

Abstract
Protein design will ultimately allow for the creation of artificial enzymes with novel functions and unprecedented stability. To test our current mastery of nature’s approach to catalysis, a Zn(II) metalloenzyme was prepared using de novo design. α3DH3 folds into a stable single-stranded three-helix bundle and binds Zn(II) with high affinity using His3O coordination. The resulting metalloenzyme catalyzes the hydration of CO2 better than any small molecule model of carbonic anhydrase and with an efficiency within 1400-fold of the fastest carbonic anhydrase isoform, CAII, and 11-fold of CAIII.
Keywords: de novo design, metalloenzyme, protein design, carbonic anhydrase, zinc enzyme
Protein design is an increasingly popular approach for studying and modeling the structure-function relationships in proteins.[1] There is a growing interest in the development of artificial enzymes that can perform with the efficiency of natural enzymes toward reactions not normally seen in nature. Specifically, artificial metalloenzymes are important design targets because over one-third of natural proteins use metal ions for structural, catalytic, and/or electron-transfer functions. There are two main metalloprotein design strategies: protein redesign and de novo design. The former approach involves the introduction of a metal-binding site into an existing, stable protein. The latter relies on first principles to design well-defined structures from amino acid sequences not found in nature. De novo design is challenging due to its requirement for complete control over folding and function, but can lead to significant insight into the nature of metal-enzyme interactions. Several recent examples showcase the power of de novo design in creating metalloenzymes with enzymatic activity for ester hydrolysis,[2-5] nitrite reduction,[6] oxidation,[7,8] and N-hydroxylation.[9]
One test of our current mastery of protein design is to model the activity of a natural enzyme. Carbonic anhydrase, ubiquitous in animals, plants, and bacteria, is essential for respiration, vision, regulation of acid-base equilibrium, and many other processes. One of the most efficient enzymes in nature, human carbonic anhydrase II (CAII) catalyzes the reversible hydration of CO2 with a catalytic efficiency approaching the diffusion limit in water. The importance of this metalloenzyme, in conjunction with its well-studied mechanism, solid-state structure, and inhibition, make it a highly appealing, yet challenging, target to assess our ability to replicate the activity of a natural metalloenzyme in a simplified system.
Previously, our group published a highly efficient CA-mimic by modeling the active site within a de novo designed three-stranded coiled-coil (3SCC). The bifunctional Hg(II)SZn(II)N(TRIL9CL23H)3 closely models the structure of the Zn(II)His3 OH primary coordination sphere of CAII, yet places it within a very different fold (α-helices vs β-sheets in CAII), and contains an additional Hg(II)Cys3 site which provides structural stability. The metalloenzyme catalyzes CO2 hydration with an efficiency comparable to some naturally occurring CAs and within 350-fold of the fastest isozyme, CAII. While this is the fastest CA-model to date, improvements to the system are limited by the inherent symmetry resulting from the self-assembly of three parallel α-helices. Not only are antiparallel helices more typical in nature,[10] but CAII contains a network of H-bonds that cannot currently be modeled in this system. Therefore, a new approach is required.
To allow for asymmetry in the secondary sphere of the active site, we have designed a metalloenzyme starting from α3D, a de novo single-stranded antiparallel three-helix bundle. Designed by DeGrado and coworkers,[13] this 73 amino acid protein folds with native protein-like stability, is tolerant of mutations within the hydrophobic core,[14] and has been structurally characterized by NMR spectroscopy.[12] Previously, our lab incorporated a Cys3 metal binding site near the C-terminus of α3D and showed that the resulting protein, α3 DIV, binds Hg(II), Pb(II), and Cd(II) with high affinity in coordination geometries previously identified within the TRI family of 3SCCs.[15] Here, we report a new metalloenzyme, α3DH3, which contains a His3 site that, upon binding Zn(II), catalyzes the hydration of CO2 (Figure 1).
Figure 1.
