

Abstract
Aims
Bioactives of Artemisia dracunculus L. (termed PMI 5011) have been shown to improve insulin action by increasing insulin signalling in skeletal muscle. However, it has not known if PMI 5011’s effects are retained during an inflammatory condition. We examined the attenuation of insulin action and whether PMI 5011 enhances insulin signalling in the inflammatory environment with elevated cytokines.
Methods
Muscle cell cultures derived from lean, overweight and diabetic obese subjects were used. Expression of pro-inflammatory genes and inflammatory response of human myotubes were evaluated by RT-PCR. Insulin signalling and activation of inflammatory pathways in human myotubes were evaluated by Multiplex protein assays.
Results
We found increased gene expression of MCP1 and TNFα, and basal activity of the NFkB pathway in myotubes derived from diabetic-obese subjects as compared to myotubes derived from normal-lean subjects. In line with this, basal Akt phosphorylation (Ser473) was significantly higher, while insulin-stimulated phosphorylation of Akt (Ser473) was lower in myotubes from normal-overweight and diabetic-obese subjects compared to normal-lean subjects. PMI 5011 treatment reduced basal phosphorylation of Akt and enhanced insulin-stimulated phosphorylation of Akt in the presence of cytokines in human myotubes. PMI 5011 treatment led to an inhibition of cytokine-induced activation of inflammatory signalling pathways such as Erk1/2 and IkBα-NFkB and moreover, NFkB target gene expression, possibly by preventing further propagation of the inflammatory response within muscle tissue.
Conclusions
PMI 5011 improved insulin sensitivity in diabetic-obese myotubes to the level of normal-lean myotubes despite the presence of pro-inflammatory cytokines.
Keywords: Skeletal muscle inflammation, insulin resistance, insulin signaling, cytokine signaling, PMI 5011, Artemisia dracunculus L, Akt, IRS1, Erk1/2, NFkB, IkBα, TNF, IL6, cytokines, chemokines
Introduction
Insulin resistance is a key pathophysiological feature of obesity and type 2 diabetes; therefore enhancing insulin sensitivity is a primary strategy for improving metabolic control in subjects with type 2 diabetes. Given that skeletal muscle is responsible for 70-90% of insulin stimulated whole body glucose uptake [1, 2], skeletal muscle insulin sensitivity plays a pivotal role in the pathogenesis of metabolic disorders such as type 2 diabetes. On the other hand, skeletal muscle inflammation contributes largely to the insulin resistant state resulting in impaired insulin signalling in skeletal muscle of obese and diabetic subjects [3-5].
Emerging data shows that in the obese state, elevated plasma free fatty acids, circulating adipose-derived cytokines, and inflammatory macrophage infiltration of muscle tissue can induce skeletal muscle insulin resistance via activation of inflammatory signalling pathways such as JNK, p38 MAPK and IKKβ, NFkB, which interfere with insulin signalling [6-8]. It is well known that pro-inflammatory cytokines such as TNFα, IL6 and IL1β are key mediators linking local tissue inflammation to insulin resistance [9, 10]. These studies suggest that therapeutic interventions aimed at reducing skeletal muscle inflammation via inhibition of inflammatory signalling in muscle could be a useful strategy for the treatment of insulin resistance. In this regard, botanicals have traditionally been used as an alternative medicine with simultaneous effects on inflammation and insulin resistance due to their complex composition [11].
Artemisia drancunculus L.or Russian tarragon, is a culinary herb that has been widely used in traditional medicine for centuries because of its potential antimicrobial and antioxidant activities. To date, the anti-diabetic effect of A. drancunculus has been extensively reported [12-15]. PMI 5011, an ethanolic extract of A. drancunculus has been shown to decrease glucose and insulin levels in animal models and to improve insulin signalling in primary human skeletal muscle culture and murine models [16-19]. Previous studies have also shown that PMI 5011 significantly down-regulates Il6ra and Ccl9 in the skeletal muscle of KK-Ay mice [20]. However, the cellular mechanism and relationship between the potential anti-inflammatory activity of PMI 5011 and preservation of insulin signalling within skeletal muscle still has not been investigated.
We hypothesized that the mechanism through which PMI 5011mediates improvements in insulin signalling involves attenuation of cytokine-induced activation of inflammatory signalling pathways in skeletal muscle cells. We utilized human skeletal muscle myotubes cultured from lean, overweight and diabetic-obese subjects. Herein we report that the PMI 5011 modulates Erk1/2 and IkBα/NFkB inflammatory pathways and cytokine-mediated inflammatory response of skeletal muscle, thus ameliorating insulin signalling in insulin resistant state.
Methods
Source and characterization of PMI 5011
PMI 5011 was produced from plants grown hydroponically under uniform and controlled conditions. The growing, quality control, phytochemical content, biochemical and bioactives characterization, and preparation of PMI 5011 have been reported [16, 20-22].
Cell culture
Cryopreserved human skeletal muscle myoblasts (HSMM) from three non-diabetic or Normal-Lean (BMI, 20.4±1.6), three Normal-Overweight (BMI, 25.7±0.4) and three Diabetic-Obese (BMI, 32.3±5.9) subjects at passage 2 were purchased from Lonza (Walkersville, MD, USA) and maintained in human skeletal growth media (SkGM-2 Bullet Kit, Lonza, Walkersville, MD). Myoblasts at passage 4 were differentiated into fused multinucleated myotubes by switching to fusion medium DMEM-F12 (Lonza) supplemented with 2% horse serum. Myotubes were pretreated with vehicle (DMSO) or PMI 5011 at a dose of 5 μg/ml overnight (16 h) before insulin and cytokine stimulation for different time points. LPS (0111:B4) (Sigma) and recombinant human TNFα and IL6 (R@D Systems) were used for treatments. LPS and cytokines were dissolved in PBS (phosphate buffered saline), therefore PBS was used as a control for LPS and cytokine treatments.
Gene expression analysis
Total RNA from HSMM were purified using RNeasy Micro Kit (Qiagen). DNAse digestion was performed on the RNeasy spin columns to remove potential genomic DNA contamination in total RNA. iScript cDNA synthesis kit (Bio-Rad) was used to synthesize cDNA, then 4 ng cDNA was used per reaction for qRT-PCR with Sybr green system. Human cyclophilin B was used as a housekeeping gene control for normalization of gene expression. Relative quantification (delta CT method) was used for data analysis. Primers used in qRT-PCR are shown in Supplementary Table 1.
