

Abstract
Biomaterial approaches for engineering orthopedic interfaces such as the tendon-bone junction (TBJ) are limited by a lack of understanding of how insoluble (microstructure, composition) and soluble regulators of stem cell fate work in concert to promote bioactivity and differentiation. One strategy for regenerating the interface is to design biomaterials containing spatially-graded structural properties sufficient to induce divergent mesenchymal stem cell (MSC) differentiation into multiple interface-specific phenotypes. This work explores the hypothesis that selective structural modification to a 3D collagen-glycosaminoglycan (CG) scaffold combined with biochemical supplementation can drive human bone marrow-derived MSC differentiation down tenogenic, osteogenic, and chondrogenic lineages. Tenogenic differentiation is enhanced in geometrically anisotropic scaffolds versus a standard isotropic control. Notably, blebbistatin treatment abrogates this microstructurally-driven effect. Further, enhanced osteogenic differentiation and new mineral synthesis is achieved by incorporation of a calcium phosphate mineral phase within the CG scaffold along with the use of osteogenic induction media. Finally, chondrogenic differentiation is optimally driven by combining chondrogenic induction media with a reduced density scaffold that promotes increased cellular condensation, significantly higher expression of chondrogenic genes, and increased GAG deposition. Together these data provide critical insight regarding design rules for elements of an integrated biomaterial platform for orthopedic interface regeneration.
Keywords: mesenchymal stem cells, differentiation, tendon, collagen scaffolds, osteotendinous junction
1. Introduction
Orthopedic junctions connect soft tissue and bone to promote joint stability and locomotion. However, these interfaces are mechanically heterogeneous and therefore common sites of injury due to the presence of stress concentrations.[1] One important class of orthopedic interface is the tendon-bone junction (TBJ). Tendons are non-mineralized tissues composed of highly aligned, anisotropic type I collagen fiber bundles that transfer muscle-generated force to the skeletal system to enable normal movement. Comparatively, bone is mineralized and more isotropic in its structural organization. Importantly, tendon and bone are connected by a narrow (100–1000 µm[2]) fibrocartilagenous interface rich in type II collagen and proteoglycans such as aggrecan that helps dissipate stress concentrations during mechanical loading.[3] This osteotendinous interface contains elegant gradations of extracellular matrix (ECM) proteins, structural alignment, mineral content, and growth factors to maintain joint patency.[4, 5] Unfortunately, TBJs such as the supraspinatus-humerus junction in the rotator cuff are still common injury sites. Current repair strategies poorly recapitulate the tendinous, cartilagenous, and osseous regions found in the native TBJ, resulting in suboptimal patient outcomes such as high rates of re-failure (in some cases > 90%).[6]
Current clinical treatments for many orthopedic interfaces such as the TBJ primarily focus on mechanical fixation of soft (tendon) and hard (bone) tissue rather than biological integration.[7] An attractive alternative strategy is to guide regeneration of the spatially-graded junction using a spatially-patterned biomaterial seeded with patient-derived progenitor cells (e.g., mesenchymal stem cells, MSCs). Here, we propose that spatially-graded instructive cues engineered into the biomaterial could promote spatially-graded stem cell lineage specification down tenogenic, chondrogenic, and osteogenic lineages respectively. Towards the design of such an instructive biomaterial, a wide range of studies have investigated the use of material biophysical properties to induce divergent MSC bioactivity and lineage specification. Seminal work from Engler et al. defined the role of substrate elasticity, without biomolecular perturbation, for driving MSCs down neurogenic, myogenic, and osteogenic lineages with increasing substrate stiffness.[8] More recent work has demonstrated that substrate geometry[9] and tethering[10] can have a profound influence on stem cell fate. While these studies were performed on planar substrates, it has been more difficult to translate these findings into design rules for 3D biomaterials. However, recent progress has been made in this arena towards understanding the roles of crosslinking, rigidity,[11] and degradation properties[12] in directing stem cell lineage, primarily using monolithic materials. In parallel, many approaches have used soluble cues in the form of induction media[13] or growth factor supplementation[14] to aid differentiation and regeneration. However, few approaches have considered the combined influence of both insoluble (mechanics, structural organization, composition) and soluble (growth factor, cytokine) cues on guiding MSC fate. The lack of understanding of how insoluble and soluble regulators of stem cell fate work in concert to promote differentiation, especially in the context of tendon tissue engineering, is a critical limiting factor to the development of improved TBJ repair strategies.
In this study we have evaluated the potential for integrating selective biophysical modification of a single collagen-glycosaminoglycan (CG) scaffold with biochemical signals to create a series of instructive biomaterials to guide separate tenogenic, osteogenic, and chondrogenic MSC differentiation. Such an effort precedes development of a single, integrated biomaterial to repair multi-tissue junctions such as the TBJ. The CG scaffold platform employed in this study possesses many advantageous properties for tissue engineering applications, including high porosity, natural ligands to support cell adhesion and bioactivity, and approval for use by various regulatory agencies.[15] As analogs of the native ECM, these materials have also served as platforms to quantitatively examine the impact of local biomaterial properties on a wide range of cell activities, notably cell adhesion,[16] migration,[17] and regenerative potential.[18] While previous efforts to drive MSC differentiation within CG scaffolds have focused on single-lineage osteogenic or chondrogenic differentiation,[19, 20] this study addresses the suitability of the CG scaffold platform for guiding MSC differentiation towards a series of osteotendinous junction phenotypes, focusing in particular on tenogenic differentiation. Unlike osteogenic or chondrogenic differentiation, there is no well-established induction media to guide tenogenic MSC differentiation. Previously described methods to induce tenogenesis, primarily using two-dimensional substrates, include co-culture with primary fibroblasts,[21] cell stretching through mechanical stimulation,[22, 23] and inducing cell alignment/elongation through the use of contact guidance cues.[24, 25] In this study we explored the ability to employ selective structural modifications to our standard CG scaffold, in conjunction with biomolecule stimulation, to bias MSC differentiation potential. Due to the selective nature of these modifications as well as our previous description of a fabrication method able to integrate multiple CG scaffold compartments into a single, spatially-graded biomaterial,[26] we believe this work is critical for informing design of CG scaffolds for a range of musculoskeletal tissue engineering applications.
