA repair in cells exposed to UVB and treated with the indole compounds was shown using the Comet assay. Finally, melatonin and its metabolites further enhanced expression of p53 phosphorylated at Ser-15 but not at Ser-46 or its non-phosphorylated form. In conclusion, melatonin, its precursor NAS, and its metabolites 6-OHM, AFMK, 5-MT, which are endogenously produced in keratinocytes, protect these cells against UVB-induced oxidative stress and DNA damage.
Keywords: melatonin, epidermal keratinocytes, UVB, DNA damage, 6-hydroxymelatonin, AFMK, N-acetylserotonin, 5-methoxytryptamine
Introduction
Ultraviolet radiation (UVR) spectrum of solar light represents the major skin stressor. UVR-related damage to the skin and epidermal keratinocytes are tightly connected with the production of reactive oxygen species (ROS), including hydrogen peroxide (H2O2) and nitric oxide (NO), which can cause oxidative damage and further reduction of the important antioxidant glutathione (GSH) and inhibition of DNA repair, in addition to genotoxic and mutagenic effects caused by these [1]. The mutagenic and genotoxic effects of UVR are predominantly related to absorption of UVB by DNA which acts as chromophores for wavelengths in the range of 290–320 nm [2]. Exposure to UVR causes DNA damage in form of primarily cyclobutane pyrimidine dimers (CPD) and to lower degree of pyrimidine photoproducts (6-4)PPs [3]. CPDs are DNA lesions produced in human skin exposed to either UVB or UVA [4], albeit lower levels for UVA exposure are required [3].
Melatonin (N-acetyl-5-methoxytryptamine) and its metabolites have multiple functions. They regulate circadian rhythms, biological responses, act as hormone, neurotransmitters, antiinflammatory agents, and as antioxidants [5–13]. These effects are secondary to activation of membrane bound receptors or are receptors independent [14, 15]. Melatonin’s main sites of production are the pineal gland and retina, where serotonin is acetylated to N-acetylserotonin (NAS), which is further methylated, to melatonin [10, 16, 17]. Melatonin is also produced in peripheral organs including the skin [12, 13, 18]. It is well documented that mammalian skin has the capability of transforming L-tryptophan to serotonin that is further metabolised in this organ to melatonin [19–21]. Also, of note is that significant amounts of NAS accumulate in the skin as the main intermediate of the melatonin pathway [12, 20–23]. Melatonin is metabolised through the indolic and kynuric pathways resulting in the production of 6-hydroxymelatonin (6-OHM), cyclic 3-hydroxymelatoni, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), and 5-methoxytryptamine (5-MT) as main metabolites along with several additional products [reviewed in [11, 13, 24, 25]]. The production of AFMK occurs via the kynuric pathway [11, 25], although it can also be generated non-enzymatically through the interaction of melatonin and H2O2 [26, 27] or through the photochemical transformation secondary to exposure to UVB [27, 28]. NAS, a precursor to melatonin, is also a product of its metabolism [29], during the process of demethylation [13, 30].
The main product of melatonin metabolism in human keratinocytes is 6-OHM the production of which is stimulated by UVB exposure [28, 31]. 5-MT is a product of melatonin deacetylation, which in addition to retina is also produced in rodent skin [19, 23] and human epidermal keratinocytes [28, 31]. UVB can both directly induce transformation of melatonin to 2-hydroxymelatonin and AFMK [28] or stimulate the endogenous production of AFMK in human epidermal keratinocytes [28, 31]. Thus, human epidermal keratinocytes produce NAS, melatonin, AFMK and 5-MT [18, 20, 28, 31]
Melatonin is recognised as an important protective factor in many different cells [11, 32–35]. Melatonin and its metabolites scavenge both ROS/RNS through a free radical scavenging cascade [11, 27, 34, 36]. Beside their antioxidative properties [37], melatonin and its metabolites exhibit antiproliferative and prodifferentiation properties in human epidermal keratinocytes [31, 38, 39]. These effects have been described mainly in intact histocultured skin or in HaCaT immortalized human keratinocytes [28, 31, 39–42]. Although, the protective effects of melatonin and AFMK have been studied extensively, there is a paucity of data supporting the protective role of NAS and other melatonin derivatives including 6-OHM and 5-MT. Furthermore, information on protective effects of melatonin’s metabolites on primary epidermal keratinocytes cultured in vitro is limited. Therefore, in the current study we investigated the antioxidative and protective effects of melatonin metabolites in comparison to melatonin in normal human epidermal keratinocytes exposed to a range of doses of UVB (25, 50, and 75 mJ/cm2). Since melatonin reportedly prevents DNA damage in cancer cells [43], in the current study we tested the ability of melatonin and its metabolites to prevent DNA damage in skin cells. p53 expression increases with UVB exposure and it initiates growth arrest and damaged DNA has been detected [44]. Melatonin induces a p53-dependent DNA damage response [45] and p53 protein expression [46]. We hypothesised that the UVR-induced increase in p53 is further augmented by melatonin derivatives and that this effect likely contributes to enhanced repair of UVR-induced thymine dimers.
Material and Methods
Chemicals
Melatonin (N-acetyl-5-methoxytryptamine) (M5250), 6-hydroxymelatonin (3-(N-acetylaminoethyl)-6-hydroxy-5-methoxyindole) (H0627), N-acetyl-5-hydroxy-tryptamine (N-acetylserotonin, normelatonin) (A1824), 5-methoxy tryptamine (2-(5-methoxyindol-3-yl) ethylamine, 3-(2-aminoethyl)-5-methoxyindole, 5-methoxyindole-3-ethanamine, 5-MT, deacetylmelatonin) (286583) were purchased from Sigma-Aldrich (St. Louis, MO); and AFMK (N1-acetyl-N2-formyl-5-methoxykynuramine, N-[3-[2-(formylamino)-5-methoxyphenyl]-3-oxypropyl]-acetamide) (10005254) from Cayman Chemical Company (Ann Arbor, MI, USA). Chemicals were prepared and used according to the protocols described elsewhere [47]. Ethanol was used as a dilutent and a vehicle.
Cell culture
HaCaT keratinocytes were grown in Dulbecco’s minimal essential media (DMEM) supplemented with 5% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA, USA) [38, 48]. Under the current experimental conditions charcoal-treated FBS was used (Atlanta Biologicals, Lawrenceville, GA, USA). Human epidermal keratinocytes (HEKn) are isolated from neonatal foreskin of African-American donors as previously described [49, 50]. Cells were grown in keratinocyte growth media (KGM) supplemented with keratinocyte growth factors (KGF) (Lonza Walkersville Inc, Walkersville, MD). Cells in their third passage were treated with melatonin or its derivatives for 24 h before UVB exposure and for various times after UVB exposure. For UVB exposure the media were replaced with phosphate buffered saline (PBS).
UVB exposure
The UV irradiation was performed with a Biorad UV transluminator 2000 (Bio-RAD Laboratories, Hercules, CA, USA) as previously described [41, 51]. For the experiments, doses of 25, 50, 75, or 200 mJ/cm2 were chosen. With the emittance of UVB by the UV-source of 4.01 mW/cm2 these doses were achieved after 20, 40, 60, or 120 s of irradiation, respectively.
Cell proliferation assays
HEKn keratinocytes cells were plated in 96-well plates, 10,000 cells/well. After overnight incubation, melatonin and its metabolites, initially dissolved in ethanol, were added to the medium to achieve final concentrations ranging from 1 mM–0.1 pM, while corresponding ethanol concentrations served as a control. After 44 h of incubation with the compounds, 20 μl of MTS/PMS solution (Promega, Madison, WI) was added to the cells. Four hours later, absorbance was recorded at 490 nm using an ELISA plate reader. The number of viable cells was measured in six replicates.