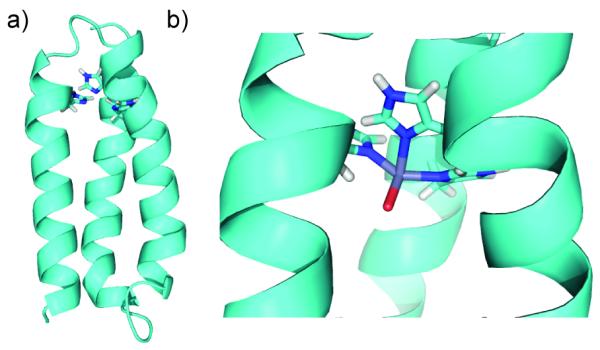
PyMol[11] models of α3DH3 showing (a) the entire bundle (last four residues removed for simplicity) and (b) the Zn(II)His3O site incorporating EXAFS Zn-N/O distances. Models are based on the NMR solution structure of α3D (PDB 2A3D).[12]
α3DH3 differs from α3D in that three leucine residues were replaced with histidine residues (L18H, L28H, L67H), a histidine residue was replaced with valine (H72V) to ensure no competition for Zn(II) binding, and four extra residues were added to the end of the chain (GSGA) which improved expression yields (Table 1). Expressed from a synthetic gene in Escherichia coli, α3DH3 was purified by high-performance liquid chromatography (HPLC) and characterized by electrospray ionization mass spectroscopy (EI-MS). The observed molecular weight of 8283.5 Da corresponds to the protein after deletion of the N-terminal methionine residue (MWcalc = 8283.1 Da). The protein is well-folded and stable according to circular dichroism spectroscopy (Figure S1). The observed double well at 208 and 222 nm is characteristic of α-helical proteins and the molar ellipticities suggest that the protein is 82% folded at pH 9. Chemical denaturations with guanidine hydrochloride were fit to a two-state unfolding model, revealing a ΔGu = 3.1 kcal/mol. Although the histidine substitutions destabilized the protein compared to native α3D (ΔGu = 5.1 kcal/mol), the construct is stable enough for use in kinetic studies. The presence of Zn(II) caused no change in the folding or stability of α3DH3 (Figure S1).
Table 1.
De Novo Designed Peptide Sequences.[a]
| Peptide | Sequence |
|---|---|
| α3DH3 | MGSWAEFKQRLAAIKTRHQALGG |
| SEAEHAAFEKEIAAFESELQAYKGKGNPE | |
| VEALRKEAAAIRDEHQAYRVNGSGA | |
| α3DH72V | MGSWAEFKQRLAAIKTRLQALGG |
| SEAELAAFEKEIAAFESELQAYKGKGNPE | |
| VEALRKEAAAIRDELQAYRVN |
Mutations with respect to α3D are in bold and underlined
Apparent Zn(II) binding constants were measured by UV-Vis spectroscopy using Zincon as a colorimetric probe (Figure S2-S4). α3DH3 binds Zn(II) with a 150 ± 40 nM affinity at pH 7.5, which strengthens to 59 ± 9 nM at pH 9.0. A control peptide, α3DH72V, which lacks the His3 binding site, binds with 20-fold weaker affinity (Kd = 1.2 ± 0.4 μM at pH 9.0), confirming that the histidine site is involved in specific Zn(II) binding. These affinities are weaker than that of CAII measured by equilibrium dialysis (Kd = 0.8 ± 0.1 pM);[16] however, recent isothermal titration calorimetry experiments suggest a three-orders of magnitude weaker affinity for CAII (Kd = 0.45 nM).[17] Based on this newer measurement, α3DH3 has an affinity only two-orders of magnitude weaker than CAII and stronger than those observed for our previously published Hg(II)SZn(II)N(TRIL9CL23H)3 (Kd = 0.8 ± 0.1 and 0.22 ± 0.06 μM at pH 7.5 and 9.0, respectively).[18]
A sample of Zn(II)α3DH3 at pH 9.0 was analyzed by extended x-ray absorption fine structure (EXAFS) spectroscopy. The EXAFS data is dominated by nearest neighbor-scattering typical of oxygen or nitrogen ligation and can be modeled with a single oxygen/nitrogen shell at ~1.98 Å, a distance consistent with a 4-coordinate Zn site. There is clear evidence of outer shell scattering typical of histidine ligation metals (i.e., features at R+α>2 Å in Figure 2). Given the relatively limited k range of these data, there are multiple models with similar fit quality, making it difficult to define the number of histidine ligands from EXAFS alone. However, the best fit, both in terms of mean-square-deviation and in terms of the presence of physically reasonable fit parameters, uses one oxygen at 1.90 Å and 3 histidines at 1.99 Å. These parameters are very similar to EXAFS distances measured for CAII (Zn-N/O of 1.98 Å).[17]
Figure 2.