2. Results
2.1 Selective modification of the scaffold microenvironment
Three structural modifications were made to a standard CG scaffold in order to explore the ability of scaffold microstructure/composition to promote tenogenic, osteogenic, or chondrogenic lineage specification (Table 1). The standard CG scaffold, used as a control throughout, has previously been shown to possess an isotropic (uniform) pore structure[27] capable of withstanding cell-mediated contraction.[28] Here we modified a single element of this base scaffold architecture in order to improve scaffold selectivity for promoting tendinous, osseous, and fibrocartilagenous differentiation. Tenogenic studies explored whether a geometrically anisotropic (aligned) scaffold could promote MSC tenogenic differentiation. We have previously identified that scaffold anisotropy could support proliferation and phenotypic stability of mature tenocytes while resisting cellular contraction, leading to this study to explore whether such an architectural modification could also promote MSC differentiation.[28, 29] Osteogenic experiments examined whether a mineralized CG (CGCaP) scaffold variant that has been shown to present a mechanically robust, hydroxyapatite-rich environment could promote osteogenesis.[30] Chondrogenic studies explored whether reducing the scaffold relative density (ρ*/ρs = 1 – % porosity), a modification previously shown to reduce scaffold mechanical properties and allow extensive cell-mediated contraction,[28, 31] could enhance MSC chondrogenic differentiation. Previous studies using CG scaffolds suggested reducing scaffold crosslink density could also increase cellular condensation and chondrogenesis,[32] so here we hypothesized that reducing scaffold relative density may lead to a similar pro-chondrogenic response.
Table 1. Study design with scaffold type and biomolecule supplementation combinations for each desired lineage.
Two scaffold types (six experimental groups total) were assayed for each lineage. CG and CGCaP scaffolds are sometimes referred to in the text as “nonmineralized” and “mineralized” respectively. Each lineage (tenogenic, chondrogenic, or osteogenic) included use of the standard scaffold group as well as a structurally-modified scaffold (anisotropic, low-density, or CGCaP, respectively). All variants were cultured in MSC growth media. Additionally, the structurally-modified scaffolds (and the standard scaffold in chondrogenic and osteogenic cases) were then cultured with differentiation media or growth media supplemented with growth factors as listed.
| Lineage | Biomolecule treatment |
Scaffold type, biomolecule dose | |
|---|---|---|---|
| Tenogenic | CG (standard) | CG (anisotropic) | |
| Growth media | Yes | Yes | |
| bFGF | 5 ng/mL | ||
| IGF-1 | 100 ng/mL | ||
| GDF-5 | 100 ng/mL | ||
| GDF-7 | 100 ng/mL | ||
| Chondrogenic | CG (standard) | CG (low density) | |
| Growth media | Yes | Yes | |
| Chondrogenic | Yes | Yes | |
| TGF-β1 | 5 ng/mL | ||
| TGF-β3 | 5 ng/mL | ||
| Osteogenic | CG (standard) | CGCaP (40 wt% mineral) | |
| Growth media | Yes | Yes | |
| Osteogenic | Yes | Yes | |
| BMP-2 | 50 ng/mL | ||
| BMP-7 | 50 ng/mL | ||
2.2 MSC metabolic activity in CG scaffolds
All scaffold variants supported sustained MSC metabolic activity over the 21 day culture period. Two-way ANOVA analyses revealed a significant effect of both culture time (p < 0.0001) and scaffold type (p < 0.0001) on MSC metabolic activity in the tenogenic, osteogenic, and chondrogenic differentiation experimental sets (Figure 1, S1).
Figure 1. MSC metabolic activity in CG scaffolds.

Metabolic activity was measured at days 7, 14, and 21 using alamarBlue® incubation on MSC-seeded scaffolds to drive A) tenogenic, B) osteogenic, and C) chondrogenic differentiation (n = 3). All results are normalized to MSC metabolic activity at day 0. *: significant difference at that time point compared to standard. ⋀: significant increase compared to previous time point. ⋁: significant decrease compared to previous time point.
2.2.1 MSC metabolic activity in tenogenic cultures
Standard (isotropic control) and anisotropic CG scaffolds both supported steadily increasing, but similar, levels of metabolic activity throughout the experiment (Figure 1(a)). All tenogenic groups showed significant increases in metabolic activity from day 7 to day 14 (p < 0.002) with the anisotropic as well as IGF-1, GDF-5, and GDF-7-supplemented anisotropic scaffolds having significantly higher activity than the standard control (p < 0.007) (Figure S1(a)). However, by day 21 these differences between groups had been eliminated. Surprisingly, the bFGF-supplemented anisotropic group displayed significantly lower metabolic activity than the other anisotropic scaffold groups as well as the standard group (p < 0.0001) (Figure S1(a)).
2.2.2 MSC metabolic activity in osteogenic cultures
Standard CG scaffolds consistently had significantly higher metabolic activity compared to selectively mineralized scaffold groups (Figure 1(b), S1(b)). Osteogenic induction media led to significantly increased MSC metabolic activity in mineralized scaffolds at all three time points (p < 0.0001). While only the two non-mineralized groups showed significant increases in metabolic activity from day 7 to day 14 (p < 0.04), all experimental groups displayed significantly increased metabolic activity on day 21 compared to day 14 (p < 0.03) (Figure S1(b)).