Comet assay
Keratinocytes were plated in 12-well plates. Cells were treated with melatonin, or its metabolites at the concentration of 5×10−5 M, or ethanol (vehicle, dilution 5:1,000) for 24 h. Then the medium was replaced with 1xPBS and cells were exposed to UVB 200 mJ/cm2. PBS was removed and replaced with fresh medium containing compounds and cells were further incubated for 3 h at 37°C. After detaching, cells were counted and used for the comet assay. The comet assay was performed following the manufacturer’s protocol (Trevigen, Gaithersburg, MD) as previously described [52]. Cell suspensions at a density of 1×105/ml were combined with molten 1.2% low-melting-point agarose, diluted 1:10, placed onto two frosted slides precoated with 0.6% normal agarose, and incubated at 4°C for 30 min. Cells were further digested in lysis solution at 4°C for 60 minutes. DNA strand breaks were separated by electrophoresis in alkaline electrophoreses solution (200 mM NaOH, 1mM EDTA, pH>13) in horizontal gel electrophoresis slide tray (Comet-10, Thistle Scientific, UK). DNA breaks were exposed to alkaline unwinding for 20 min at room temperature after which electrophoresis was performed at 25 V for 1 h. Following neutralization in neutralizing buffer (0.4 M Tris, pH7), comets are visualized by ethidium bromide (Sigma-Aldrich, St. Louis, MO) or Sybr Green I (Trevigen, Gaithersburg, MD) staining. The slides were examined and images were captured using Nikon fluorescent microscope and the Leica digital DM 4000b fluorescent microscope equipped with image analysis software. Approximately 60 comet images were taken for each condition. DNA damage was measured by the tail length [53] using Comet Score software available from http://www.scorecomets.com/
6-4PP and CPD dimers
Removal of pyrimidine (6-4) pyrimidone photoproducts (6-4 photoproducts) and cyclobutane pyrimidine dimers (CPDs) were measured by immunoslot blot assay [54, 55]. For this experiment HaCaT keratinocytes were grown in DMEM +5% FBS, in 60 mm dishes, in duplicates, until they have reached 70% confluency. Cells were treated with melatonin or its metabolites at the concentration of 5×10−5 M, or ethanol (vehicle, dilution 5:1,000) for 30 before the UVB exposure (50 mJ/cm2) and for 3 h or 6 h after the exposure. Cells were harvested and DNA was isolated (DNeasy, Qiagen), heat-denatured at 100°C for 10 minutes, applied to a Hybond nitrocellulose membrane (Amersham) using a vacuum-driven slot blot apparatus (100 ng and 50 ng/slot for (6-4) photoproduct and CPD analysis, respectively), and fixed by baking for 1 h at 80°C. Membranes were incubated with mouse monoclonal antibodies specific for 6–4 photoproducts (1/10,000) and CPDs (1/10,000) (Cosmo Bio Co. Cat# NMDND002 and Cat#NMDNDOO1, respectively). Peroxidase-conjugated anti-mouse secondary antibody was used at a dilution of 1/10,000 in blocking buffer. Equal loading of DNA was confirmed by DAPI (Invitrogen) staining.
CPDs in cultured normal keratinocytes were visualised using immunofluorescence. Briefly, HEKn were plated onto chamber slides, treated with melatonin or its metabolites at the concentration of 5×10−5 M, or ethanol (vehicle, dilution 5:1,000) for 24 h prior UVB exposure, and further for 3 h after the exposure. Following the manufacturer protocol, cells were stained with anti-CPD (clone TDM2) (Cosmo Bio Co Ltd., Tokyo, Japan) (dil 1:100) and subsequently with goat anti-mouse IgG-FITC (Santa Cruz Biotechnology, Santa Cruz, CA) (1:100). Nuclei were stained red with Vectashield mounting media with propidium iodide (Vector Laboratories, Burlingame, CA). Stained cells were imaged with a fluorescence microscope. Pictures recorded were analysed using ImageJ software (NIH free download).
Immunofluorescence for p53
Keratinocytes were plated onto chamber slides in duplicates. Cells were treated with melatonin, or its metabolites at the concentration of 5×10−5 M, or ethanol (vehicle, dilution 5:1,000) for 24 h prior UVB exposure and for 3 h for detection of p53 or phosphor-p53 Ser-15 or 16 h for detection of phosphor p53 Ser-46 [43]. Immunofluorescence and analysis are performed as described previously [56]. Cells were further fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature and washed three times with 0.1% Triton X-100 (BioRad, Hercules, CA) in PBS to permebealize membrane. Blocking was performed in 10% FBS in PBS for 1 h at RT after quenching of endogenous peroxidase with 1% H2O2 (BioRad, Hercules, CA) in PBS. Cells were incubated in primary antibodies diluted in blocker 1:100 overnight at 4°C as follows: anti-p53 (ab4060, Cell Signaling, Danvers, MA), phospho p53 Ser-15 (9284 Cell Signaling, Danvers, MA) or anti-p53 (phosphor S-46) (ab131348, Abcam, Cambridge, MA). The following day cells were washed and incubated with secondary antibody Alexa-Fluor 488 goat anti-rabbit IgG (Invitrogen Molecular Probes, Eugene, Oregon, USA) diluted in blocker 1:100 for 1 h at RT. After washing with PBS, nuclei were stained red with Vectashield mounting media with propidium iodide (Vector Laboratories, Burlingame, CA). Stained cells were imaged with a fluorescence microscope. Pictures recorded were analysed using ImageJ software (NIH free download).
Reactive oxygen species (ROS) assays
ROS levels were measured using a fluorescence method [57]. Keratinocytes were plated onto 96-well plate and treated with melatonin, or its metabolites at the concentration of 5×10−5 M, or ethanol (vehicle, dilution 5:1,000) for 24 h. After preincubation with CM-H2DCFDA dye, cells were irradiated with 200 mJ/cm2 of UVB. After 2 h of incubation cells were washed with PBS. The generation of ROS was determined by measuring the fluorescence at 480 nm excitation and 528 nm emission, on SpectraMax M2e instrument (Molecular Devices, Sunnyvale, CA). Data are presented as percentile of control (EtOH-treated cells).
Nitric oxide (NO−) assays
Nitrite levels (NO−) levels were measured using Griess reagent (1% sulfanilamide-0.1% N-1-naphthyl-ethylenediamine dihydrochloride in 2.5% phosphoric acid) (Sigma, St. Louis, MO) as previously described [57]. Keratinocytes were treated with melatonin Cells were treated with melatonin, or its metabolites, at the concentration of 5×10−5 M, or ethanol (vehicle, dilution 5:1,000) for 24 h. After incubation, cells in 1xPBS were irradiated with the UV of different intensity: 0, 25, 50, and 75 mJ/cm2. After incubation cells were harvested by scraping. Cells pellets were further centrifuged at 3000 g for 5 min, supernatant was collected and mixed with an equal amount of Griess reagent. The generation of NO was determined by measuring the absorbance at 540 nm in a spectrophotometer of purple azo compound formed from the reaction between nitrates formed in samples and Griess reagent. Potassium nitrate (Sigma, St. Louis, MO) dilutions, ranging from 0–35 μM, were used to create standard curve. Data are presented as concentration of nitite (μmol) per protein amount (mg), and further as percentile of control (EtOH-treated cells).
H2O2 assays
Hydrogen peroxide (H2O2) levels were measured using a luminescence method [58]. Keratinocytes were treated with melatonin, its derivatives at the concentration of 5×10−5 M, or ethanol (vehicle, dilution 5:1,000) for 24 h. After incubation, cells in 1xPBS were irradiated with the UVB of different intensity: 0, 25, 50, and 75 mJ/cm2. The generation of H2O2 was determined by measuring the luminescence of luminol (Sigma, St. Louis, MO) by H2O2 that is released by keratinocytes 1–2 min until 1 h after UV exposure. Aliquots of PCS from each dish were mixed with lumiol and horse radish peroxidase (Sigma, St. Louis, MO) in respiratory buffer and luminescence was measured in a Turner Luminometer (TD20/20) (Promega, Madison, WI). The specificity of the reaction was determined by treating separate UV-irradiated cells with 300 units/mL of catalase (Sigma, St. Louis, MO), which degraded H2O2 to H2O and O2. Data are presented as concentration of H2O2 (pmol) per 0.1 million cells and further as percentile of control (EtOH treated cells).
Reduced glutathione (GSH) assay
Reduced glutathione (GSH) levels were measured using a fluorometric method [57]. Keratinocytes were treated with melatonin, or its metabolites at the concentration of 5×10−5 M, or ethanol (vehicle, dilution 5:1,000) for 24 h. After incubation, cells were irradiated with the UVB of different intensity: 25, 50, or 75 mJ/cm2. After UV exposure cells were further incubated for 1 h. Cells were harvested with trypsinization, washed with 1x PBS-EDTA, pH8. After centrifugation, cell pellets were resuspended in ice cold 5% meta-phosphoric acid (MPA) (Sigma, St. Louis, MO), sonicated, and centrifuged at 12,000 rpm for 5 min. After collection of supernatant, aliquot were taken for protein determination. Supernatant was further mixed with 1x PBS-EDTA buffer and OPAME (o-phthaldialdehyde (Sigma, St. Louis, MO) in methanol (Fisher, Pittsburgh, PA) and borate buffer (potassium-tetraborate, Sigma, St Lois, MO). Mixtures are further aliquoted in 96-well plate, incubated for 15 min at RT and reduced GSH levels were determined by measuring the fluorescent signal at 350nm excitation and 420nm emission, on SpectraMax M2e instrument (Molecular Devices, Sunnyvale, CA). Data are presented as percentile of control (EtOH-treated cells).