Multiplex analysis
Treated myotubes were harvested in Cell Signalling Lysis Buffer (Millipore). The following Map mates were used: p-Akt (Ser473), p-IRS1 (Tyr) and total Akt, IRS1 (Millipore) for insulin signalling analysis; p-IkBα (Ser32), p-JNK (Thr183/Tyr705), p-p38 MAPK (Thr180/Tyr182), p-Erk1/2 (Thr185/Tyr187), p-STAT3 (Tyr705) (all Millipore), p-NFkB p65 (Ser536) (Bio-Rad) and total IkBα, JNK, p38 MAPK, Erk1/2, STAT3 (Millipore) for cytokine signalling; human GAPDH (Millipore) in all multiplex assays for normalization of protein data analysis. Map mates were prepared and combined according to the manufacturer instructions. Phospho- and total Map mates were used in separate assays, but human GAPDH was used in each assay. First, mean fluorescence intensity (MFI) of phospho- and total map mates were normalized to MFI of GAPDH separately, than secondly ratios of phospho-protein to total protein were calculated to compare activation of the signalling pathways.
Statistical Analyses
A two–way ANOVA (GraphPad Prism 5) was used to determine significance in differences between subjects or treatments. All data are presented as the mean ± SE. p < 0.05 was considered significant.
Results
Human skeletal muscle culture retains in vivo characteristics of the insulin resistant phenotype
To understand the mechanism of cytokine action on skeletal muscle cells and the inflammatory response of skeletal muscle cells to cytokine stimuli and the link to insulin signalling, we utilized human skeletal muscle myotubes (HSMM, Lonza). Previous reports have shown that myotubes derived from type 2 diabetic subjects display diminished insulin signalling compared to myotubes from insulin sensitive individuals [23, 24]. Elevated basal Akt phosphorylation [25, 26] and reduced Akt phosphorylation with insulin stimulation were observed in obese [27] or insulin resistant [28] mice and human myotubes [29]. In agreement with these reports, we found that basal Ser473-phosphorylation of Akt in myotubes was significantly elevated in overweight, diabetic-obese subjects compared with lean subjects (Fig. 1A). Notably, insulin-stimulated phosphorylation of Akt was significantly lower in myotubes from diabetic-obese subjects compared to non-diabetic lean subjects, whereas in overweight subjects, it was not significant (Fig. 1C).
Fig. 1. Basal insulin sensitivity and insulin-stimulated phosphorylation of Akt in human myotubes.
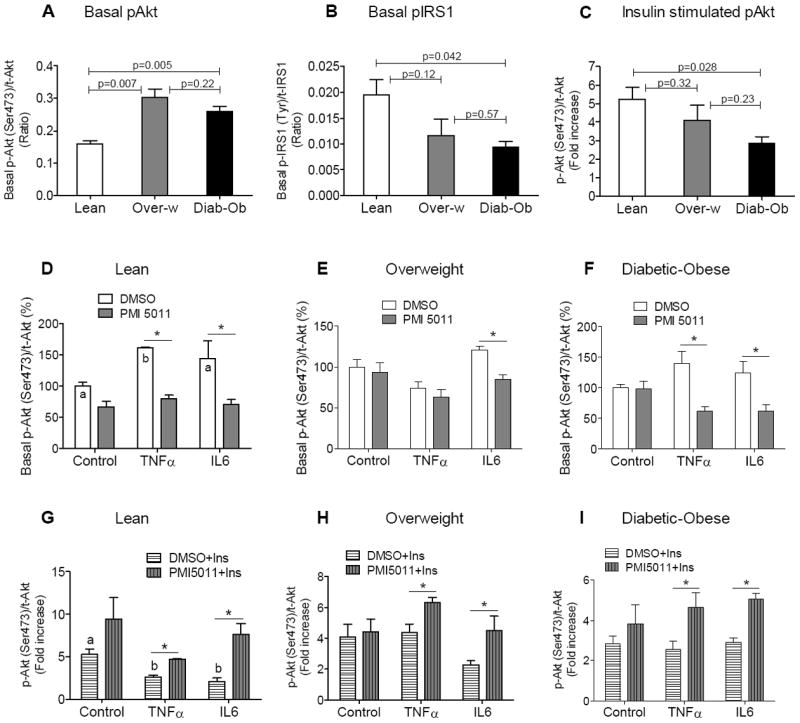
(A) Basal level of Akt phosphorylation (Ser473) and (B) IRS1 phosphorylation at Tyr (Pan) in human myotubes presented as ratio of phospho-Akt to total Akt protein. (C) Insulin-stimulated phosphorylation of Akt (Ser473) in human myotubes presented as fold increase over basal level for each group. (D-F) Phosphorylation of Akt at baseline in myotubes from lean (D), overweight (E) and diabetic-obese (F) subjects treated with vehicle and PMI 5011 in the absence or presence of cytokines. Phosphorylation of Akt was normalized to GAPDH and t-Akt first, and then basal activity of Akt in control (DMSO + PBS-treated) myotubes was counted as 100% for each subject. (G-I) Insulin-stimulated phosphorylation of Akt in myotubes from lean (G), overweight (H) and diabetic-obese (I) subjects treated with vehicle and PMI 5011 in the absence or presence of cytokines. Phosphorylation of Akt was normalized to GAPDH and t-Akt first, and then fold increase in insulin-stimulated phosphorylation of Akt was calculated as fold over basal phosphorylation of Akt for PBS and TNFα, IL6 in each subject. Multiplex Mapmate signaling assay was used to quantify levels of phospho- and total proteins of insulin signaling. First, mean fluorescence intensity (MFI) of phospho- and total proteins was normalized to MFI of GAPDH separately and then the ratio of phospho-to total was calculated (second normalization). Myotubes were pretreated with DMSO or PMI 5011 (5 μg/ml) overnight (16h) in the absence or presence of TNFα (50 ng/ml), IL6 (80 ng/ml). Then after serum starvation for 4h, myotubes were treated with PBS or insulin (100 nM) and at 5 min of incubation, protein lysates were prepared. Results shown are representative of three independent culture of HSMM per subject. All data are presented as means ± SEM, n=3 subjects per group. *p < 0.05 and higher significance between DMSO vs PMI 5011 treatment, and letter “a” or “b” is p < 0.05 and higher significance between control (PBS) vs cytokine treatment.
IRS1 serine-phoshporylation is often elevated in insulin resistant models [30]. Once serine-phosphorylated, IRS1 cannot be appropriately tyrosine-phosphorylated in response to an insulin signal. Our data demonstrate that basal Tyr-phosphorylation of IRS1 is significantly decreased in diabetic-obese subjects and trends lower in overweight subjects, compared to lean subjects (Fig. 1B), which is consistent with other reports on insulin resistant and obese murine models [25].
These results are indicative of attenuated insulin signalling in vivo in skeletal muscle tissue of overweight and diabetic-obese subjects.
PMI 5011 modulates basal Akt phosphorylation in the presence of cytokines
Elevated levels of circulating pro-inflammatory cytokines such as TNFα and IL6 secreted from inflamed adipose tissue induces insulin resistance in other insulin-sensitive organs, such as skeletal muscle. To understand the mechanisms through which PMI 5011 ameliorates inflammation-associated insulin resistance in skeletal muscle, we evaluated phosphorylation of Akt/PKB in human myotubes exposed to TNFα and IL6 in the presence or absence of PMI 5011 using Multiplex Mapmate signalling technology.