2.2.3 MSC metabolic activity in chondrogenic cultures
The chondrogenic media supplemented groups showed significantly higher metabolic activity than the standard control at day 7 (p < 0.04) (Figures 1(c)). However, on day 14 the low density chondrogenic as well as the low density TGF-β1 and TGF-β3-supplemented groups displayed significantly lowered metabolic activity compared to day 7 (p < 0.04) (Figure S1(c)). By day 21, all groups had significantly lower metabolic activity than the standard group (p < 0.003).
2.3 Effect of scaffold anisotropy on tenogenic gene expression profiles
The expression of the tenogenic genes scleraxis (SCXB) and tenascin-C (TNC) was quantified at days 7, 14, and 21 of culture (Figure 2). There was a significant effect of both culture time (p < 0.0001) and scaffold treatment (type, growth factor supplementation; p = 0.01) on SCXB expression. While no statistically significant differences in SCXB expression were observed at day 7, anisotropic scaffolds were sufficient to induce elevated SCXB expression levels at both days 14 and 21 compared to the standard scaffolds (p < 0.03) (Figure 2(a)). Although bFGF and GDF-7-supplemented anisotropic scaffolds also significantly up-regulated SCXB at days 14 and 21 (p < 0.03) (Figure 2(a), S2(a)), there was no apparent synergistic effect of scaffold anisotropy and growth factor supplementation on SCXB expression. However, while there was no significant effect of scaffold anisotropy alone on TNC expression (p = 0.52), culture time did have a significant effect (p < 0.0001). Interestingly, GDF-5 and GDF-7-supplemented anisotropic scaffolds showed late up-regulation of TNC between days 14 and 21 (p < 0.04) (Figure 2(b)).
Figure 2. MSC tenogenic gene expression in response to scaffold anisotropy and soluble factor supplementation.

Expression of tenogenic genes A) SCXB and B) TNC was measured at days 7, 14, and 21 (n = 3). Scaffold anisotropy significantly improved MSC SCXB expression. While GDF supplementation did not improve SCXB expression, it is elicited significant up-regulation of TNC at day 21 in anisotropic scaffolds. *: significant difference at that time point compared to standard. ⋀: significant up-regulation compared to previous time point.
Type I collagen (COL1A1) expression was also quantified for all experimental groups (Figure S2(c)), with two-way ANOVA showing a significant effect of both culture time and scaffold type (p < 0.0001). Notably, COL1A1 expression was initially up-regulated in the IGF-1, GDF-5, and GDF-7-supplemented anisotropic groups at day 7 (p < 0.002), but down-regulated in the bFGF supplemented anisotropic group (p = 0.01), compared to the standard control scaffold. Following down-regulation in the IGF-1, GDF-5, and GDF-7-supplemented anisotropic groups at day 14 (p < 0.005) there were no significant differences between any experimental groups at later time points.
2.4 MSC morphology and expression of tenogenic genes and proteins in response to blebbistatin
While initially unclear how scaffold anisotropy may impact MSC early tenogenic markers, we examined canonical mechanotransduction pathways. Based on previous work in our lab that suggested the importance of scaffold structural anisotropy for supporting tenocyte alignment and phenotypic maintenance[28, 29] as well as the SCXB expression results reported here, we first explored whether scaffold anisotropy impacted MSC cytoskeletal organization. We examined MSC response to scaffold anisotropy in the presence of blebbistatin, a myosin II inhibitor, for 7 days. Confocal microscopy revealed a drastic reduction in MSC elongation and spreading within CG scaffolds when treated with blebbistatin (Figure 3(a–c)). Blebbistatin-treated MSC-seeded anisotropic scaffolds also showed a significant increase in cell shape index (increased cell roundness) compared to non-treated isotropic and anisotropic scaffolds (p < 0.0001) (Figure 3(d)). Importantly, gene expression analyses showed trends towards down-regulation of COL1A1, SCXB, and TNC expression in anisotropic scaffolds in response to blebbistatin treatment (Figure 3(e)).
Figure 3. MSC cytoskeletal organization and gene expression in response to blebbistatin.

Confocal micrographs of MSC-seeded anisotropic CG scaffolds reveal the influence of blebbistatin treatment (50 µM) on actin organization. A) MSCs stretch and elongate when cultured without blebbistatin (−) in anisotropic scaffolds. B) However, they become less spread and more rounded in response to blebbistatin (+). Green channel: actin (Alexa Fluor® 488 phalloidin). Blue channel: nuclei (DAPI). Scale bar: 50 µm. C) Cell shape index results show significantly higher (*) circularity (reduced spreading) in MSCs treated with blebbistatin compared to non-treated groups. D) Expression of tenogenic genes COL1A1, SCXB, and TNC was measured at day 7 (n = 3).
We subsequently examined activation of the RhoA/ROCK, mitogen-activated protein kinase (MAPK), and canonical Wnt signaling pathways in response to scaffold anisotropy and blebbistatin treatment by measuring protein levels of ROCK1, pERK1/2, and β-catenin respectively (Figure 4). Notably, anisotropic scaffolds displayed significantly higher levels of ROCK1 compared to standard CG scaffolds, and this effect was subsequently abrogated with blebbistatin treatment (p < 0.05). Further, blebbistatin treatment also led to significant down-regulation of pERK1/2 expression (p = 0.03), while neither scaffold anisotropy nor blebbistatin treatment were observed to impact β-catenin expression.
Figure 4. Protein expression in standard versus anisotropic scaffolds with and without blebbistatin treatment.

A) Quantified protein levels of ROCK1, pERK1/2, and β-catenin. ROCK1 levels are significantly higher in anisotropic scaffolds compared to other groups. B) Western blots of ROCK1, pERK1/2, β-catenin, and β-actin (loading control) proteins. *: significantly higher than all other experimental groups.