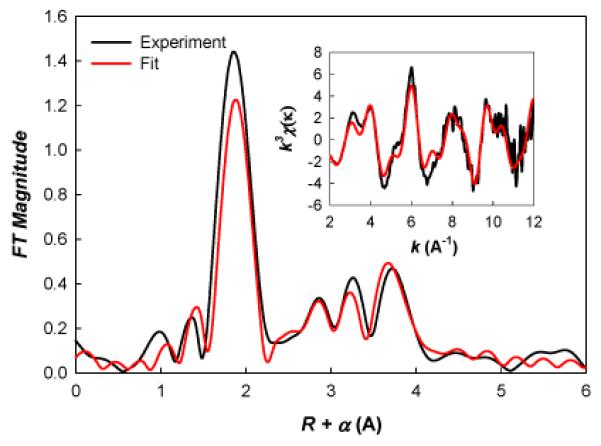
Fourier transform (FT) of the EXAFS spectra for Zn(II)α3DH3. Inset is k3-weighted EXAFS spectra used to calculate the FT. Red is the best fit (see text); black is raw data.
The true indication of a successfully designed metalloenzyme mimic is catalytic activity toward the physiological reaction of the natural enzyme, in this case, the hydration of CO2. Using Khalif’s[19] stopped flow indicator technique, Zn(II)α3DH3 was found to be an efficient catalyst with activities that increase with pH (Table 2). Testing the activity at a pH higher than 9.5 is not feasible because the stability of the bundle is compromised when interhelical salt bridges are deprotonated. Fitting the catalytic efficiencies for pH 8–9.5, a maximal efficiency of 6.9 × 104 M−1s−1 and a kinetic pKa of 9.4 were derived (Figure 3). CAII has a pKa of 6.820 for the deprotonation of Zn(II)-bound water to give the active hydroxide complex. The pKa measured here might be for the same process (as was suggested for the observed pKa (8.8) for p-nitrophenylacetate hydrolysis by Hg(II)SZn(II)N(TRIL9CL23H)3); however, it could also be for a lysine residue near the active site, which, upon deprotonation, would open up the bundle and allow for improved substrate access. In either case, catalysis occurs at the Zn(II)His3O site; even in the presence of Zn(II) the control peptide α3DH72V shows significantly lower activity (Figure S5).
Table 2.
Kinetic parameters for the catalysis of CO2 hydration by enzymes and model complexes.