Interestingly, in lean subjects, phosphorylation of Akt at baseline was significantly increased in myotubes exposed to TNFα compared to non-cytokine treated control myotubes, whereas it was not significantly higher in myotubes exposed to IL6 (Fig. 1D). PMI 5011 significantly decreased basal phosphorylation of Akt in the presence of TNFα and IL6. Surprisingly, in overweight and diabetic-obese subjects, cytokine treatments did not result in a further increase in phosphorylation of Akt at baseline, which was already significantly elevated in these myotubes in vivo (Fig. 1A,E,F). However, PMI 5011 still significantly reduced basal Akt phosphorylation in myotubes treated with IL6 in overweight (Fig. 1E), and in the presence of both cytokines in diabetic-obese subjects (Fig. 1F). Notably, the reduced levels of basal Akt phosphorylation in diabetic-obese myotubes were comparable with the basal level of Akt phosphorylation in control myotubes from lean subjects (Fig. 1D,F).
PMI 5011 enhances insulin signalling in human myotubes in the presence of pro-inflammatory cytokines
To determine whether reduced basal Akt phosphorylation sensitized insulin response in PMI 5011-treated myotubes, we compared the induction of Akt phosphorylation by insulin to basal phosphorylation levels for each treatment. Notably, in all groups of subjects, the insulin signalling enhancing effect of PMI 5011 in the absence of cytokines was not significant, however, the effect was more apparent in myotubes exposed to cytokines (Fig. 1G-I). In lean subjects, TNFα- and IL6-treatment resulted in significant reduction of Akt phosphorylation with insulin stimulation compared to control myotubes, however, PMI 5011-treatment rescued insulin signalling in lean myotubes in the presence of cytokines (Fig. 1G).
Interestingly, in overweight and diabetic-obese subjects, treatments with pro-inflammatory cytokines did not result in a further decrease in insulin-stimulated phosphorylation of Akt compared to non-cytokine treated control myotubes (Fig. 1H,I). Notably, it was lower in these groups of subjects compared to lean in the absence of cytokines (Fig. 1C). PMI 5011-treatment resulted in significantly increased insulin-stimulated phosphorylation of Akt in the presence of TNFα and IL6 in overweight and in diabetic-obese myotubes (Fig. 1G-I).
Human skeletal muscle culture retains characteristics of the inflammatory phenotype of skeletal muscle cells observed in vivo
Next to understand the relationship between baseline insulin sensitivity and activation of inflammatory signalling, we examined inflammatory pathways in human myotubes using Multiplex Mapmate assays. Notably, activity of the NFkB signalling cascade at baseline was significantly higher in myotubes from diabetic-obese subjects compared to lean subjects, whereas it was not changed in overweight myotubes compared to lean subjects (Fig. 2B). Basal activity of Erk1/2 and IkBα signalling pathways was not different between myotubes from all three groups of subjects (Fig. 2A,C).
Fig. 2. The inflammatory phenotype of skeletal muscle cells derived from diabetic-obese subjects.
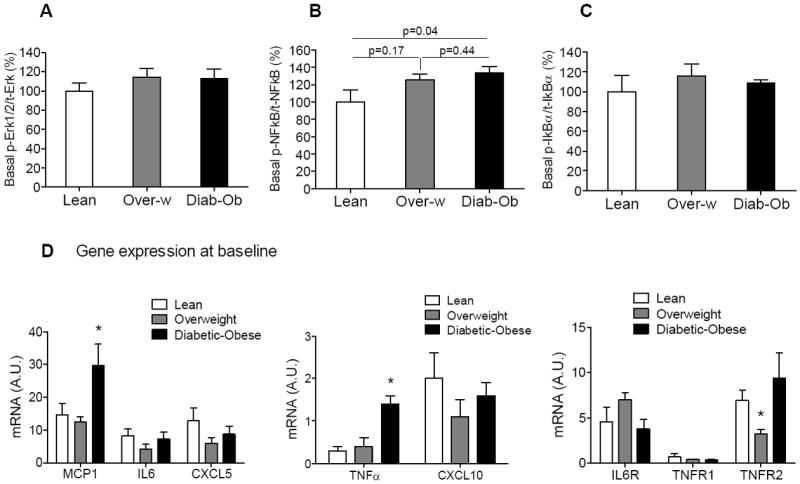
(A-C) Basal level of activity of inflammatory signaling pathways in human myotubes determined by Multiplex cell signaling protein analysis and presented as percentage, where the values for ratio of phospho-protein to total protein from myotubes cultured from normal-lean subjects set to 100%. The following inflammatory signaling pathways were evaluated: Erk1/2 by phosphorylation at Thr185/Tyr187 (A), NFkB by phosphorylation at Ser536 on p65 (B), IkBα by phosphorylation at Ser32 (C). (D) Inflammatory gene expression analysis in human myotubes at baseline as determined by quantitative RT-PCR. The mRNA expression was normalized to cyclophilin B and shown as relative unite (A.U.) Results shown are representative of four independent culture of HSMM per subject. All data are presented as means ± s.e.m., n=3 subjects per group, *p < 0.05 and higher significance compared to lean.
Maintenance of the pro-inflammatory phenotype induced in vivo by circulating cytokines in the obese-diabetic condition is further confirmed by gene expression analysis that showed enhanced expression of the pro-inflammatory genes, MCP1 and TNFα in myotubes from diabetic-obese subjects compared to lean at baseline (Fig. 2D). Interestingly, expression of the pro-inflammatory cytokine IL6, chemokines CXCL5, CXCL10 and cytokine receptors IL6R, TNFR1 were similar in myotubes from all three groups of subjects. Surprisingly, TNFR2 was decreased in myotubes from overweight subjects compared to lean at baseline, whereas TNFR2 expression in myotubes from diabetic-obese subjects was comparable with lean subjects.
Thus, higher basal activity of the NFkB signalling pathway and increased expression of inflammatory markers MCP1 and TNFα in myotubes from diabetic-obese subjects compared to lean subjects are indicative of the inflammatory phenotype seen in vivo, even without cytokine stimulation in culture (Fig. 2B,D).
PMI 5011 attenuates cytokine-induced inflammatory signalling
We demonstrated that PMI 5011 enhances insulin signalling through activation of Akt in human myotubes in an inflammatory environment. To understand how PMI 5011 prevents inflammation-mediated decreases to insulin signalling, we examined its effects on cytokine-induced inflammatory signalling in human myotubes treated with LPS, TNFα, IL6 and vehicle using Multiplex Mapmate assays. Interestingly, we observed no effect of PMI 5011 on basal activity of inflammatory signalling in myotubes in the absence of cytokine induction.