2.5 Osteogenic gene expression and histology
Significant effects of both culture time and scaffold mineral content/biomolecule supplementation were found for the expression of all three osteogenic genes examined (ALP, RUNX2, OCN, p < 0.05) (Figure 5, S3). Notably, ALP expression was significantly up-regulated at all time points for both the standard (non-mineralized) and mineralized scaffold groups cultured in osteogenic induction media (p < 0.02) (Figure 5(a), S3(a)). Although the standard scaffold plus osteogenic media group showed higher ALP expression than the mineralized osteogenic group at day 7 by about a factor of 3, the mineralized osteogenic group displayed significant ALP up-regulation from day 7 to day 14 and day 14 to day 21 (p < 0.05), finishing with an ALP expression level about twice that of the standard osteogenic group.
Figure 5. MSC osteogenic gene expression in response to selective scaffold mineralization and soluble factor supplementation.

Day 21 expression of osteogenic genes A) ALP, B) RUNX2, and C) OCN was measured (n = 3) with osteogenic induction media promoting significantly elevated expression levels. *: significant difference compared to standard.
Similarly, expression levels of osteogenic marker RUNX2 were significantly elevated in both of the osteogenic induction groups at day 21 (p < 0.003) (Figure 5(b)). Although the standard scaffold group had higher expression than its mineralized counterpart at day 7 (Figure S3(b)), this trend was again reversed by day 21. The osteogenic marker OCN was initially (day 7) up-regulated in the non-supplemented and BMP-2-supplemented mineralized groups compared to the standard (non-mineralized) scaffolds (p < 0.009) (Figure S3(c)). While BMP-2 and BMP-7 supplementation elicited a significant up-regulation of OCN in mineralized scaffolds compared to the standard control at day 14 (p < 0.0003), this trend had disappeared by day 21 (p > 0.05). However, by the final time point only the osteogenic media-supplemented standard scaffold displayed significant OCN up-regulation compared to the non-supplemented standard scaffold (p = 0.02) (Figure 5(c)).
Histological sections were taken after day 21 to evaluate MSC distribution and mineralization via H&E and Alizarin red staining respectively (Figure 6, S5(a)). While there did not appear to be major differences in MSC infiltration based on H&E sections, Alizarin red staining revealed notable differences between groups. Significant mineralization was observed in all groups with the combination of osteogenic media and a mineralized scaffold leading to the greatest level of mineralization (Figure 6(d)). BMP-2 supplementation did not appear to improve mineral deposition over the non-supplemented mineralized group (Figure 6(c), S5(a)).
Figure 6. Alizarin red and H&E analysis of mineral synthesis and cell distribution.

Representative histology sections from day 21 time point reveal the highest mineral content in the Mineralized scaffold + Osteogenic group, with similar cellular distribution observed in all groups. A) Standard, B) Standard + Osteogenic, C) Mineralized, D) Mineralized + Osteogenic. Scale bars: 500 µm (4× objective images on left), 100 µm (20× objective images on right).
2.6 Chondrogenic gene expression and histology
Finally, we evaluated the combined influence of scaffold relative density and biomolecule supplementation on MSC chondrogenic differentiation. Expression levels for the fibrocartilage marker type II collagen (COL2A1) were significantly elevated at day 21 by the combination of a reduced relative density scaffold and chondrogenic induction media (p < 0.0001) (Figure 7(a)). SOX9 expression was significantly affected by culture time (p < 0.0001), but not scaffold type (p = 0.41). While SOX9 was up-regulated in the standard chondrogenic group at the intermediate time point (day 14, p = 0.004), the low density scaffold plus chondrogenic media group showed significant SOX9 up-regulation at the final time point (day 21, p = 0.02), leading to higher expression than the other experimental groups (Figure 7(b), S4(b)). The gene encoding for the proteoglycan aggrecan (ACAN) was significantly down-regulated in the low density TGF-β-supplemented groups at day 7 (p < 0.03) (Figure S4(c)). Notably, the chondrogenic induction groups had significantly higher ACAN transcript levels than the standard scaffold control at the day 14 time point (p < 0.03). However, from day 14 to day 21 ACAN was significantly down-regulated in the standard (higher density) scaffold plus chondrogenic media group (p < 0.0001). Meanwhile, the low density scaffolds with chondrogenic media maintained significantly higher expression compared to the standard control (p = 0.02) (Figure 7(c)).
Figure 7. MSC chondrogenic gene expression in response to reducing scaffold relative density as well as soluble factor supplementation.
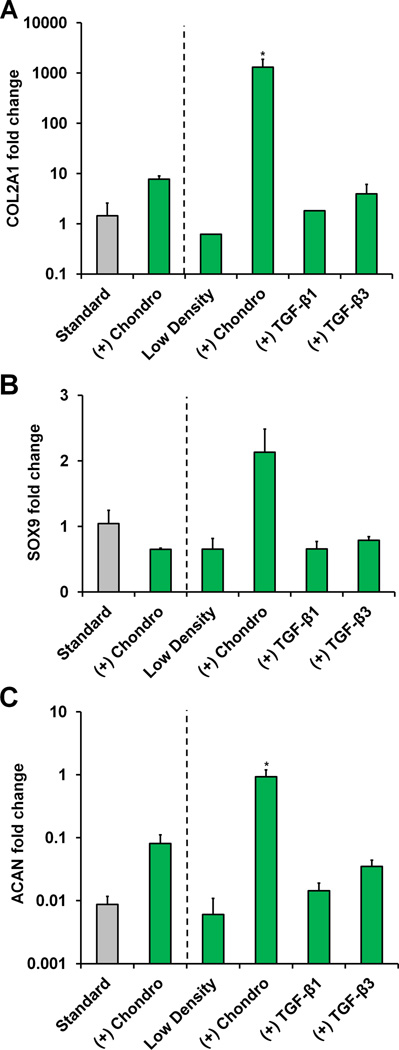
Day 21 expression of chondrogenic genes A) COL2A1, B) SOX9, and C) ACAN was measured (n = 3) with the combination of a low density scaffold and chondrogenic induction media promoting significant up-regulation of COL2A1 and ACAN. *: significant difference compared to standard.