Statistical analysis
Data are presented as means ± SEM and were analyzed with Student’s t-test (*) and appropriate post hoc test (for more than two groups), using Microsoft Excel and Prism 4.00 (GraphPad Software, San Diego, CA). Statistically significant differences are denoted in the figures and corresponding figure legends.
Results
UVB-induced damage was investigated in human epidermal keratinocytes cultured in vitro. HEKn cells were preincubated with melatonin and its derivatives at graded concentrations from 10−13 to 10−5 M prior to and after UVB irradiation at doses of 25, 50, or 75 mJ/cm2. It had been shown previously that treatment with melatonin prior UV exposure protects cells more from UVB [41, 59]. MTS was used as biological test of cellular viability. There was a significant reduction in cell viability and cell number after UVB exposure (data not shown), which was significantly reduced by treatment with melatonin or its derivatives. Figure 1 shows the strong dose-dependent protective effect of melatonin and its derivatives with maximal protection achieved at ligand concentration of 10−5M. NAS and melatonin exhibited lower potency than AFMK, 6-OHM or 5-MT. The data were presented after subtraction from non-irradiated cells, since melatonin and its derivatives have anti-proliferative effect on normal human keratinocytes; thus, fully recovered cells have 0 on y-axis
Figure 1.

Melatonin, NAS, 6-OHM, AFMK, and 5-MT increase viability of HEKn keratinocytes exposed to UVB. Data are calculated by subtracting viability from non-irradiated cells which means we considered the anti-proliferative effect of melatonin and its derivatives on this cell. Data were analyzed using student t-test, * p<0.05, ** p<0.01, and *** p<0.001.
UVB irradiated human keratinocytes produce ROS and induce DNA damage [1], which can be prevented by melatonin, as tested in HaCaT cells [40, 41]. To determine if melatonin or its metabolites also prevent cell damage and have a protective role, UVB-irradiated keratinocytes, both HEKn and HaCaT, were treated with or without melatonin or its metabolites, were tested for oxidative stress markers i.e., ROS including NO and H2O2 formation, GSH levels, CPD dimers formation, and DNA damage.
Figure 2 shows significant reduction (30–50%) in ROS levels produced by UVB irradiated keratinocytes compared with control in cells treated with melatonin or its derivatives. All metabolites had a similar potency in ROS reduction compared to melatonin in both keratinocytes, HEKn and HaCaT (Fig 2 A and B).
Figure 2.

Melatonin, NAS, 6-OHM, AFMK, and 5-MT decrease UVB-induced ROS generation. UVB-irradiated (200 mJ/m2) keratinocytes (HEKn (A) and HaCaT (B)) were treated with melatonin or its metabolites for 24 h prior UV irradiation and further for 2 h post UV at the concentration 5×10−5 M. Data were analyzed using student t-test, *** p<0.001.
Melatonin decreases the toxicity of NO [60]. To test the ability of melatonin metabolites to reduce the amount of NO, HaCaT keratinocytes were incubated with melatonin or its metabolites for 24 h before the UVB exposure. Cells in PBS were irradiated with UVB doses of 25, 50, or 75 mJ/cm2. Cells were harvested by scraping and collected after 1–2, 30, 45, and 60 min of irradiation to measure azo compounds formed in the reaction between NO and Griess reagent. Release of NO from irradiated cells is compared to non-irradiated cells. There was no NO release in non-irradiated cells in the presence or absence of melatonin or its metabolites (Fig 3). Keratinocytes produced NO in response to UVB irradiation [1] in a UVB-dose dependent manner (Fig 3, insert). The production of NO occurred shortly after irradiation, reaching a peak after 30 min, and returning to low levels after 1 h (data not shown). Treatment of cells with melatonin or its metabolite before UVB irradiation reduced NO production. The most prominent decrease in NO occurred 30 min after UVB exposure (Fig 3). The strongest inhibition of NO release was observed at 50 mJ/cm2 (Fig 3). Melatonin and AFMK strongly inhibited NO after 25 or 50 mJ/cm2 (p<0.001). Other metabolites showed strong inhibition of NO after 25 mJ/cm2, 5-MT and NAS (p<0.001) and 6-OHM (p<0.05), but were much weaker after 50 mJ/cm2, 5-MT and NAS (p<0.05). Only melatonin inhibited NO after 75 mJ/cm2 of UVB (p<0.05) (Fig 3).
Figure 3.
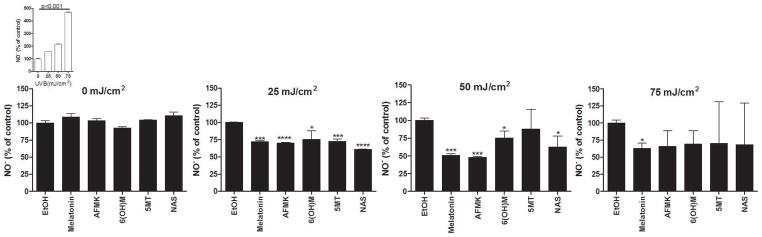
Melatonin, NAS, 6-OHM, AFMK, and 5-MT reduce levels of NO-produced by UVB-irradiated keratinocytes. HaCaT keratinocytes were treated with melatonin, AFMK, 6-OHT, 5-MT, NAS, or EtOH for 24 h and subsequently irradiated with 0, 25, 50, or 75 mJ/cm2 of UVB. NO-produced in keratinocytes was determined 30 min after UVB irradiation. The differences in NO-production under different UVB intensities are shown as an insert. Data are presented as percent and were analyzed using t-test, * p<0.05 and *** p<0.001.
Melatonin scavenges H2O2 directly [26]. To test the ability of melatonin metabolites to scavenge H2O2 produced in cells, HaCaT keratinocytes were incubated with melatonin and its metabolites for 24 h before the UVB exposure. Cells in PBS were irradiated with UVB doses of 25, 50, or 75 mJ/cm2. Supernatants were collected after 1–2, 30, 45, and 60 min of irradiation to measure the release of H2O2. Release of H2O2 from irradiated cells was compared to non-irradiated cells. While there was no H2O2 release in non-irradiated cells (Fig 4), keratinocytes produced H2O2 in response to UVB irradiation [1], and this production is UVB dose-dependent (Fig 4, insert). The production of H2O2 occurred 1–2 min after irradiation, reaching a peak after 30 min, and returned to low levels after 1 h (data not shown). Treatment of cells with melatonin or its metabolite reduced H2O2 release induced by UVB. This decrease was the most prominent 30 min after UVB exposure, when H2O2 release reached its peak (Fig 4). The strongest inhibition of H2O2 release was observed at the lowest UVB dose (25 mJ/cm2), while the lowest inhibition was at the highest UVB dose (75 mJ/cm2) (Fig 4). AFMK exhibited the strongest inhibitory effect on H2O2 release, followed by melatonin and 6-OHM, while the weakest effect was seen after treatment with 5-MT or NAS. The inhibition of H2O2 release was observed at 25 and 50, but not after 75 mJ/cm2 (Fig 4). AFMK showed the strongest inhibition of H2O2 by approximately 70% (p<0.001). Melatonin and 6-OHM exhibited stronger inhibition of NO after 25 mJ/cm2, (p<0.001), when compared to 50 mJ/cm2 inhibition, ~30–50% (p<0.05). Interestingly, 5-MT and NAS showed stronger activity after higher dose of UVB, ~20% (p<0.01 at 50 mJ and p<0.05 at 25 mJ/cm2). None of the compounds tested inhibited H2O2 production at a UVB intensity of 75 mJ/cm2 (Fig 4).
Figure 4.