LPS, TNFα and IL6 induced activation of Erk1/2 inflammatory signalling in myotubes from all groups of subjects (Fig. 3A). PMI 5011 treatment reduced Erk1/2 activation to near basal levels in myotubes from lean and diabetic-obese subjects. It also inhibited TNFα- and IL6-induced activation of Erk1/2 in myotubes from overweight subjects, but did not change LPS-induced activation of Erk1/2.
Fig. 3. Effect of PMI 5011 on cytokine-induced inflammatory signaling in human myotubes.
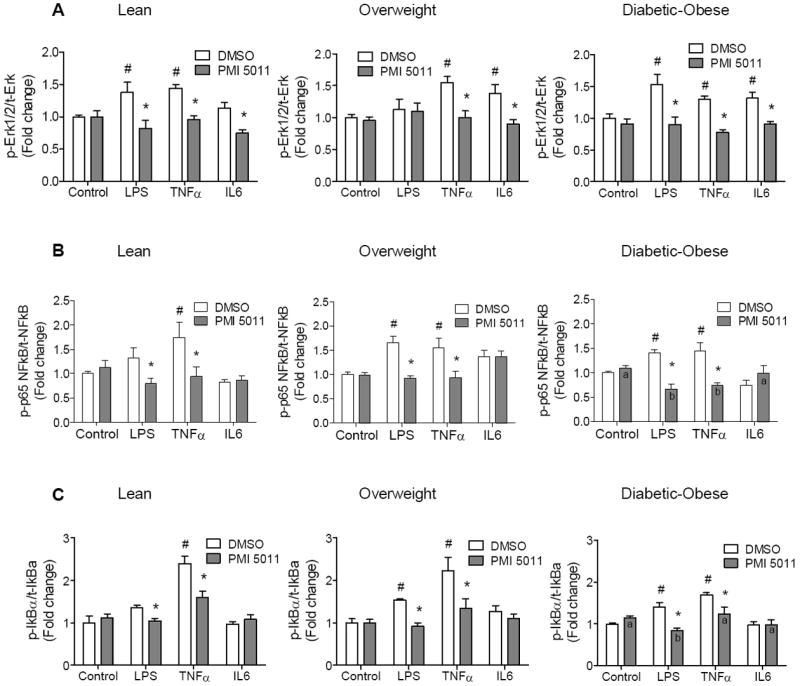
Inhibitory effect of PMI 5011 on activation of inflammatory signaling pathways such as Erk1/2 (A), NFkB (B) and IkBα (C) in response to cytokine induction in myotubes from lean (left), overweight (center) and diabetic-obese (right) subjects. Activities of the signaling pathways were evaluated by Multiplex Mapmate protein assays and ratios of phospho-proteins to total-proteins were calculated after GAPDH normalization. The values for control myotubes (DMSO + PBS-treated) set to 1 and fold change was calculated as fold over control (DMSO + PBS-treated) for each subject. Myotubes were pretreated with DMSO or PMI 5011 (5 μg/ml) overnight (16h). Then after serum starvation for 5h, myotubes were treated with one of the following PBS, LPS (100 ng/ml), TNFα (10 ng/ml), IL6 (40 ng/ml) and at 30 min of incubation, protein lysates were prepared. Results shown are representative of three independent culture of HSMM per subject. All data are presented as means ± SEM, n=3 subjects per group. *p < 0.05 and higher significance between DMSO vs PMI 5011 treatment, # p < 0.05 and higher significance between control (PBS) vs cytokine stimulation in vehicle (DMSO)-treated myotubes, letter “a” or “b” is p < 0.05 and higher significance between control vs cytokine stimulation in PMI 5011 treated myotubes.
LPS and TNFa, but not IL6, treatment resulted in an activation of the NFkB signalling pathway in myotubes from all groups of subjects (Fig. 3B). PMI 5011 demonstrated an inhibitory effect toward the NFkB inflammatory signalling pathway when it is activated with LPS and TNFα. In lean subjects, LPS-induced NFkB activation was not significant compared to unstimulated control myotubes; however, PMI 5011 still inhibited LPS-induced activation of NFkB. PMI 5011 inhibition of TNFα-induced activation of NFkB was significant. In diabetic-obese subjects, inhibition of LPS- and TNFα-induced NFkB activation with PMI 5011 resulted in NFkB activity that was significantly lower than even non-cytokine treated control myotubes in this group. Notably, diabetic-obese myotubes have higher basal NFkB activity than lean myotubes (Fig 2B). Indeed, the reduced levels of cytokine-induced NFkB activation in myotubes from diabetic-obese subjects were comparable with the basal level of activation of NFkB in control myotubes from lean subjects (Fig. 3B).
Interestingly, PMI 5011 also modulated LPS- and TNFα-induced activation of IkBα inflammatory signalling, the canonical upstream signalling system of NFkB cascade, although it did not affect IL6-induced activation of IkBα (Fig. 3C). In lean subjects, LPS-induced IkBα activation was not significant compared to unstimulated control myotubes, whereas PMI 5011-mediated inhibition of LPS-induced activation of IkBα was significant. Notably, the inhibitory effect of PMI 5011 on LPS-induced activation of IkBα in myotubes from diabetic-obese subjects resulted in a level of IkBα activity comparable with basal level in lean subjects.
Thus PMI 5011 modulates activation of cytokine-induced Erk1/2 and NFkB inflammatory signalling pathways in the inflammatory condition. However, pSTAT3, pJNK, and p-p38 MAPK signalling pathways were unchanged with PMI 5011-treatment in our HSMM models and experimental conditions when evaluated by Multiplex Map mate assays (data not shown).
Inflammatory response of human myotubes to cytokine stimulation
In response to pro-inflammatory cytokine stimulation that comes from circulation, skeletal muscle secretes factors including cytokines, chemokines and myokines, which exert their action via auto-, para-, and endocrine mechanisms. To evaluate muscle inflammatory response, we studied the influence of LPS, TNFα and IL6 on inflammatory gene expression in human myotubes using quantitative RT-PCR.
Expression of MCP1, TNFα, IL6, CXCL5, CXCL10 and TNFR2 was significantly increased in all myotubes with LPS-stimulation (Table 1). Interestingly, LPS-induced levels of pro-inflammatory gene expression of MCP1, CXCL5, CXCL10 and TNFR2 were significantly higher in myotubes from diabetic-obese compared to lean subjects.
Table 1.