Scaffold-mediated induction of pro-chondrogenic phenotype was further evaluated by staining histology sections with H&E and Alcian blue to assess MSC distribution and GAG deposition respectively (Figure 8, S5(b)). In agreement with gene expression profiles, the low density scaffolds supplemented with chondrogenic media or TGF-β3 showed significant scaffold contraction and cellular condensation as demonstrated by H&E staining (Figure 8(d), S5(b)). Further, Alcian blue staining appeared to be strongest in the low density scaffold with the addition of chondrogenic media (Figure 8(d)) while no major differences in GAG content were apparent in any other experimental groups.
Figure 8. Alcian blue and H&E analysis of GAG synthesis and cell distribution.

Representative histology sections from day 21 time point reveal higher GAG content and cellular condensation in the Low Density + Chondrogenic group. A) Standard, B) Standard + Chondrogenic, C) Low Density, D) Low Density + Chondrogenic. Scale bars: 500 µm (4× objective images on left), 100 µm (20× objective images on right).
3. Discussion
This study aimed to identify whether select alterations to the structural properties of a model 3D CG scaffold under development for osteotendinous repair, in conjunction with biomolecule supplementation, could impact multi-lineage MSC differentiation potential. Here we used a series of monolithic scaffolds tailored for the osseous, tendinous, and fibrocartilagenous compartments across the osteotendinous junction in order to screen a large experimental space. This approach was designed to identify relevant instructive cues able to selectively enhance discrete osteotendinous differentiation paths. Since spatially-selective presentation of differentiation media is not plausible we also explored the use of single factors (e.g., GDF-5, BMP-2, TGF-β1) that could potentially be tethered to discrete scaffold compartments. Moving forward, we will leverage strategies previously developed in our laboratory that enable spatially-defined control of scaffold structural properties[26] (via layering of precursor scaffold suspensions prior to fabrication) and biochemical supplementation[27] (via photolithography) in order to integrate the findings outlined here into a spatially-graded material for TBJ repair applications.
While tendon injuries represent an important class of orthopedic trauma, comprehensive evaluation of strategies to drive MSC tenogenic differentiation have not been explored in the context of designing a 3D biomaterial implant. We first hypothesized that a combination of geometric anisotropy and tenogenic growth factor supplementation would improve tenogenic MSC differentiation. Previous work in our lab has identified scaffold pore anisotropy as a key regulator of tenocyte alignment[29] and resultant maintenance of tenogenic phenotypic stability.[28] Other studies suggested that inducing cell alignment and elongation via two-dimensional substrate contact guidance cues can impact tenocyte phenotype[24] and drive MSC fibroblastic differentiation.[23, 33] Growth factors such as bFGF, IGF-1, GDF-5, and GDF-7 can also influence the tenogenic response.[34, 35, 36] Therefore, our aim was to evaluate whether 3D scaffold anisotropy increased MSC tenogenic specification, and further whether selective addition of individual growth factors improved this response. We chose factor dosages based on previous work in our lab with tenocytes[35] as well as from the literature,[36, 37] although we acknowledge subsequent studies may optimize factor dosing.
Notably, scaffold anisotropy supported significant up-regulation of the tenogenic phenotype marker scleraxis (Figure 2(a)), suggesting aligned contact guidance cues in a 3D microenvironment could enhance tenogenic MSC differentiation. Although anisotropic scaffolds supplemented with growth factors (bFGF and GDF-7) also supported SCXB up-regulation, there did not appear to be a synergistic effect of anisotropy and growth factor supplementation (Figure 2(a), S2(a)). In contrast, growth factors did not seem to affect TNC expression until day 21 where expression in the GDF-5 and GDF-7 groups was up-regulated (Figure 2(b)). These results indicate that for some tendon markers (SCXB) cell alignment/anisotropy may be more important while for others (TNC) growth factor cues may be the critical instructive signal. Moving forward, it may also be important to explore gene targets related to alternative lineages (e.g., myogenic differentiation).
While our gene expression results indicated that scaffold anisotropy plays a role in early tenogenic differentiation, it remained unclear what pathways MSCs might utilize to sense scaffold anisotropy given that scaffold pores were larger (> 150 µm) than the MSCs themselves. Given the structural modification of the scaffold, and known impact of scaffold strut organization on cell activity within CG scaffolds,[38] we hypothesized that canonical mechanotransduction pathways may play a role. Therefore we designed a series of experiments where MSC-seeded anisotropic scaffolds were exposed to the myosin II inhibitor blebbistatin to remove the capability of MSCs to stretch out and “feel” the microstructure of their environment. Indeed, blebbistatin treatment had a significant effect on actin organization, with MSCs in anisotropic scaffolds remaining rounded (Figure 3(a–d)), as well as tenogenic specification, with MSCs showing downward trends in COL1A1, SCXB, and TNC expression (Figure 3(e)).