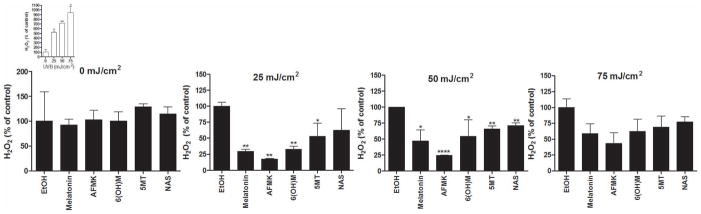
Melatonin, NAS, 6-OHM, AFMK, and 5-MT reduce levels of H2O2 produced by UVB-irradiated HaCaT keratinocytes. Keratinocytes were treated with melatonin, AFMK, 6-OHT, 5-MT, NAS, or EtOH for 24 h and further irradiated with 0, 25, 50, or 75 mJ/cm2 UVB. H2O2 produced in keratinocytes was determined 30 min after UVB irradiation. The differences in H2O2 production under different UVB intensities are shown as an insert. Data are presented as percent of control and were analyzed using t-test, * p<0.05, ** p<0.01, *** p<0.001, and **** p<0.0001.
Exposure to UVB causes immediate reduction of GSH [61]. Previously it was documented that melatonin can increase reduced GSH in human vascular endothelial cells [62] The effects of melatonin and its metabolites on the relative GSH content after treatment of keratinocytes for 24 h before and 1 h after UVB exposure are shown in figure 5. The level of reduced GSH decreased with the exposure to UVB light of different intensities (Fig 5, insert). At different intensities of UVB light tested (25, 50, or 75 mJ/cm2), melatonin and its metabolites caused a significant rise in reduced GSH after 1 h of UVB exposure. GSH content increased by 25% after 25 mJ/cm2, and 30–70% after 50, or 75 mJ/cm2 exposure (fig 5). All melatonin metabolites exhibited statistically significant increase in the levels of GSH (p<0.05-p<0.001).
Figure 5.
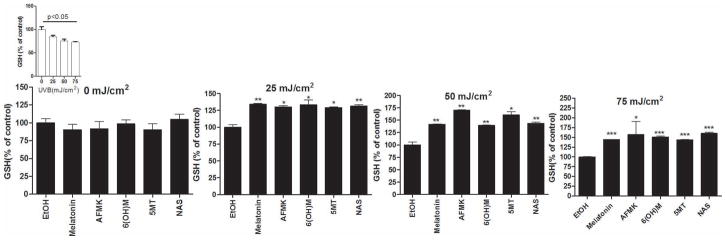
Melatonin, NAS, 6-OHM, AFMK, and 5-MT increase GSH in UVB-exposed keratinocytes. HaCaT keratinocytes were treated with melatonin, AFMK, 6-OHM, 5-MT, NAS, or EtOH for 24 h and further irradiated with 0, 25, 50, or 75 mJ/cm2 UVB. GSH produced in keratinocytes was determined 1 h after UVB irradiation. The differences in GSH production under different UVB intensities are shown as an insert. Data are presented as percentile of control and were analyzed using t-test, * p<0.05, ** p<0.01, *** p<0.001.
To measure DNA repair capacity, elimination of DNA damage in the form of UVB-induced 6-4 photoproducts was measured by immunoslot-blot analysis, as previously described [54]. Nucleotide excision repair (NER) is the principal mechanism for repair of UVR generated (6-4) photoproducts; the majority of 6-4PP was removed by 6 h after irradiation (Fig 6A). Densitometric analysis of the bands (Fig 6B) showed that melatonin or melatonin derivatives enhanced the rate of removal of 6-4PP dimers in HaCaT cells. This effect was similar at both concentrations tested (10−5 M vs 10−6 M) (data not shown). NAS had no effect on removal of 6-4PP dimers from keratinocytes irradiated with UVB (data not shown).
Figure 6.

Melatonin, NAS, 6-OHM, AFMK, and 5-MT treated keratinocytes exhibit enhanced UVR-induced DNA repair. HaCaT cells were treated with melatonin or its metabolites for 30 min before UVB exposure (50 mJ/cm2) and for 3 or 6 h after. DNA was measured by immunoslot blot analysis following UVB exposure. The relative DNA repair efficiency was compared to non-treated cells (EtOH).
Immunoslot-blot analysis failed to show any effect on CPDs after 3 or 6 h (not shown). Therefore, reduction in CPDs following UVB exposure in keratinocytes was assessed by immunohistochemistry followed by image analysis (Fig 7). UVB-irradiated HEKn cells were immediately treated with or without melatonin or its derivatives for 3 h, and CPD-positive cells were evaluated with CPD-specific antibody. CPD-positive cells were not detected in untreated cells (Fig 7A). CPD-staining intensity of UVB-irradiated cells was weaker in cells treated with melatonin or its derivatives (Fig 7B,C), as compared to untreated cells, indicating that melatonin or its derivatives might accelerate the repair of UVB-induced CPD in keratinocytes. The differences between melatonin, melatonin metabolite-treated cells, and non-treated cells after the exposure to UVB was statistically significant and more pronounced after UVB of 25 mJ/cm2 (p<0.01 or p<0.001) (Fig 7B), then after 50 mJ/cm2 (p<0.05) (Fig 7C). Melatonin, AFMK, and 6-OHM were the strongest inhibitors of CPD formation, but they showed no significant effects in cells exposed to UVB dose of 75 mJ/cm2 (data not shown).
Figure 7.
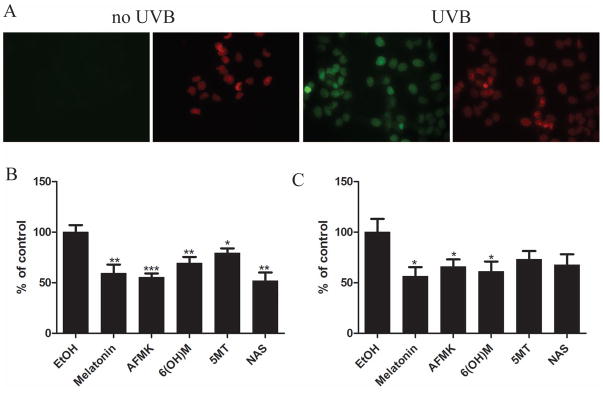
Melatonin, NAS, 6-OHM, AFMK, and 5-MT treated keratinocytes decrease CPD formation after UVB exposure. HEKn keratinocytes were treated with melatonin or its derivative for 24 h before UVB exposure. Cells were exposed to UVB intensities of 25 or 50 mJ/cm2 and immediately treated again with melatonin or its derivatives for 3 h. Cells were further fixed and stained with anti-CPD antibody (green) and propidium iodine (PI) (red) (A). Stained cells were imaged with a fluorescence microscope, fluorescence intensity was analysed using ImageJ software, and data are analysed using Graph Pad Prizm after 25 mJ/cm2 (B) and 50 mJ/cm2 (C). Data are presented as percentile of control and were analyzed using t-test, * p<0.05, ** p<0.01, *** p<0.001
To quantify the extent of the DNA damage, we employed the Comet assay which allows DNA of the damaged cells to migrate from the nucleus in agarose under an electric current, and further using fluorescent microscopy for visualisation of DNA migration. We observed an extensive DNA damage when HEKn cells were exposed to UVB (Fig 8, insert). There was a significant difference between UVB-induced DNA damage in control cells (non-treated) vs. cells treated with melatonin or NAS (p<0.01) or 6-OHM and 5-MT (p<0.05), but not AFMK, as measured by mean comet tail moment (Fig 8).
Figure 8.

Melatonin, NAS, 6-OHM, AFMK, and 5-MT enhance repair of DNA damage induced by UVB as indicated by comet assay. HEKn keratinocytes were treated with melatonin or its derivatives before and after UV-irradiation (200 mJ/cm2) for 3 h. Photographs show comets in UV-irradiated and non-irradiated keratinocytes. UV caused strand breaks in DNA which migrated in the electrophoretic field to form comet tails. Tail moment is used to measure DNA damage. Data are analysed using student t-test and labelled as * p<0.05, ** p<0.01.
Tumor suppressor p53 has very important role in DNA repair of UV-induced DNA damage [63]. Following UV irradiation or oxidative stress p53 accumulates in nucleus (Fig 9A). While phosphorylation of p53 can occur at different sites, the most important are the Ser-15 site, promoting accumulation and activation of p53 and DNA repair, and Ser-46, regulating apoptosis following DNA damage [64]. Previously it had been shown that melatonin induces a p53-dependent DNA damage response phosphorylation at Ser-15 [45, 46]. Therefore, we tested the expression of p53 and its phosphorylated forms (Ser-15 and Ser-46) in HEKn keratinocytes irradiated with UVB and treated with melatonin and its metabolites. UVB-irradiated cells showed positive nuclear signal (Fig 9A). P53 accumulation was proportional to the UVB intensity (Fig 9B).