| Enzyme/Model | pKa | pH | kcat (s−1) | Km (mM) | kcat/KM (M−1s−1) | k2 (M−1s−1) | Reference |
|---|---|---|---|---|---|---|---|
| CAII | 6.8[a] | 8.8 | 8.2 × 105 | 8.9 | 9.2 × 107 | [29] | |
| CAIII | 8.5 | 9.0 | 8 × 103 | 20 | 4 × 105 | [30] | |
| Hg(II)sZn(II)N(TRIL9CL23H)3n+ | 8.82[b] | 9.5 | (1.8 ± 0.4) × 103 | 10.0 ± 2.4 | (1.8 ± 0.3) × 105 | [2] | |
| Zn(II)α3DH3 | 9.4 | 9.5 | 134 ± 8 | 3.5 ± 0.6 | (3.8 ± 0.5) × 104 | this work | |
| 9.25 | 137 ± 8 | 4.1 ± 0.7 | (3.2 ± 0.3) × 104 | ||||
| 9.0 | 103 ± 8 | 4.9 ± 1.0 | (2.1 ± 0.3) × 104 | ||||
| 8.75 | 107 ± 12 | 7.3 ± 1.8 | (1.5 ± (0.2) × 104 | ||||
| 8.5 | 82 ± 6 | 7.2 ± 1.1 | (1.1 ± 0.1) × 104 | ||||
| 8.0 | 39 ± 4 | 6.2 ± 1.7 | (0.63 ± 0.1) × 104 | ||||
| Zn(II)([14]aneN4) | 9.8 | ind[c] | 5040[c] | [35] | |||
| Zn(II)([12]aneN4) | 8.1 | 9.1 | 3012± 193 | [27] | |||
| Zn(II)(nitrilotris(2-benzimidazolylmethyl- 6-sulfonate) |
8.3 | 9.5 | 3180[d] | [28] | |||
| Zn(II)(tris(4,5-di-n-propyl-2- imidazolyl)phosphine) |
8.0 | 6.55 | 2480[e] | [25] |
Taken from Ref20.
pKa measured for the hydrolysis of pNPA.
pH-independent second-order rate constant (maximal rate) calculated using pKa of 9.8 and measured rate of 690 M−1s−1 at pH 9.0.
Second-order rate constant measured at 15 °C.
Second-order rate constant was measured for the dehydration of HCO3− + H+ in 80% EtOH/H2O.
Figure 3.
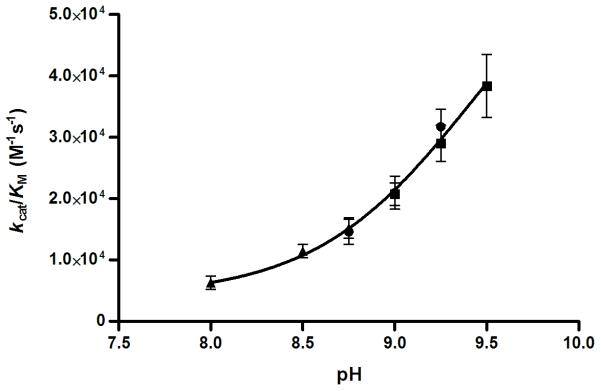
Plot of pH dependency of the catalytic efficiency of CO2 hydration by Zn(II)-α3DH3. Standard errors for the fit of the Michaelis-Menten equation to each plot of initial rate versus initial [CO2]. The buffer/indicator pairs used were (▲) TAPS/m-cresol purple (pH 8-8.75), (•) AMPSO/thymol blue (pH 8.75-9.25), and (■) CHES/thymol blue (pH 9.25-9.5).
Numerous small molecule models for CAII can be found in the literature, with varying degrees of structural similarity to the enzyme’s active site.[21-28] Many of these show some catalytic activity toward the hydration of CO2, with the fastest catalysts listed in Table 2. Both Zn(II)(tris(4,5-di-n-propyl-2-imidazolyl)phosphine) and Zn(II)(nitrilotris(2-benzimidazolylmethyl-6-sulfonate) have N3O coordination of the Zn(II) ion and show moderate activity.[25,28] Zn(II)α3DH3 outperforms each of these with a maximal catalytic efficiency that is at least 14-fold higher than their fastest second-order rate constants. Its efficiency is only 2.6-fold slower than that of Hg(II)SZn(II)N(TRIL9CL23H)3 (Table 2), the only other designed enzyme for CO2 hydration.[2] Compared to the native enzymes, Zn(II)α3DH3 is 1400-fold slower than CAII and only 11-fold slower than CAIII.[29,30]
A frequent problem with small molecule models of enzymes is product inhibition.[31-33] Using acetate as a more tractable mimic of the reaction product bicarbonate, product inhibition was tested (Figure S5). Addition of 300 mM KOAc to the assay at pH 8.5 resulted in a decrease in kcat from 82 ± 6 to 66 ± 4 s−1. With an enzyme concentration of 100 μM and HCO−3 concentrations never exceeding 25 mM, no significant loss in catalytic activity is expected due to product inhibition. This shows that the peptide is well-designed to avoid product inhibition, highlighting one benefit of de novo design over small molecule catalysts.