Further, signaling pathways associated with RhoA/ROCK, MAPK, and canonical Wnt signaling were also impacted by scaffold pore anisotropy and blebbistatin treatment. Previously, RhoA/ROCK was found to be integral for stretch-induced fibroblastic MSC differentiation,[39] and increased ROCK activation was observed for cells on aligned versus non-aligned 2D substrates.[40] Further, ERK1/2 activation was previously shown to be critical for stretch-induced collagen synthesis in tendon fibroblasts.[41] Finally, while differential Wnt signaling has been more prominently linked to osteogenic/chondrogenic development,[42] it has also been shown to be involved in tendon morphogenesis,[43] with tendon ECM components such as biglycan modulating canonical Wnt activity.[44] However, few of these results have been translated to a fully 3D biomaterial construct. While there was no apparent effect of substrate anisotropy on ERK1/2 or canonical Wnt signaling observed here, ROCK1 expression was significantly higher in the anisotropic scaffold group compared to the isotropic control (Figure 4), replicating the trend previously observed on a 2D substrate.[40] Blebbistatin treatment abrogated ROCK1 up-regulation in the anisotropic group and reduced ERK1/2 activation. Taken together our results suggest that microstructural contact guidance cues presented by 3D anisotropic scaffolds can alter intracellular signaling paths in a similar manner to mechanical stimuli, already known to be an important regulator of tenogenic activity.[39, 45]
While primarily focused on examining pathways to increase CG scaffold-mediated tenogenic MSC differentiation, we also examined modifications to our standard CG scaffold to enhance osteogenic and chondrogenic MSC differentiation. While differentiation down these lineages is far better characterized than tenogenic differentiation, we were interested in exploring the combined influence of CG scaffold structural properties and biomolecule supplementation on MSC fate. We hypothesized that osteogenic differentiation would be enhanced by the combination of a scaffold incorporating a biomimetic calcium phosphate (CaP) phase and further supplementation with factors in the BMP family (BMP-2 and 7) or with standard osteogenic induction media. Mineralized CG scaffolds used in this study, in addition to providing a more mechanically robust microenvironment[46] that may drive osteogenic differentiation itself,[8] also display a biomimetic calcium phosphate phase that has been shown in a range of previous studies to improve osteogenic outcomes in terms of new matrix deposition[20, 47] and osteogenic gene expression.[48]
Osteogenic differentiation was tracked by measuring the expression of the osteogenic genes ALP, RUNX2, and OCN and through subsequent histological analysis. Osteogenic induction media had a greater effect on osteogenic gene expression than select BMP-2/7 supplementation, leading to significant up-regulation of all three genes by day 21 (Figure 5). While BMP supplementation led to ALP and OCN up-regulation at the intermediate time point, the positive effects of supplementation did not reach that of the osteogenic induction media (Figure S3). Incorporation of a mineral phase improved ALP and RUNX2 expression by day 21. Although ALP expression in the mineralized osteogenic group initially lagged behind that of its non-mineralized counterpart, likely due to reduced MSC metabolic activity in the more dense and less permeable[46] mineralized scaffold (Figure 1(b)), the mineralized osteogenic scaffold showed the highest degree of osteogenesis. By day 21 ALP expression was twice as high in the mineralized osteogenic group. Histological analysis confirmed that the combination of osteogenic induction media and the mineralized scaffold promoted osteogenesis, evidenced by increased staining for mineral via Alizarin red compared to other groups (Figure 6).
Finally, we hypothesized that MSC chondrogenic specification would be ideally achieved in CG scaffolds by lowering scaffold relative density in conjunction with soluble supplementation of TGF-β or with chondrogenic induction media. Increasing scaffold relative density and crosslinking density have both been shown to create structures able to resist cell-mediated contraction.[28, 32, 49] In particular, lowering the degree of CG scaffold crosslinking led to increased contraction, cellular condensation, and MSC chondrogenesis as measured by type II collagen and GAG synthesis.[32] Here we aimed to replicate this effect in a lower density scaffold, and possibly enhance it through the use of soluble biomolecular supplementation. This effort was also motivated by features of the native osteotendinous junction where there is reduced stiffness in a small part of the fibrocartilagenous transition between tendon and bone. This dip in stiffness has been hypothesized to be critical for alleviating stress concentrations and maintaining joint patency.[4, 50]
Lower density scaffolds consistently displayed lower metabolic activity than the standard scaffold group after day 7 (Figure 1(c)), most likely due to a combination of extensive MSC-mediated contraction, reduced metabolite diffusion, and differentiation into less metabolically-active cells (Figure 8). Chondrogenic phenotype was initially tracked through the expression of chondrogenic genes COL2A1, SOX9, and ACAN (Figure 7, S4). As with osteogenic differentiation, the chondrogenic induction media had the most profound effect on lineage specification while soluble TGF-β1 or 3 alone had little effect. However, the combination of the low density scaffold with chondrogenic induction media clearly led to a significantly enhanced chondrogenic transcriptomic profile with significantly increased expression of all three markers tested after day 21. Histology also revealed strong staining for GAG deposition in the low density chondrogenic group compared to other experimental groups (Figure 8). Additionally, H&E-stained sections showed extensive pore contraction and cellular condensation in the lower density groups, which likely promoted chondrogenesis (Figure 8).
4. Conclusions
This work explored simple modifications to a standard CG scaffold to investigate the roles of scaffold structure (geometric anisotropy, mineral content, relative density) in combination with biomolecule supplementation on driving tenogenic, osteogenic, and chondrogenic MSC differentiation respectively. The importance of scaffold microstructural anisotropy for enhancing tenogenic differentiation was shown by increased expression of the phenotype marker SCXB as well as increased ROCK1 levels in anisotropic compared to isotropic scaffolds. Notably, small molecule cytoskeletal inhibition abrogated these effects. MSC osteogenic differentiation was enhanced by osteogenic induction media, although the inclusion of a mineral phase in the scaffold led to elevated ALP and RUNX2 expression as well as increased mineralization after 21 days. Chondrogenic differentiation was enhanced by combining classic chondrogenic induction media with a low density scaffold that enhanced cellular condensation and led to significant up-regulation of COL2A1 and ACAN as well as increased GAG production. Together, these data provide insight into critical scaffold instructive cues that should inform the development of CG scaffolds for a variety of single and multi-tissue musculoskeletal tissue engineering applications. In particular, these data suggest a scaffold with an anisotropic tendon region and a mineralized bone region, joined with lower density interface and coupled with spatially-graded biomolecular cues, may be optimal for osteotendinous repair.