Figure 9.

UVB irradiation increases p53 levels. HEKn keratinocytes were exposed to UVB intensities of 0, 25, 50, or 75 mJ/cm2. Cells were subsequently fixed and stained with anti-p53 antibody (green) and propidium iodine (PI) (red) (A). UVR-irradiated cells showed positive nuclear signal for p53 (A). Stained cells were imaged with a fluorescence microscope; fluorescence intensity was analysed using ImageJ software. Data are analysed using Graph Pad Prizm and expressed as fold change compared to control (0 mJ/cm2). P53 levels increased proportionally to UVB intensity (B). Data are presented as percentile of control and were analyzed using t-test, * p<0.05, ** p<0.01, *** p<0.001.
To determine whether a reduction of UVB-induced cell damage by treatment with melatonin and its metabolites allows keratinocytes with DNA damage to survive, we compared p53 accumulation in irradiated-treated vs. non-treated cells. The phosphorylation of keratinocytes of the Ser-15 site occurs earlier (3 h after UVB exposure), while phosphorylation of the Ser-46 site occurs later (12 h after UVB exposure) (data not shown), which is in accordance with previous report in MCF7 breast carcinoma cells [43]. While melatonin and its metabolites did not change significantly the expression of p53 and p53 phosphorylated form (Ser-46) (data not shown), the expression of p53 phosphorylated form of the Ser-15 increased after treatment with melatonin or its metabolites (Fig 10). The strongest response was after AFMK treatment (p<0.001), followed by melatonin, 6-OHM and NAS (p<0.01 or p<0.05), and 5-MT (p<0.05) after exposure to 25 mJ/cm2 UVB (Fig 10A) or 50 mJ/cm2 (figure 10B). At UVB 75 mJ/cm2, only 6-OHM and 5-MT showed effect on p53 levels (p<0.05) (Fig 10C).
Figure 10.

Melatonin, NAS, 6-OHM, AFMK, and 5-MT treated HEKn keratinocytes show increase of p53 phosphorylated at Serine 15 after UVB exposure. Keratinocytes were treated with melatonin or its derivative for 24 h before UVB exposure. Cells were exposed to UVB intensities of 25 (A), 50 (B), or 75 mJ/cm2 (C) and immediately treated again with melatonin or its derivatives for 12 h. Cells were further fixed and stained with anti-phosphorylated p53S15 antibody. Stained cells were imaged with a fluorescence microscope; fluorescence intensity was analysed using ImageJ software. Data are analysed using Graph Pad Prizm. Data are presented as percentile of control and were analyzed using t-test, * p<0.05, ** p<0.01, *** p<0.001
Discussion
UVB damages skin keratinocytes causing: oxidative DNA damage and the production of reactive oxygen species, including hydrogen peroxide and nitric oxide, and changing levels of glutathione [1]. UVB also plays a role in melatonin production in the skin [12], which further protects skin from oxidative damage induced by UV irradiation [12, 18, 30, 40, 65, 66]. Melatonin’s protective actions from UVR-induced oxidative damage is a results of its stimulation of antioxidative enzyme activities and scavenging free radicals [12, 18, 30, 40, 65, 66]. Stress promotes production of melatonin [11] and it as well as its metabolites scavenge free radicals and stimulate antioxidative enzymes, including glutathione peroxidase, glutathione reductase, and superoxide dismutase [5, 8, 26, 35, 67–73]. Under physiological conditions melatonin can stimulate the production of radical oxygen species (ROS) in a small amount, while when the cell is exposed to stress, melatonin protects cells by limiting the leakage of ROS out of cell [32] and maintaining normal membrane potential [42, 74], thus preventing cell injury. To determine if melatonin metabolites also prevent cell damage and exhibit protective role, UVB-irradiated keratinocytes treated with or without melatonin or its metabolites were tested for oxidative stress markers, including NO and H2O2 formation, GSH levels, CPD dimers formation, and DNA damage. Herein, we report protective effects of melatonin and its metabolites in to reducing oxidative cell damage in human keratinocytes elicited by UVB, as determined by attenuation of each of these oxidative stress parameters (ROS, NO, H2O2, rGSH, CPD). Thus, like melatonin, its derivatives also attenuate oxidative damage induced by UVB.
Nitric monoxide (NO−) toxicity is created by its coupling of respiratory produced O2, thus forming a non-radical, but highly toxic peroxynitrite anion (ONOO−), which may be further metabolized to –OH [6]. Melatonin also detoxifies NO [60], thereby lowering ONOO− formation. Since melatonin promotes the activities of antioxidative enzymes: including glutathione peroxidase (GPx) and glutathione reductase (GPr) [71, 75], damage by free radicals is limited when H2O2 is metabolised to non-radical species and glutathione is maintained in its reduced form. GSH is a potent antioxidant and plays an important role in preventing protein damage [61]. In general, melatonin plays an important role in maintaining glutathione homeostasis [76]. The current studies are thus in agreement with other reports showing that melatonin increases concentration of intracellular GSH [62]. Moreover, we show that melatonin metabolites are equally as potent as melatonin in increasing GSH levels (Figure 5), decreasing ROS levels (Fig 2), NO (Fig 3), and H2O2 levels (Fig 4) in UVB-exposed keratinocytes.
Irradiation of human keratinocytes with UVB resulted in the generation of H2O2 (Fig 4). H2O2, although a weakly toxic ROS, can be converted to highly toxic -OH [6]. The levels of released H2O2 were markedly reduced by melatonin or its metabolites, suggesting that they modulate the UV-induced oxidative stress in keratinocytes. Numerous studies have shown that melatonin prevents DNA damage by scavenging H2O2 directly or indirectly through production of AFMK [26]. The scavenging of H2O2 happens rapidly, within first few minutes and is melatonin mediated; while a slower phase of scavenging is mediated by AFMK and other metabolites [26]. Our data show that melatonin and its metabolites limit H2O2 generationin UVB-irradiated keratinocytes in a UVB dose-dependent manner, suggesting that the best scavenging effects occur after exposure to a lower dose of UVB (Fig 4). This data is in accordance with the findings that melatonin’s action is proportional to the amount of stress the cell is under [67, 77]. The strongest scavenging effects are seen 30 min after UVB exposure, when the release of H2O2 by keratinocytes was the highest (Fig 4).
In this study, the comet assay was used to examine the actions of melatonin and its metabolites on DNA repair (Fig 8). Comet assay involves single-cell electrophoresis that enables the DNA of the damaged cells to migrate from the nucleus, thus forming a tail moment. It is clear that melatonin or its derivatives reduced UVB-induced DNA-damage (Fig 8). Our data also shows that UV-induced DNA repair in keratinocytes was also enhanced by melatonin and its derivatives (Fig 6 and 7).
Previously, it was shown that melatonin prevents UVB-mediated keratinocyte damage [41]. Tumor suppressor protein p53 mediates the cell response to stress by activating cell-cycle arrest or apoptosis [63]. Different stressors, including gamma and UV irradiation, hypoxia, some chemicals, induce DNA damage and activation of p53 [78]. The extend of DNA damage and the rise in p53 are proportional [78]. UV irradiation causes transient DNA damage in the cell, such as DNA breaks and excision repair of the DNA dimers, thus inducing a moderate elevation in p53 level and cell-cycle arrest that allows repair to occur [63]. Phosphorylation plays an important role in p53 functioning, including binding to DNA [79, 80]. Phosphorylation of p53 occurs at different sites. P53 phosphorylation at the Ser-15 and Ser-20 sites promotes accumulation and activation of p53 and DNA repair, while p53 phosphorylation at the Ser-46 site closely regulates apoptosis after DNA damage [64]. Melatonin promotes phosphorylation of p53 at Ser-15 position, thus activating p53 protein and consecutively inhibiting cell growth, preventing accumulation of DNA damage and promoting antitumor activity [14, 43]. This action of melatonin is mediated by its MT1 and MT2 receptors [45]. Since phosphorylation of p53 at Ser-46 site is required for p53-induced apoptosis, we tested the ability of melatonin and its metabolites to promote p53-phosphorylation, thus inducing p53-dependent apoptosis. Upregulation of nuclear p53 and phosphorylated form of p53 after UVB was measured by immunohistochemistry and image analysis. As indicated by the preliminary data, there was no difference in p53 expression in sham-irradiated keratinocytes treated with vehicle compared with cells treated with melatonin or its derivatives, as measured by immunohistochemistry (data not shown). Exposure to UVR significantly augmented p53 expression in human keratinocytes compared with sham vehicle (control cells) (Fig 9). This effect was further enhanced in UVB-exposed keratinocytes treated with melatonin and its derivatives as shown in figure 10. Melatonin and its derivatives significantly induced the level of phosphorylated form of p53 at the Serine 15 site (Fig 10). Thus, our data indicate that the protective effects of melatonin and its metabolites against UVB induced damage are associated with p53-phosphorilation at Ser-15.