Cytokine induced gene expression (Fold increase over basal)
| Gene | Lean | Overweight | Diabetic-Ob |
|---|---|---|---|
| LPS-induced | |||
| MCP1 | 4.9 * | 5.1 * | 6.2 *# |
| TNFα | 19.1 * | 3.3 * | 1.6 * |
| IL6 | 3.0 * | 4.3 * | 4.0 * |
| CXCL5 | 2.8 * | 4.5 * | 10.8 *# |
| CXCL10 | 7.3 * | 13.5 * | 51.4 *# |
| TNFR1 | NC | NC | 1.5 |
| TNFR2 | 1.8 * | 2.2 * | 2.6 *# |
| IL6R | NC | NC | 1.4 |
| TNFα-induced | |||
| MCP1 | 43.5 * | 61.9 * | 36.2 * |
| TNFα | 84.6 * | 44.3 * | 18.5 * |
| IL6 | 4.3 * | 7.4 * | 6.0 * |
| CXCL5 | 10.0 * | 34.7 * | 16.0 * |
| CXCL10 | 233 * | 521 * | 901 *# |
| TNFR1 | NC | NC | 1.3 |
| TNFR2 | 4.5 * | 11.7 *# | 5.7 *# |
| IL6R | NC | NC | NC |
| IL6-induced | |||
| MCP1 | 1.7 | 3.5 * | 1.2 |
| TNFα | 4.0 * | 3.3 * | 1.3 |
| IL6 | 1.5 | 2.5 | 2.4 |
| CXCL5 | 1.9 | 2.5 * | 1.3 |
| CXCL10 | 9.6 * | 14.3 * | 19.9 *# |
| TNFR1 | NC | NC | 1.3 |
| TNFR2 | NC | NC | 1.1 |
| IL6R | NC | NC | 1.4 |
p<0.05, significance between basal and cytokine-induced gene expression levels;
p<0.05, significance between cytokine-induced gene expression levels in overweight, diabetic-obese myotubes compared to lean myotubes;
NC, gene expression is not changed (the same with basal level)
TNFα-induction resulted in a dramatic increase in expression of MCP1, CXCL5, IL6 and TNFα in myotubes from all subjects, although these levels were not different between three groups of subjects (Table 1). Notably, TNFα-induced expression of CXCL10 was significant higher in myotubes from diabetic-obese subjects compared to lean and it was the highest among cytokine-stimulated genes from all subjects. Also, the increase in TNFα-induced expression of TNFR2 was significant higher in myotubes from overweight, diabetic-obese subjects compared to lean.
The IL6-induced expression of CXCL10 was significantly higher in myotubes from diabetic-obese subjects as compared to lean subjects. Expression of IL6, CXCL5 and TNFR2 was not induced in myotubes treated with IL6.
Additionally, expression of cytokine receptors IL6R and TNFR1 was not changed with LPS- and cytokine-induction (Table 1). Expression of IL10 and NOS2 was not detectable in any myotubes at baseline and with LPS- and cytokine-treatments (data not shown).
PMI 5011 attenuates the inflammatory response to cytokine stimuli in human myotubes
When presented with the inflammatory ligand LPS or cytokines TNFα and IL6, we observed a resultant induction of inflammatory markers such as MCP1, TNFα, CXCL5, CXCL10 and TNFR2 in human myotubes. This induction is reflective of the chronic-low grade inflammatory response to cytokine stimuli observed in insulin resistant skeletal muscle tissue in vivo. We found that PMI 5011 attenuates activation of the cytokine-induced signalling axis IkBα-NFkB. In the absence of pro-inflammatory stimuli, IkBα binds to NFkB and keeps NFkB inactive. Upon activation, IkBα (phosphorylation at Ser32) releases NFkB and the p65 subunit of NFkB gets activated through phosphorylation at Ser536. This consequently results in translocation of NFkB to the nucleus and initiation of transcription of pro-inflammatory genes that in turn determine inflammatory response of cells [31-33].
To determine whether PMI 5011 modulates inflammatory response of myotubes through inhibition of NFkB signalling, we examined changes in LPS- and cytokine-induced gene expression with PMI 5011-treatment. PMI 5011 inhibits expression of inflammatory genes that are induced with LPS, TNFα and IL6 in a dose dependent manner in myotubes from all three groups (Fig. 4A-C). Remarkably, PMI 5011-treatment significantly decreased expression of TNFR2 that was upregulated upon LPS- and TNFα-stimulation in all myotubes (Fig. 4A,B and Table 1). Notably, expression of TNFR1 was not changed with cytokine- (Table 1) and PMI 5011-treatments in myotubes from lean and overweight subjects (data not shown). However, expression of TNFR1 was significantly reduced with PMI 5011-treatment in myotubes from diabetic-obese subjects, although it was not induced with cytokine treatments (data not shown). Thus, PMI 5011 reduces responsiveness of myotubes to increased environmental TNFα in obesity-associated insulin resistant state and modulates activation of inflammatory signalling and inflammatory response of myotubes in the inflammatory condition. Interestingly, expression of IL6R was not changed with PMI 5011-treatment in myotubes from all subjects (data not shown). Also, we did not find significant changes in basal gene expression in myotubes with PMI 5011-treatment in the absence of cytokines (data not shown).
Fig. 4. Inhibitory effect of PMI 5011 on cytokine-induced inflammatory response of human myotubes.

Reduction in inflammatory gene expression with PMI 5011 treatment in human myotubes in the presence of cytokine stimulation such as LPS (A), TNFα (B) and IL6 (C) as determined by qRT-PCR and presented in percentage, where cytokine-induced gene expression level in vehicle (DMSO)-treated myotubes for each subject set to 100%. Myotubes were pretreated with DMSO or PMI 5011 (2.5 μg/ml, 5.0 μg/ml, and 10.0 μg/ml) overnight (16h). After serum starvation for 1h, myotubes were treated with one of the following PBS, LPS (100 ng/ml), TNFα (10 ng/ml), IL6 (40 ng/ml) and at 6h of incubation cell lysates were prepared for further RNA isolation. The mRNA expression was normalized to cyclophilin B. Results shown are representative of four independent culture of HSMM per subject. All data are presented as means ± s.e.m., n=3 subjects per group, *p < 0.05 and higher significance between DMSO- and PMI 5011 treated myotubes in each group of subjects.
Discussion
Adipose tissue inflammation as a major contributor to systemic insulin resistance in obese mice and obese-diabetic subjects is well documented [34-36]. However, the role of chronic inflammation in skeletal muscle is less characterized. TNFα was previously reported to attenuate insulin signalling in human cultured skeletal muscle cells, while IL6 stimulates insulin signalling after short-term exposure and inhibits it after long-term incubation ([37, 38]). Using HSSM myotubes (Lonza, MD) we show that TNFα- and IL6-treatment resulted in significantly increased basal Akt phosphorylation and decreased insulin-stimulated Akt phosphorylation in myotubes from lean subjects, which is indicative of insulin resistance in these myotubes. Thus our data demonstrate that incubation of lean myotubes with cytokines was sufficient to attenuate insulin signalling, perhaps mimicking the insulin resistant state in skeletal muscle tissue within an inflammatory environment. Interestingly, diabetic-obese myotubes showed significantly decreased insulin-stimulated Akt phosphorylation in the absence of cytokines and TNFα-, IL6-treatment did not decrease phosphorylation further, which suggests that in vivo chronic exposure to cytokines converts myotubes to an insulin resistant state that cannot be further exacerbated by additional cytokine-treatment.