5. Experimental Section
*All reagents purchased from Sigma-Aldrich (St Louis, MO) unless otherwise specified.
Precursor suspension preparation
Low and high density CG suspensions (0.5 and 1.5 collagen w/v% respectively) were prepared by homogenizing type I microfibrillar collagen and chondroitin sulfate in 0.05 M acetic acid as previously described.[51] CGCaP suspension was prepared via triple co-precipitation of type I collagen (1.9 w/v%) and chondroitin sulfate as before with the addition of calcium salts (calcium hydroxide, calcium nitrate tetrahydrate) in phosphoric acid to create 40 wt% mineralized scaffolds.[30]
Scaffold fabrication via freeze-drying
All scaffolds were fabricated using a VirTis freeze-dryer (Gardiner, NY). All studies used an isotropic CG scaffold (1.5 w/v%) as a standard control. Tenogenic differentiation studies used geometrically-anisotropic CG scaffolds (1.5 w/v%) fabricated via directional solidification at a freezing temperature of −10°C in a thermally-mismatched mold.[28, 29] Isotropic low density (0.5 w/v%) CG scaffolds were used in chondrogenesis experiments while mineralized CGCaP scaffolds were examined in osteogenesis studies. Isotropic low density and high density CG scaffolds as well as mineralized CGCaP scaffolds were fabricated using a constant cooling method at a final freezing temperature of −10°C.[51] The freeze-drying process resulted in dry, macroporous scaffolds.[29, 30, 51]
Scaffold hydration and crosslinking
Dry CG scaffolds underwent dehydrothermal crosslinking in a vacuum oven (Welch, Niles, IL) at 105°C for 24 h. All scaffolds were then sterilized in ethanol for 1 h, hydrated in PBS overnight, and crosslinked for 1.5 h in a solution of 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (NHS) at a molar ratio of 5:2:1 EDC:NHS:COOH where COOH represents the amount of collagen in the scaffold.[31, 52]
MSC culture in scaffolds
Human bone marrow-derived mesenchymal stem cells (MSCs) were purchased from Lonza (Walkersville, MD) and cultured in complete growth media as supplied by the manufacturer. Cells were used at passage 6 for all experiments. 7.5 × 104 MSCs were seeded onto scaffold discs (6 mm diameter, 3 mm thickness) using a previously validated static seeding method.[16] All groups were cultured in complete growth media (Lonza) except as noted in Table 1. All soluble factors were human recombinant proteins from ProSpec (Israel). Proteins were diluted to experimental concentrations (Table 1) in complete media at doses selected based on the literature.[35, 36, 37] Blebbistatin (concentration: 50 µM[8]) was used for experiments examining the linkage between scaffold anisotropy and MSC tenogenesis. Scaffolds were cultured at 37°C and 5% CO2 and fed twice a week for all experiments.
Quantification of MSC metabolic activity
MSC mitochondrial metabolic activity on scaffolds was quantified using an alamarBlue® fluorescent assay (Invitrogen, Carlsbad, CA) previously used to determine the metabolic health of cells grown on CG scaffolds.[53] Briefly, MSC-seeded scaffolds were incubated in alamarBlue® solution for 2.5 h, where viable cells continuously reduce the alamarBlue® dye to a fluorescent byproduct (resorufin). Fluorescence was measured (excitation: 540 nm, emission: 580 nm) on a fluorescent spectrophotometer (Tecan, Switzerland).
Confocal microscopy
MSC-seeded scaffolds were fixed in 10% neutral buffered formalin and stored at 4°C until staining. Following permeabilization in 0.1 % Triton ×100 in PBS for 10 min, MSC were stained with Alexa Fluor® 488 phalloidin (F-actin, Invitrogen) for 20 min and DAPI (nuclei, Invitrogen) for 5 min. Scaffolds were imaged with a Zeiss 710 multiphoton confocal microscope (10× objective) equipped with a Spectraphysics Mai-Tai Ti-Sapphire laser. Cell shape index (CSI) was quantified for at least 50 cells per experimental group using ImageJ.[54]
RNA isolation, reverse transcription, and real-time PCR
Total RNA was isolated from cell-seeded scaffolds using an RNeasy Plant Mini kit (Qiagen, Valencia, CA).[28, 55] RNA was reverse transcribed to cDNA using the QuantiTect Reverse Transcription kit (Qiagen) in a Bio-Rad S1000 thermal cycler. Primer sets for PCR reactions were mined from the literature[56, 57] (Table S1) and synthesized by Integrated DNA Technologies (Coralville, IA) with the exception of the scleraxis (SCXB) primer set, which was purchased from Qiagen. Real-time PCR reactions were executed in triplicate using SYBR green chemistry (QuantiTect SYBR Green PCR kit, Qiagen) in an Applied Biosystems 7900HT Fast Real-Time PCR system (Applied Biosystems, Carlsbad, CA). Data were analyzed using Sequence Detection Systems software v2.4 (Applied Biosystems) via the delta-delta Ct method with GAPDH serving as a housekeeping gene. Results were expressed as fold changes normalized to the expression levels of MSCs cultured in standard scaffolds at day 7.
Protein isolation and Western blotting
Protein lysates were obtained by immersing scaffolds in RIPA buffer supplemented with protease and phosphatase inhibitors for 30 min on ice. Lysates were separated on 10% polyacrylamide gels via electrophoresis and transferred to nitrocellulose membranes (Fisher Scientific, Pittsburgh, PA) using standard techniques. Antibodies for Rho-associated protein kinase 1 (ROCK1), β-catenin, double phosphorylated extracellular signal-regulated protein kinase 1/2 (pERK-1/2), and β-actin were purchased from Cell Signaling Technology (Beverly, MA) and used at 1:2000 dilution in 5% non-fat milk. Horseradish peroxide (HRP)-conjugated secondary antibodies (Cell Signaling) were added following primary antibody incubation. Signal was developed using SuperSignal West Pico Chemiluminescent Substrate solutions (Thermo Scientific, Rockford, IL) and visualized on an Image Quant LAS 4010 (GE Healthcare, Pittsburgh, PA). Band intensities were quantified using ImageJ and expression levels were normalized to β-actin expression.