Although the role melatonin and AFMK play in protection against oxidative damage from different stressors including UVB, is well established [35, 81–84], we believe our findings are novel in the areas of skin biology and photobiology. Thus, using immortalized and most importantly normal epidermal primary keratinocytes we compared protective role of both melatonin and its metabolites including NAS, 6-OHM, AFMK, and 5-MTover wide range of parameters (oxidative stress, cell viability, DNA damage, p53 phosphorylation) that was differential and dependent on the molecule structure in the context of parameters tested. Taking into consideration that melatonin is both produced and metabolised in epidermal keratinocytes [13, 31], we propose that activity of the local melatoninergic system define protection of human epidermis against UVB exposure by attenuating DNA and metabolic damage via several mechanism, including reduction in H2O2 levels, reduction of NO levels, promotion of GSH, enhancement of DNA repair, and promotion of keratinocytes survival, in a context dependent fashion.
Acknowledgments
This paper is dedicated to Dr. Aaron B Lerner, a discoverer of melatonin, and who has trained the senior author (AS) of this manuscript. The work was supported by NIH grant 1R01AR056666-01A2 to AS.
Footnotes
Author Contributions
Z.J. designed and carried out most of the experiments, analysed the data, and wrote the manuscript. A.S. and R.J.R. designed experiments, analysed data and wrote the paper. Z.P. performed comet assay and data analysis. S.H. performed microscopy for some IF experiments and contributed data analysis. S.J. performed and analysed dot-blot experiments. T.K. performed and analysed the MTS experiments.
References
- 1.HECK DE, VETRANO AM, MARIANO TM, et al. UVB light stimulates production of reactive oxygen species: unexpected role for catalase. J Biol Chem. 2003;278:22432–224326. doi: 10.1074/jbc.C300048200. [DOI] [PubMed] [Google Scholar]
- 2.WONDRAK GT, ROBERTS MJ, CERVANTES-LAUREAN D, et al. Proteins of the extracellular matrix are sensitizers of photo-oxidative stress in human skin cells. J Invest Dermatol. 2003;121:578–586. doi: 10.1046/j.1523-1747.2003.12414.x. [DOI] [PubMed] [Google Scholar]
- 3.PFEIFER GP, BESARATINIA A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem Photobiol Sci. 2012;11:90–97. doi: 10.1039/c1pp05144j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MOURET S, BAUDOUIN C, CHARVERON M, et al. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci U S A. 2006;103:13765–13770. doi: 10.1073/pnas.0604213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.TAN DXCL, POEGGELER B, MANCHESTER LC, REITER RJ. Melatonin: a potent endogenous hydroxyl radical scavenger. Endocr J. 1993;1:57–60. [Google Scholar]
- 6.REITER RJ, TAN DX, MAYO JC, et al. Melatonin as an antioxidant: biochemical mechanisms and pathophysiological implications in humans. Acta Biochim Pol. 2003;50:1129–1146. [PubMed] [Google Scholar]
- 7.REITER RJ, TAN DX, FUENTES-BROTO L. Melatonin: a multitasking molecule. Prog Brain Res. 2010;181:127–151. doi: 10.1016/S0079-6123(08)81008-4. [DOI] [PubMed] [Google Scholar]
- 8.GALANO A, TAN DX, REITER RJ. Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res. 2011;51:1–16. doi: 10.1111/j.1600-079X.2011.00916.x. [DOI] [PubMed] [Google Scholar]
- 9.MAURIZ JL, COLLADO PS, VENEROSO C, et al. A review of the molecular aspects of melatonin’s anti-inflammatory actions: recent insights and new perspectives. J Pineal Res. 2013;54:1–14. doi: 10.1111/j.1600-079X.2012.01014.x. [DOI] [PubMed] [Google Scholar]
- 10.TAN DX, MANCHESTER LC, HARDELAND R, et al. Melatonin: a hormone, a tissue factor, an autocoid, a paracoid, and an antioxidant vitamin. J Pineal Res. 2003;34:75–78. doi: 10.1034/j.1600-079x.2003.02111.x. [DOI] [PubMed] [Google Scholar]
- 11.TAN DX, MANCHESTER LC, TERRON MP, et al. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. 2007;42:28–42. doi: 10.1111/j.1600-079X.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 12.SLOMINSKI A, FISCHER TW, ZMIJEWSKI MA, et al. On the role of melatonin in skin physiology and pathology. Endocrine. 2005;27:137–148. doi: 10.1385/ENDO:27:2:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.SLOMINSKI A, TOBIN DJ, ZMIJEWSKI MA, et al. Melatonin in the skin: synthesis, metabolism and functions. Trends Endocrinol Metab. 2008;19:17–24. doi: 10.1016/j.tem.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 14.SLOMINSKI RM, REITER RJ, SCHLABRITZ-LOUTSEVITCH N, et al. Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol. 2012;351:152–166. doi: 10.1016/j.mce.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.KILIC U, YILMAZ B, UGUR M, et al. Evidence that membrane-bound G protein-coupled melatonin receptors MT1 and MT2 are not involved in the neuroprotective effects of melatonin in focal cerebral ischemia. J Pineal Res. 2012;52:228–235. doi: 10.1111/j.1600-079X.2011.00932.x. [DOI] [PubMed] [Google Scholar]
- 16.REITER RJ. Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr Rev. 1991;12:151–180. doi: 10.1210/edrv-12-2-151. [DOI] [PubMed] [Google Scholar]
- 17.STEHLE JH, SAADE A, RAWASHDEH O, et al. A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J Pineal Res. 2011;51:17–43. doi: 10.1111/j.1600-079X.2011.00856.x. [DOI] [PubMed] [Google Scholar]
- 18.SLOMINSKI A, WORTSMAN J, TOBIN DJ. The cutaneous serotoninergic/melatoninergic system: securing a place under the sun. FASEB J. 2005;19:176–194. doi: 10.1096/fj.04-2079rev. [DOI] [PubMed] [Google Scholar]
- 19.SLOMINSKI A, BAKER J, ROSANO TG, et al. Metabolism of serotonin to N-acetylserotonin, melatonin, and 5-methoxytryptamine in hamster skin culture. J Biol Chem. 1996;271:12281–12286. doi: 10.1074/jbc.271.21.12281. [DOI] [PubMed] [Google Scholar]
- 20.SLOMINSKI A, PISARCHIK A, SEMAK I, et al. Serotoninergic and melatoninergic systems are fully expressed in human skin. FASEB J. 2002;16:896–898. doi: 10.1096/fj.01-0952fje. [DOI] [PubMed] [Google Scholar]
- 21.SLOMINSKI A, PISARCHIK A, SEMAK I, et al. Serotoninergic system in hamster skin. J Invest Dermatol. 2002;119:934–942. doi: 10.1046/j.1523-1747.2002.00156.x. [DOI] [PubMed] [Google Scholar]
- 22.SLOMINSKI A, PISARCHIK A, SEMAK I, et al. Characterization of the serotoninergic system in the C57BL/6 mouse skin. Eur J Biochem. 2003;270:3335–3344. doi: 10.1046/j.1432-1033.2003.03708.x. [DOI] [PubMed] [Google Scholar]
- 23.SEMAK I, KORIK E, NAUMOVA M, et al. Serotonin metabolism in rat skin: characterization by liquid chromatography-mass spectrometry. Arch Biochem Biophys. 2004;421:61–66. doi: 10.1016/j.abb.2003.08.036. [DOI] [PubMed] [Google Scholar]
- 24.TAN DX, HARDELAND R, MANCHESTER LC, et al. Cyclic-3-hydroxymelatonin (C3HOM), a potent antioxidant, scavenges free radicals and suppresses oxidative reactions. Curr Med Chem. 2013;21:1557–1565. doi: 10.2174/0929867321666131129113146. [DOI] [PubMed] [Google Scholar]