Here, we successfully modified an existing de novo designed protein to contain a Zn(II)His3 binding site while retaining the desired fold. We report the first pH profile for CO2 hydration by a de novo designed protein and show that Zn(II)α3DH3 catalyzes this reaction better than any small molecule model and within 1-3 orders of magnitude of natural CA isozymes. The efficiency of our single stranded construct is slightly lower than our previously reported 3SCC Hg(II)SZn(II)N(TRIL9CL23H)3 and the Zn(II) affinity is stronger, likely a consequence of differences in the peptide scaffolds. The antiparallel bundle is expected to have a weaker dipole than the coiled-coil, imidazole rings in less-symmetric orientations, and different electrostatics around the binding site. Compared to Hg(II)SZn(II)N(TRIL9CL23H)3, Zn(II)α3DH3 has greater potential for improving catalysis because it is far easier to make asymmetric protein modifications in this system. Furthermore, being an antiparallel construct, Zn(II)α3DH3 more faithfully reproduces helical structures found in nature. In CAII, mutation of Thr199 to alanine lowers the activity 100-fold, showcasing the importance of second coordination sphere residues involved in hydrogen bonds.[34] Future work will focus on site selective incorporation of acid-base catalytic residues or design of water channels that may tune the Lewis acidity of Zn(II) to optimize activity.
Experimental Section
Please see the Supporting Information for experimental details.
Supplementary Material
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- [1].Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Chem. Rev. 2014;114:3495–3578. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL. Nat. Chem. 2012;4:118–123. doi: 10.1038/nchem.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Der BS, Edwards DR, Kuhlman B. Biochemistry. 2012;51:3933–3940. doi: 10.1021/bi201881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Khare SD, Kipnis Y, Greisen P, Takeuchi R, Ashani Y, Goldsmith M, Song Y, Gallaher JL, Silman I, Leader H, Sussman JL, Stoddard BL, Tawfik DS, Baker D. Nat. Chem. Biol. 2012;8:294–300. doi: 10.1038/nchembio.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rufo CM, Moroz YS, Moroz OV, Stöhr J, Smith TA, Hu X, DeGrado WF, Korendovych IV. Nat. Chem. 2014;6:303–309. doi: 10.1038/nchem.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL. Proc. Natl. Acad. Sci. USA. 2012;109:21234–21239. doi: 10.1073/pnas.1212893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Faiella M, Andreozzi C, de Rosales RTM, Pavone V, Maglio O, Nastri F, DeGrado WF, Lombardi A. Nat. Chem. Bio. 2009;5:882–884. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaplan J, DeGrado WF. Proc. Natl. Acad. Sci. USA. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, Kulp DW, Butch SE, Calhoun JR, Szyperski T, Solomon EI, DeGrado WF. Nat. Chem. 2012;4:900–906. doi: 10.1038/nchem.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Richardson JS. Adv. Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- [11].The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; [Google Scholar]