PMI 5011-treatment lowers the elevated basal Akt phosphorylation in myotubes from lean and diabetic-obese subjects in the presence of cytokines. Thus PMI 5011 improves sensitivity to insulin at baseline, which in turn amplifies insulin signalling in muscle in response to insulin induction. In agreement with these results, PMI 5011-treatment resulted in increased insulin-stimulated Akt phosphorylation in all three groups of myotubes despite the presence of TNFα and IL6. Indeed, PMI 5011-treatment restores insulin-stimulated Akt phosphorylation in cytokine-treated myotubes from all subjects to the levels of control lean myotubes.
Higher basal activity of the NFkB signalling pathway in myotubes from diabetic-obese subjects compared to lean subjects might explain the decreased insulin sensitivity at baseline in myotubes of diabetic-obese subjects. LPS is a well known inflammatory ligand that broadly activates pro-inflammatory signalling cascades, including phosphorylation of IKK and MAPKs such as JNK, p38, and extracellular signal-regulated kinases (ERK) [10]. TNFα stimulates inflammation by activating NFkB and MAPKs signalling pathways [39]. In agreement with these reports, we found that LPS and TNFα activated Erk1/2, NFkB, IkBα pathways, whereas IL6 induced only activation of Erk1/2 signalling in our HSMM model. PMI 5011 inhibits activation of cytokine-induced signalling pathways Erk1/2, NFkB, and IkBα in myotubes from all subjects. Moreover, in overweight and diabetic-obese myotubes, PMI 5011 modulates the level of activation of inflammatory signalling cascades NFkB and IkBα by equalizing these levels with lean myotubes. Interestingly, effect of PMI 5011 on NFkB and IkBα profiles are similar, which suggests that PMI 5011 acts on the NFkB pathway through the IkBα cascade (Fig. 5).
Fig. 5. Schematic presentation of the inhibitory effect of PMI 5011 on cytokine-induced inflammatory signaling pathways in human skeletal muscle cells.
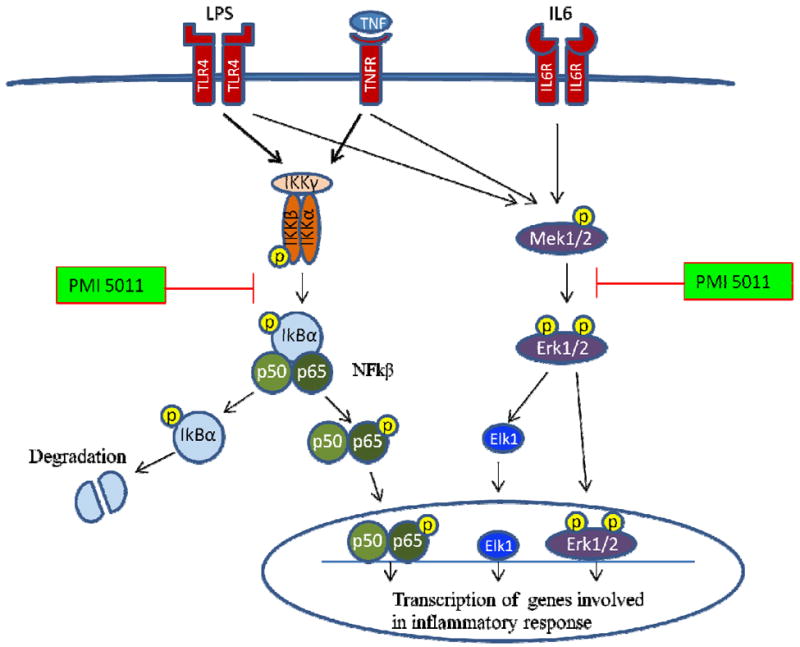
PMI 5011 inhibits activity of IkBα-NFkB axis and Erk1/2 signaling pathways when pro-inflammatory ligands and cytokines are present in the environment, which leads to a reduced transcription of inflammatory marker genes.
Recent reports demonstrate that NFkB and JNK pathways in skeletal muscle are regulated by pro-inflammatory stimuli, and high fat diet, and muscle contraction. Activation of these signalling systems in skeletal muscle cells results in secretion of inflammatory mediators such as TNFα, IL6, iNOS, CXCL1, CXCL5, CXCL9, CXCL10 and MCPs [29, 40-42]. Consistent with these reports and in vivo NFkB pathway activation in diabetic-obese muscle, we found elevated expression of genes such as MCP1 and TNFα at baseline in myotubes from diabetic-obese subjects compared to lean and overweight subjects. Basal expression of TNFR2 is associated with secondary TNF release in man [43]. Notably, TNFR2 expression was decreased in myotubes from overweight subjects, which might explain reduced responsiveness of myotubes to TNFα stimulation and enhanced sensitivity to insulin despite the presence of TNFα.
Furthermore, LPS- and TNFα were capable to stimulate gene expression of cytokines and chemokines such as MCP1, CXCL5, CXCL10, TNFα and TNFR2 co-ordinately in myotubes from all subjects. Remarkably, myotubes from diabetic-obese subjects showed the greatest magnitude of response to LPS- and TNFα-stimulation. Specifically, LPS- and cytokine-induced gene expression of CXCL10, and LPS-induced CXCL5 expression were significantly higher in myotubes from diabetic-obese subjects compared to lean subjects. CXCL10 is inducible by TNFα and its up-regulation is associated with up-regulation of TNFR2 in human skeletal muscle cells [44], however CXCL10 receptor, CXCR3 is not expressed in myofibers [40]. On the other hand, CXCR3 is present widely on Th1 pro-inflammatory T cells, while CXCL5 is a chemokine that attracts neutrophils. Thus inflammatory response in vivo in diabetic-obese subjects could result in recruitment of more pro-inflammatory monocyte/macrophages, T cells and other immune cells to skeletal muscle. This could result in chronic inflammation that in turn may contribute largely to impaired insulin signalling in muscle. Notably, PMI 5011 showed remarkable anti-inflammatory effects, inhibiting LPS-, TNFα- and IL6-induced inflammatory gene expression in a dose-dependent manner in myotubes from all subjects. Thus PMI 5011 attenuated classical NFkB target gene expression in response to cytokine stimuli.