- 25.SEMAK I, NAUMOVA M, KORIK E, et al. A novel metabolic pathway of melatonin: oxidation by cytochrome C. Biochemistry. 2005;44:9300–9307. doi: 10.1021/bi050202d. [DOI] [PubMed] [Google Scholar]
- 26.TAN DX, MANCHESTER LC, REITER RJ, et al. Melatonin directly scavenges hydrogen peroxide: a potentially new metabolic pathway of melatonin biotransformation. Free Radic Biol Med. 2000;29:1177–1185. doi: 10.1016/s0891-5849(00)00435-4. [DOI] [PubMed] [Google Scholar]
- 27.HARDELAND R, TAN DX, REITER RJ. Kynuramines, metabolites of melatonin and other indoles: the resurrection of an almost forgotten class of biogenic amines. J Pineal Res. 2009;47:109–126. doi: 10.1111/j.1600-079X.2009.00701.x. [DOI] [PubMed] [Google Scholar]
- 28.FISCHER TW, SWEATMAN TW, SEMAK I, et al. Constitutive and UV-induced metabolism of melatonin in keratinocytes and cell-free systems. FASEB J. 2006;20:1564–1566. doi: 10.1096/fj.05-5227fje. [DOI] [PubMed] [Google Scholar]
- 29.YOUNG IM, LEONE RM, FRANCIS P, et al. Melatonin is metabolized to N-acetyl serotonin and 6-hydroxymelatonin in man. J Clin Endocrinol Metab. 1985;60:114–119. doi: 10.1210/jcem-60-1-114. [DOI] [PubMed] [Google Scholar]
- 30.REITER RJ, REITER MN, HATTORI A, et al. The pineal melatonin rhythm and its regulation by light in a subterranean rodent, the valley pocket gopher (Thomomys bottae) J Pineal Res. 1994;16:145–153. doi: 10.1111/j.1600-079x.1994.tb00094.x. [DOI] [PubMed] [Google Scholar]
- 31.KIM TK, KLESZCZYNSKI K, JANJETOVIC Z, et al. Metabolism of melatonin and biological activity of intermediates of melatoninergic pathway in human skin cells. FASEB J. 2013;27:2742–2755. doi: 10.1096/fj.12-224691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LUCHETTI F, CANONICO B, BETTI M, et al. Melatonin signaling and cell protection function. FASEB J. 2010;24:3603–3624. doi: 10.1096/fj.10-154450. [DOI] [PubMed] [Google Scholar]
- 33.LANOIX D, LACASSE AA, REITER RJ, et al. Melatonin: the smart killer: the human trophoblast as a model. Mol Cell Endocrinol. 2012;348:1–11. doi: 10.1016/j.mce.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 34.HARDELAND R, CARDINALI DP, SRINIVASAN V, et al. Melatonin--a pleiotropic, orchestrating regulator molecule. Prog Neurobiol. 2011;93:350–384. doi: 10.1016/j.pneurobio.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 35.GALANO A, TAN DX, REITER RJ. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J Pineal Res. 2013;54:245–257. doi: 10.1111/jpi.12010. [DOI] [PubMed] [Google Scholar]
- 36.FISCHER TW, SCHOLZ G, KNOLL B, et al. Melatonin suppresses reactive oxygen species induced by UV irradiation in leukocytes. J Pineal Res. 2004;37:107–112. doi: 10.1111/j.1600-079X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 37.PERIANAYAGAM MC, OXENKRUG GF, JABER BL. Immune-modulating effects of melatonin, N-acetylserotonin, and N-acetyldopamine. Ann N Y Acad Sci. 2005;1053:386–393. doi: 10.1111/j.1749-6632.2005.tb00046.x. [DOI] [PubMed] [Google Scholar]
- 38.SLOMINSKI A, PISARCHIK A, ZBYTEK B, et al. Functional activity of serotoninergic and melatoninergic systems expressed in the skin. J Cell Physiol. 2003;196:144–153. doi: 10.1002/jcp.10287. [DOI] [PubMed] [Google Scholar]
- 39.FISCHER TW, SLOMINSKI A, ZMIJEWSKI MA, et al. Melatonin as a major skin protectant: from free radical scavenging to DNA damage repair. Exp Dermatol. 2008;17:713–730. doi: 10.1111/j.1600-0625.2008.00767.x. [DOI] [PubMed] [Google Scholar]
- 40.FISCHER TW, KLESZCZYNSKI K, HARDKOP LH, et al. Melatonin enhances antioxidative enzyme gene expression (CAT, GPx, SOD), prevents their UVR-induced depletion, and protects against the formation of DNA damage (8-hydroxy-2′-deoxyguanosine) in ex vivo human skin. J Pineal Res. 2013;54:303–312. doi: 10.1111/jpi.12018. [DOI] [PubMed] [Google Scholar]
- 41.FISCHER TW, ZBYTEK B, SAYRE RM, et al. Melatonin increases survival of HaCaT keratinocytes by suppressing UV-induced apoptosis. J Pineal Res. 2006;40:18–26. doi: 10.1111/j.1600-079X.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 42.FISCHER TW, ZMIJEWSKI MA, WORTSMAN J, et al. Melatonin maintains mitochondrial membrane potential and attenuates activation of initiator (casp-9) and effector caspases (casp-3/casp-7) and PARP in UVR-exposed HaCaT keratinocytes. J Pineal Res. 2008;44:397–407. doi: 10.1111/j.1600-079X.2007.00542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.SANTORO R, MARANI M, BLANDINO G, et al. Melatonin triggers p53Ser phosphorylation and prevents DNA damage accumulation. Oncogene. 2012;31:2931–2942. doi: 10.1038/onc.2011.469. [DOI] [PubMed] [Google Scholar]
- 44.CAMPBELL C, QUINN AG, ANGUS B, et al. Wavelength specific patterns of p53 induction in human skin following exposure to UV radiation. Cancer Res. 1993;53:2697–2699. [PubMed] [Google Scholar]
- 45.SANTORO R, MORI F, MARANI M, et al. Blockage of melatonin receptors impairs p53-mediated prevention of DNA damage accumulation. Carcinogenesis. 2013;34:1051–1061. doi: 10.1093/carcin/bgt025. [DOI] [PubMed] [Google Scholar]
- 46.MEDIAVILLA MD, COS S, SANCHEZ-BARCELO EJ. Melatonin increases p53 and p21WAF1 expression in MCF-7 human breast cancer cells in vitro. Life Sci. 1999;65:415–420. doi: 10.1016/s0024-3205(99)00262-3. [DOI] [PubMed] [Google Scholar]
- 47.NICKEL A, WOHLRAB W. Melatonin protects human keratinocytes from UVB irradiation by light absorption. Arch Dermatol Res. 2000;292:366–368. doi: 10.1007/s004030000141. [DOI] [PubMed] [Google Scholar]
- 48.ZBYTEK B, JANJETOVIC Z, TUCKEY RC, et al. 20-Hydroxyvitamin D3, a product of vitamin D3 hydroxylation by cytochrome P450scc, stimulates keratinocyte differentiation. J Invest Dermatol. 2008;128:2271–2280. doi: 10.1038/jid.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.JANJETOVIC Z, TUCKEY RC, NGUYEN MN, et al. 20,23-dihydroxyvitamin D3, novel P450scc product, stimulates differentiation and inhibits proliferation and NF-kappaB activity in human keratinocytes. J Cell Physiol. 2010;223:36–48. doi: 10.1002/jcp.21992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.JANJETOVIC Z, ZMIJEWSKI MA, TUCKEY RC, et al. 20-Hydroxycholecalciferol, product of vitamin D3 hydroxylation by P450scc, decreases NF-kappaB activity by increasing IkappaB alpha levels in human keratinocytes. PLoS One. 2009;4:e5988. doi: 10.1371/journal.pone.0005988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.PISARCHIK A, SLOMINSKI AT. Alternative splicing of CRH-R1 receptors in human and mouse skin: identification of new variants and their differential expression. FASEB J. 2001;15:2754–2756. doi: 10.1096/fj.01-0487fje. [DOI] [PubMed] [Google Scholar]