- [12].Walsh STR, Cheng H, Bryson JW, Roder H, DeGrado WF. Proc. Natl. Acad. Sci. USA. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. Protein Sci. 1998;7:1404–1414. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Walsh ST, Sukharev VI, Betz SF, Vekshin NL, DeGrado WF. J. Mol. Biol. 2001;305:361–373. doi: 10.1006/jmbi.2000.4184. [DOI] [PubMed] [Google Scholar]
- [15].Chakraborty S, Kravitz JY, Thulstrup PW, Hemmingsen L, DeGrado WF, Pecoraro VL. Angew. Chem. Int. Ed. Engl. 2011;50:2049–2053. doi: 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hunt JA, Fierke CA. J. Biol. Chem. 1997;272:20364–20372. doi: 10.1074/jbc.272.33.20364. [DOI] [PubMed] [Google Scholar]
- [17].Song H, Wilson DL, Farquhar ER, Lewis EA, Emerson JP. Inorg. Chem. 2012;51:11098–11105. doi: 10.1021/ic301645j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zastrow ML, Pecoraro VL. J. Am. Chem. Soc. 2013;135:5895–5903. doi: 10.1021/ja401537t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Khalifah RG. J. Biol. Chem. 1971;246:2561–2573. [PubMed] [Google Scholar]
- [20].Fierke CA, Calderone TL, Krebs JF. Biochemistry. 1991;30:11054–11063. doi: 10.1021/bi00110a007. [DOI] [PubMed] [Google Scholar]
- [21].Woolley P. Nature. 1975;258:677–682. doi: 10.1038/258677a0. [DOI] [PubMed] [Google Scholar]
- [22].Huguet J, Brown RS. J. Am. Chem. Soc. 1980;102:7571–7572. [Google Scholar]
- [23].Brown RS, Curtis NJ, Huguet J. J. Am. Chem. Soc. 1981;103:6953–6959. [Google Scholar]
- [24].Brown RS, Salmon D, Curtis NJ, Kusuma S. J. Am. Chem. Soc. 1982;104:3188–3194. [Google Scholar]
- [25].Slebocka-Tilk H, Cocho JL, Frakman Z, Brown RS. J. Am. Chem. Soc. 1984;106:2421–2431. [Google Scholar]
- [26].Zhang X, van Eldik R, Koike T, Kimura E. Inorg. Chem. 1993;32:5749–5755. [Google Scholar]
- [27].Zhang X, van Eldik R. Inorg. Chem. 1995;34:5606–5614. [Google Scholar]
- [28].Nakata K, Shimomura N, Shiina N, Izumi M, Ichikawa K, Shiro M. J. Inorg. Biochem. 2002;89:255–266. doi: 10.1016/s0162-0134(01)00419-6. [DOI] [PubMed] [Google Scholar]
- [29].Jackman JE, Merz KM, Fierke CA. Biochemistry. 1996;35:16421–16428. doi: 10.1021/bi961786+. [DOI] [PubMed] [Google Scholar]
- [30].Jewell DA, Tu C, Paranawithana SR, Tanhauser SM, LoGrasso PV, Laipis PJ, Silverman DN. Biochemistry. 1991;30:1484–1490. doi: 10.1021/bi00220a006. [DOI] [PubMed] [Google Scholar]
- [31].Looney A, Han R, McNeill K, Parkin G. J. Am. Chem. Soc. 1993;115:4690–4697. [Google Scholar]
- [32].Nakata K, Uddin MK, Ogawa K, Ichikawa K. Chem. Lett. 1997;26:991–992. [Google Scholar]
- [33].Yamaguchi S, Tokairin I, Wakita Y, Funahashi Y, Jitsukawa K, Masuda H. Chem. Lett. 2003;32:406–407. [Google Scholar]
- [34].Krebs JF, Ippolito JA, Christianson DW, Fierke CA. J. Biol. Chem. 1993;268:27458–27466. [PubMed] [Google Scholar]
- [35].Koziol L, Valdez CA, Baker SE, Lau EY, Floyd WC, III, Wong SE, Satcher JH, Jr., Lightstone FC, Aines RD. Inorg. Chem. 2012;51:6803–6812. doi: 10.1021/ic300526b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.