Obesity-associated insulin resistance is characterized by elevated circulating fatty acids and cytokines. Accumulation of toxic intracellular lipids such as ceramides and DAG interferes with insulin signalling in skeletal muscle, thus plays an important role in skeletal muscle insulin resistance ([45]). Previously, it has been shown that PMI 5011 enhances insulin signalling in primary human skeletal muscle culture and murine cells despite the presence of palmitate and ceramides [16-18]. Here we demonstrate that PMI 5011 enhances insulin action through Akt insulin signalling in human myotubes in an inflammatory environment and the effect was related to its anti-inflammatory effect. It is well known that IKKβ is a central coordinator of inflammatory responses through IkBα-NFkB, where NFkB activation induces inflammatory mediators that cause insulin resistance [46]. Although our results suggest that PMI 5011 may modulate inflammatory signalling through IkBα, perhaps phosphorylation of IKK should be investigated further. Given that NFkB activation plays essential role in function of immune cells during both innate and adaptive immune responses, the modulating effect of PMI 5011 on activation of inflammatory signalling gives proper quality to PMI 5011 as a potential drug. Thus PMI 5011 could be a very promising treatment agent for obese-diabetic patients to simultaneously increase muscle insulin sensitivity and reduce skeletal muscle inflammation.
Supplementary Material
Acknowledgments
We thank Dieyun Ding, Jingying Zhang, Heather Kirk-Ballard, Gail Effler-Braymer, Elizabeth Floyd and Rob Noland (all from Pennington Biomedical Research Center) for technical assistance and helpful advice. This work was supported by the pilot grant to B.V. from the Botanical Research Center, which funded from NCAMM (P50AT002776 NIH). The research in the Mynatt laboratory is supported by the grants from ADA (#1-10-BS-129) and NIH (RO1DK089641) to R.L.M. This work used the PBRC facilities of the Transgenic, Comparative Biology, Genomics Core and Clinical Research Laboratory supported in part by COBRE (NIH 8P20GM103528) and the NORC (NIH 2P30-DK072476) center grants from the National Institutes of Health.
Footnotes
Conflict of interest
The authors declare no potential conflicts of interest relevant to this article.
Conflict of interest details: Design: Vandanmagsar B., Mynatt R.L. Conduct/data collection: Vandanmagsar B., Bermudez E., Mendoza T., Ribnicky D. Analysis: Vandanmagsar B., Cefalu W., Mynatt R.L. Writing manuscript: Vandanmagsar B., Haynie K., Wicks S., Mynatt R.L.
Authorship details: Vandanmagsar B: no conflict of interest Haynie K: no conflict of interest Wicks S: no conflict of interest Bermudez E: no conflict of interest Mendoza T: no conflict of interest Ribnicky D: no conflict of interest Cefalu W: no conflict of interest Mynatt R: no conflict of interest
References
- 1.Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322:223–228. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 2.DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- 3.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barma P, Bhattacharya S, Bhattacharya A, Kundu R, Dasgupta S, Biswas A, et al. Lipid induced overexpression of NF-kappaB in skeletal muscle cells is linked to insulin resistance. Biochim Biophys Acta. 2009;1792:190–200. doi: 10.1016/j.bbadis.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 5.Feve B, Bastard JP. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:305–311. doi: 10.1038/nrendo.2009.62. [DOI] [PubMed] [Google Scholar]
- 6.Bilan PJ, Samokhvalov V, Koshkina A, Schertzer JD, Samaan MC, Klip A. Direct and macrophage-mediated actions of fatty acids causing insulin resistance in muscle cells. Arch Physiol Biochem. 2009;115:176–190. doi: 10.1080/13813450903079314. [DOI] [PubMed] [Google Scholar]
- 7.Samokhvalov V, Bilan PJ, Schertzer JD, Antonescu CN, Klip A. Palmitate- and lipopolysaccharide-activated macrophages evoke contrasting insulin responses in muscle cells. Am J Physiol Endocrinol Metab. 2009;296:E37–46. doi: 10.1152/ajpendo.90667.2008. [DOI] [PubMed] [Google Scholar]
- 8.Kewalramani G, Bilan PJ, Klip A. Muscle insulin resistance: assault by lipids, cytokines and local macrophages. Curr Opin Clin Nutr Metab Care. 2010;13:382–390. doi: 10.1097/MCO.0b013e32833aabd9. [DOI] [PubMed] [Google Scholar]
- 9.Varma V, Yao-Borengasser A, Rasouli N, Nolen GT, Phanavanh B, Starks T, et al. Muscle inflammatory response and insulin resistance: synergistic interaction between macrophages and fatty acids leads to impaired insulin action. Am J Physiol Endocrinol Metab. 2009;296:E1300–1310. doi: 10.1152/ajpendo.90885.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu M, Patsouris D, Li P, Flores-Riveros J, Frincke JM, Watkins S, et al. A new antidiabetic compound attenuates inflammation and insulin resistance in Zucker diabetic fatty rats. Am J Physiol Endocrinol Metab. 2010;298:E1036–1048. doi: 10.1152/ajpendo.00668.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt B, Ribnicky DM, Poulev A, Logendra S, Cefalu WT, Raskin I. A natural history of botanical therapeutics. Metabolism. 2008;57:S3–9. doi: 10.1016/j.metabol.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Govorko D, Logendra S, Wang Y, Esposito D, Komarnytsky S, Ribnicky D, et al. Polyphenolic compounds from Artemisia dracunculus L. inhibit PEPCK gene expression and gluconeogenesis in an H4IIE hepatoma cell line Am J Physiol Endocrinol Metab. 2007;293:E1503–1510. doi: 10.1152/ajpendo.00420.2007. [DOI] [PubMed] [Google Scholar]