- 52.HOSNI-AHMED A, BARNES JD, WAN J, et al. Thiopurine methyltransferase predicts the extent of cytotoxicty and DNA damage in astroglial cells after thioguanine exposure. PLoS One. 2011;6:e29163. doi: 10.1371/journal.pone.0029163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DUAN J, DUAN J, ZHANG Z, et al. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int J Biochem Cell Biol. 2005;37:1407–1420. doi: 10.1016/j.biocel.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 54.JARRETT SG, NOVAK M, HARRIS N, et al. NM23 deficiency promotes metastasis in a UV radiation-induced mouse model of human melanoma. Clin Exp Metastasis. 2013;30:25–36. doi: 10.1007/s10585-012-9495-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.FORD JM, HANAWALT PC. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J Biol Chem. 1997;272:28073–28080. doi: 10.1074/jbc.272.44.28073. [DOI] [PubMed] [Google Scholar]
- 56.GUPTA R, DIXON KM, DEO SS, et al. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J Invest Dermatol. 2007;127:707–715. doi: 10.1038/sj.jid.5700597. [DOI] [PubMed] [Google Scholar]
- 57.TOBON-VELASCO JC, VAZQUEZ-VICTORIO G, MACIAS-SILVA M, et al. S-allyl cysteine protects against 6-hydroxydopamine-induced neurotoxicity in the rat striatum: involvement of Nrf2 transcription factor activation and modulation of signaling kinase cascades. Free Radic Biol Med. 2012;53:1024–1040. doi: 10.1016/j.freeradbiomed.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 58.KADEKARO AL, KAVANAGH R, KANTO H, et al. alpha-Melanocortin and endothelin-1 activate antiapoptotic pathways and reduce DNA damage in human melanocytes. Cancer Res. 2005;65:4292–4299. doi: 10.1158/0008-5472.CAN-04-4535. [DOI] [PubMed] [Google Scholar]
- 59.IZYKOWSKA I, CEGIELSKI M, GEBAROWSKA E, et al. Effect of melatonin on human keratinocytes and fibroblasts subjected to UVA and UVB radiation In vitro. In Vivo. 2009;23:739–745. [PubMed] [Google Scholar]
- 60.BLANCHARD B, POMPON D, DUCROCQ C. Nitrosation of melatonin by nitric oxide and peroxynitrite. J Pineal Res. 2000;29:184–192. doi: 10.1034/j.1600-079x.2000.290308.x. [DOI] [PubMed] [Google Scholar]
- 61.LARSSON P, ANDERSSON E, JOHANSSON U, et al. Ultraviolet A and B affect human melanocytes and keratinocytes differently. A study of oxidative alterations and apoptosis. Exp Dermatol. 2005;14:117–123. doi: 10.1111/j.0906-6705.2005.00238.x. [DOI] [PubMed] [Google Scholar]
- 62.URATA Y, HONMA S, GOTO S, et al. Melatonin induces gamma-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic Biol Med. 1999;27:838–847. doi: 10.1016/s0891-5849(99)00131-8. [DOI] [PubMed] [Google Scholar]
- 63.CHEN X, CHEN J, GAN S, et al. DNA damage strength modulates a bimodal switch of p53 dynamics for cell-fate control. BMC Biol. 2013;11:73. doi: 10.1186/1741-7007-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.ODA K, ARAKAWA H, TANAKA T, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 65.KLESZCZYNSKI K, HARDKOP LH, FISCHER TW. Differential effects of melatonin as a broad range UV-damage preventive dermato-endocrine regulator. Dermatoendocrinol. 2011;3:27–31. doi: 10.4161/derm.3.1.14842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.REITER RJ, TAN DX, POEGGELER B, et al. Melatonin as a free radical scavenger: implications for aging and age-related diseases. Ann N Y Acad Sci. 1994;719:1–12. doi: 10.1111/j.1749-6632.1994.tb56817.x. [DOI] [PubMed] [Google Scholar]
- 67.MATUSZAK Z, BILSKA MA, RESZKA KJ, et al. Interaction of singlet molecular oxygen with melatonin and related indoles. Photochem Photobiol. 2003;78:449–455. doi: 10.1562/0031-8655(2003)078<0449:iosmow>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 68.XIMENES VF, SILVA SO, RODRIGUES MR, et al. Superoxide-dependent oxidation of melatonin by myeloperoxidase. J Biol Chem. 2005;280:38160–38169. doi: 10.1074/jbc.M506384200. [DOI] [PubMed] [Google Scholar]
- 69.RODRIGUEZ C, MAYO JC, SAINZ RM, et al. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res. 2004;36:1–9. doi: 10.1046/j.1600-079x.2003.00092.x. [DOI] [PubMed] [Google Scholar]
- 70.REITER RJ, TAN DX. Melatonin: a novel protective agent against oxidative injury of the ischemic/reperfused heart. Cardiovasc Res. 2003;58:10–19. doi: 10.1016/s0008-6363(02)00827-1. [DOI] [PubMed] [Google Scholar]
- 71.BARLOW-WALDEN LR, REITER RJ, ABE M, et al. Melatonin stimulates brain glutathione peroxidase activity. Neurochem Int. 1995;26:497–502. doi: 10.1016/0197-0186(94)00154-m. [DOI] [PubMed] [Google Scholar]
- 72.PABLOS MI, REITER RJ, ORTIZ GG, et al. Rhythms of glutathione peroxidase and glutathione reductase in brain of chick and their inhibition by light. Neurochem Int. 1998;32:69–75. doi: 10.1016/s0197-0186(97)00043-0. [DOI] [PubMed] [Google Scholar]
- 73.REITER RJ, PAREDES SD, MANCHESTER LC, et al. Reducing oxidative/nitrosative stress: a newly-discovered genre for melatonin. Crit Rev Biochem Mol Biol. 2009;44:175–200. doi: 10.1080/10409230903044914. [DOI] [PubMed] [Google Scholar]
- 74.ARESE M, MAGNIFICO MC, MASTRONICOLA D, et al. Nanomolar melatonin enhances nNOS expression and controls HaCaT-cells bioenergetics. IUBMB Life. 2012;64:251–258. doi: 10.1002/iub.603. [DOI] [PubMed] [Google Scholar]
- 75.PABLOS MI, REITER RJ, CHUANG JI, et al. Acutely administered melatonin reduces oxidative damage in lung and brain induced by hyperbaric oxygen. J Appl Physiol (1985) 1997;83:354–358. doi: 10.1152/jappl.1997.83.2.354. [DOI] [PubMed] [Google Scholar]
- 76.MARTIN M, MACIAS M, ESCAMES G, et al. Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J. 2000;14:1677–1679. doi: 10.1096/fj.99-0865fje. [DOI] [PubMed] [Google Scholar]
- 77.POEGGELER B, REITER RJ, TAN DX, et al. Melatonin, hydroxyl radical-mediated oxidative damage, and aging: a hypothesis. J Pineal Res. 1993;14:151–168. doi: 10.1111/j.1600-079x.1993.tb00498.x. [DOI] [PubMed] [Google Scholar]
- 78.LEVINE AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 79.MILCZAREK GJ, MARTINEZ J, BOWDEN GT. p53 Phosphorylation: biochemical and functional consequences. Life Sci. 1997;60:1–11. doi: 10.1016/s0024-3205(96)00479-1. [DOI] [PubMed] [Google Scholar]
- 80.STEEGENGA WT, VAN DER EB AJ, JOCHEMSEN AG. How phosphorylation regulates the activity of p53. J Mol Biol. 1996;263:103–113. doi: 10.1006/jmbi.1996.0560. [DOI] [PubMed] [Google Scholar]
- 81.MATUSZAK Z, RESZKA K, CHIGNELL CF. Reaction of melatonin and related indoles with hydroxyl radicals: EPR and spin trapping investigations. Free Radic Biol Med. 1997;23:367–372. doi: 10.1016/s0891-5849(96)00614-4. [DOI] [PubMed] [Google Scholar]
- 82.LEON J, ESCAMES G, RODRIGUEZ MI, et al. Inhibition of neuronal nitric oxide synthase activity by N1-acetyl-5-methoxykynuramine, a brain metabolite of melatonin. J Neurochem. 2006;98:2023–2033. doi: 10.1111/j.1471-4159.2006.04029.x. [DOI] [PubMed] [Google Scholar]
- 83.GARCIA JJ, LOPEZ-PINGARRON L, ALMEIDA-SOUZA P, et al. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: a review. J Pineal Res. 2014;56:225–237. doi: 10.1111/jpi.12128. [DOI] [PubMed] [Google Scholar]
- 84.WU CL, QIANG L, HAN W, et al. Role of AMPK in UVB-induced DNA damage repair and growth control. Oncogene. 2013;32:2682–2689. doi: 10.1038/onc.2012.279. [DOI] [PMC free article] [PubMed] [Google Scholar]