- 13.Watcho P, Stavniichuk R, Tane P, Shevalye H, Maksimchyk Y, Pacher P, et al. Evaluation of PMI-5011, an ethanolic extract of Artemisia dracunculus L., on peripheral neuropathy in streptozotocin-diabetic mice. Int J Mol Med. 2011;27:299–307. doi: 10.3892/ijmm.2011.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scherp P, Putluri N, LeBlanc GJ, Wang ZQ, Zhang XH, Yu Y, et al. Proteomic analysis reveals cellular pathways regulating carbohydrate metabolism that are modulated in primary human skeletal muscle culture due to treatment with bioactives from Artemisia dracunculus L. J Proteomics. 2012;75:3199–3210. doi: 10.1016/j.jprot.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopes-Lutz D, Alviano DS, Alviano CS, Kolodziejczyk PP. Screening of chemical composition, antimicrobial and antioxidant activities of Artemisia essential oils. Phytochemistry. 2008;69:1732–1738. doi: 10.1016/j.phytochem.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Wang ZQ, Ribnicky D, Zhang XH, Raskin I, Yu Y, Cefalu WT. Bioactives of Artemisia dracunculus L enhance cellular insulin signaling in primary human skeletal muscle culture. Metabolism. 2008;57:S58–64. doi: 10.1016/j.metabol.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obanda DN, Hernandez A, Ribnicky D, Yu Y, Zhang XH, Wang ZQ, et al. Bioactives of Artemisia dracunculus L. mitigate the role of ceramides in attenuating insulin signaling in rat skeletal muscle cells Diabetes. 2012;61:597–605. doi: 10.2337/db11-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirk-Ballard H, Wang ZQ, Acharya P, Zhang XH, Yu Y, Kilroy G, et al. An Extract of Artemisia dracunculus L. Inhibits Ubiquitin-Proteasome Activity and Preserves Skeletal Muscle Mass in a Murine Model of Diabetes. PLoS One. 2013;8:e57112. doi: 10.1371/journal.pone.0057112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kheterpal I, Coleman L, Ku G, Wang ZQ, Ribnicky D, Cefalu WT. Regulation of insulin action by an extract of Artemisia dracunculus L. in primary human skeletal muscle culture: a proteomics approach. Phytother Res. 2010;24:1278–1284. doi: 10.1002/ptr.3093. [DOI] [PubMed] [Google Scholar]
- 20.Wang ZQ, Ribnicky D, Zhang XH, Zuberi A, Raskin I, Yu Y, et al. An extract of Artemisia dracunculus L. enhances insulin receptor signaling and modulates gene expression in skeletal muscle in KK-A(y) mice. J Nutr Biochem. 2011;22:71–78. doi: 10.1016/j.jnutbio.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ribnicky DM, Poulev A, Watford M, Cefalu WT, Raskin I. Antihyperglycemic activity of Tarralin, an ethanolic extract of Artemisia dracunculus L. Phytomedicine. 2006;13:550–557. doi: 10.1016/j.phymed.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Ribnicky DM, Kuhn P, Poulev A, Logendra S, Zuberi A, Cefalu WT, et al. Improved absorption and bioactivity of active compounds from an anti-diabetic extract of Artemisia dracunculus L. Int J Pharm. 2009;370:87–92. doi: 10.1016/j.ijpharm.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry RR, Abrams L, Nikoulina S, Ciaraldi TP. Insulin action and glucose metabolism in nondiabetic control and NIDDM subjects. Comparison using human skeletal muscle cell cultures. Diabetes. 1995;44:936–946. doi: 10.2337/diab.44.8.936. [DOI] [PubMed] [Google Scholar]
- 24.Gaster M, Petersen I, Hojlund K, Poulsen P, Beck-Nielsen H. The diabetic phenotype is conserved in myotubes established from diabetic subjects: evidence for primary defects in glucose transport and glycogen synthase activity. Diabetes. 2002;51:921–927. doi: 10.2337/diabetes.51.4.921. [DOI] [PubMed] [Google Scholar]
- 25.Huisamen B, Dietrich D, Bezuidenhout N, Lopes J, Flepisi B, Blackhurst D, et al. Early cardiovascular changes occurring in diet-induced, obese insulin-resistant rats. Mol Cell Biochem. 2012;368:37–45. doi: 10.1007/s11010-012-1340-9. [DOI] [PubMed] [Google Scholar]
- 26.Salinari S, Debard C, Bertuzzi A, Durand C, Zimmet P, Vidal H, et al. Jejunal Proteins Secreted by db/db Mice or Insulin-Resistant Humans Impair the Insulin Signaling and Determine Insulin Resistance. PLoS One. 2013;8:e56258. doi: 10.1371/journal.pone.0056258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lumeng CN, Deyoung SM, Saltiel AR. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. Am J Physiol Endocrinol Metab. 2007;292:E166–174. doi: 10.1152/ajpendo.00284.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen N, Liu L, Zhang Y, Ginsberg HN, Yu YH. Whole-body insulin resistance in the absence of obesity in FVB mice with overexpression of Dgat1 in adipose tissue. Diabetes. 2005;54:3379–3386. doi: 10.2337/diabetes.54.12.3379. [DOI] [PubMed] [Google Scholar]
- 29.Sell H, Eckardt K, Taube A, Tews D, Gurgui M, Van Echten-Deckert G, et al. Skeletal muscle insulin resistance induced by adipocyte-conditioned medium: underlying mechanisms and reversibility. Am J Physiol Endocrinol Metab. 2008;294:E1070–1077. doi: 10.1152/ajpendo.00529.2007. [DOI] [PubMed] [Google Scholar]
- 30.Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55:2565–2582. doi: 10.1007/s00125-012-2644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 32.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 33.Whiteside ST, Epinat JC, Rice NR, Israel A. I kappa B epsilon, a novel member of the I kappa B family, controls RelA and cRel NF-kappa B activity. EMBO J. 1997;16:1413–1426. doi: 10.1093/emboj/16.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol. 2010;185:1836–1845. doi: 10.4049/jimmunol.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sell H, Eckel J, Dietze-Schroeder D. Pathways leading to muscle insulin resistance--the muscle--fat connection. Arch Physiol Biochem. 2006;112:105–113. doi: 10.1080/13813450600711540. [DOI] [PubMed] [Google Scholar]
- 38.Sell H, Dietze-Schroeder D, Eckel J. The adipocyte-myocyte axis in insulin resistance. Trends Endocrinol Metab. 2006;17:416–422. doi: 10.1016/j.tem.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 39.Lombardo E, Alvarez-Barrientos A, Maroto B, Bosca L, Knaus UG. TLR4-mediated survival of macrophages is MyD88 dependent and requires TNF-alpha autocrine signalling. J Immunol. 2007;178:3731–3739. doi: 10.4049/jimmunol.178.6.3731. [DOI] [PubMed] [Google Scholar]
- 40.Raju R, Vasconcelos O, Granger R, Dalakas MC. Expression of IFN-gamma-inducible chemokines in inclusion body myositis. J Neuroimmunol. 2003;141:125–131. doi: 10.1016/s0165-5728(03)00218-2. [DOI] [PubMed] [Google Scholar]
- 41.Nedachi T, Hatakeyama H, Kono T, Sato M, Kanzaki M. Characterization of contraction-inducible CXC chemokines and their roles in C2C12 myocytes. Am J Physiol Endocrinol Metab. 2009;297:E866–878. doi: 10.1152/ajpendo.00104.2009. [DOI] [PubMed] [Google Scholar]
- 42.Henningsen J, Pedersen BK, Kratchmarova I. Quantitative analysis of the secretion of the MCP family of chemokines by muscle cells. Mol Biosyst. 2011;7:311–321. doi: 10.1039/c0mb00209g. [DOI] [PubMed] [Google Scholar]
- 43.Fairfax BP, Davenport EE, Makino S, Hill AV, Vannberg FO, Knight JC. A common haplotype of the TNF receptor 2 gene modulates endotoxin tolerance. J Immunol. 2011;186:3058–3065. doi: 10.4049/jimmunol.1001791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crescioli C, Sottili M, Bonini P, Cosmi L, Chiarugi P, Romagnani P, et al. Inflammatory response in human skeletal muscle cells: CXCL10 as a potential therapeutic target. Eur J Cell Biol. 2012;91:139–149. doi: 10.1016/j.ejcb.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 45.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.