Histology
Scaffolds from the osteogenic and chondrogenic experimental sets were fixed in 10% neutral buffered formalin for histological analysis after 21 days in culture. Scaffold discs were embedded in paraffin wax, serially cut into 5 µm sections, and mounted on microscope slides. Slides were deparaffinized and stained with either hematoxylin and eosin (H&E) to assess cellular distribution, Alizarin red to evaluate mineralization, or Alcian blue to visualize GAG content.
Statistical analysis
Two-way analysis of variance (ANOVA, independent variables time and scaffold/biomolecule treatment) was performed on metabolic activity and gene expression data sets followed by Tukey-HSD post-hoc tests. Significance was set at p < 0.05. Metabolic activity and gene expression experiments used at least n = 3 scaffolds per group while Western blotting and histological analyses used n = 2 scaffold per group. Error is reported in figures as the standard error of the mean.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Daniel Weisgerber (MSE, UIUC) for fabrication of mineralized CG scaffolds, Donna Epps (IGB, UIUC) for assistance with histology, William Grier and Ji Sun Choi (CHBE, UIUC) for assistance with Western blots, Dr. Mayandi Sivaguru (IGB, UIUC) for assistance with confocal microscopy, and the IGB Core Facilities for assistance with real-time PCR. This material is based upon work supported by the National Science Foundation under Grant No. 1105300. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers R21 AR063331 and R03 AR062811. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We are also grateful for the funding for this study provided by the Chemistry-Biology Interface Training Program NIH NIGMS T32GM070421 (SRC) as well as the Chemical and Biomolecular Engineering Dept. (BAH), and the Institute for Genomic Biology (BAH) at the University of Illinois at Urbana-Champaign.
Footnotes
Supporting Information
Supporting Information is available online from the Wiley Online Library or from the author.
Contributor Information
Steven R. Caliari, 104 Roger Adams Laboratory, 600 S. Mathews St, Urbana, IL, 61801, USA
Brendan A.C. Harley, Email: bharley@illinois.edu, 110 Roger Adams Laboratory, 600 S. Mathews St, Urbana, IL, 61801, USA.
References
- 1.Thomopoulos S, Marquez JP, Weinberger B, Birman V, Genin GM. J Biomech. 2006;39:1842. doi: 10.1016/j.jbiomech.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 2.Moffat KL, Sun WH, Pena PE, Chahine NO, Doty SB, Ateshian GA, Hung CT, Lu HH. Proc Natl Acad Sci U S A. 2008;105:7947. doi: 10.1073/pnas.0712150105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galatz L, Rothermich S, VanderPloeg K, Petersen B, Sandell L, Thomopoulos S. J Orthop Res. 2007;25:1621. doi: 10.1002/jor.20441. [DOI] [PubMed] [Google Scholar]
- 4.Genin GM, Kent A, Birman V, Wopenka B, Pasteris JD, Marquez PJ, Thomopoulos S. Biophys J. 2009;97:976. doi: 10.1016/j.bpj.2009.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wopenka B, Kent A, Pasteris JD, Yoon Y, Thomopoulos S. Appl Spectrosc. 2008;62:1285. doi: 10.1366/000370208786822179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Millar NL, Wu X, Tantau R, Silverstone E, Murrell GA. Clin Orthop Relat Res. 2009;467:966. doi: 10.1007/s11999-009-0706-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; Boileau P, Brassart N, Watkinson DJ, Carles M, Hatzidakis AM, Krishnan SG. J Bone Joint Surg Am. 2005;87:1229. doi: 10.2106/JBJS.D.02035. [DOI] [PubMed] [Google Scholar]
- 7.Moffat KL, Wang IN, Rodeo SA, Lu HH. Clin Sports Med. 2009;28:157. doi: 10.1016/j.csm.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 9.Kilian KA, Bugarija B, Lahn BT, Mrksich M. Proc Natl Acad Sci U S A. 2010;107:4872. doi: 10.1073/pnas.0903269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trappmann B, Gautrot JE, Connelly JT, Strange DG, Li Y, Oyen ML, Cohen Stuart MA, Boehm H, Li B, Vogel V, Spatz JP, Watt FM, Huck WT. Nat Mater. 2012;11:642. doi: 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- 11.Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ. Nat Mater. 2010;9:518. doi: 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA. Nat Mater. 2013;12:458. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao JZ, Dennis JE, Solchaga LA, Awadallah AS, Goldberg VM, Caplan AI. Tissue Eng. 2001;7:363. doi: 10.1089/10763270152436427. [DOI] [PubMed] [Google Scholar]; Sheehy EJ, Vinardell T, Buckley CT, Kelly DJ. Acta Biomater. 2013;9:5484. doi: 10.1016/j.actbio.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Yilgor P, Tuzlakoglu K, Reis RL, Hasirci N, Hasirci V. Biomaterials. 2009;30:3551. doi: 10.1016/j.biomaterials.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 15.Harley BAC, Gibson LJ. Chem Eng J. 2008;137:102. [Google Scholar]
- 16.O'Brien FJ, Harley BA, Yannas IV, Gibson LJ. Biomaterials. 2005;26:433. doi: 10.1016/j.biomaterials.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 17.Harley BAC, Kim HD, Zaman MH, Yannas IV, Lauffenburger DA, Gibson LJ. Biophys. J. 2008;95:4013. doi: 10.1529/biophysj.107.122598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harley BA, Spilker MH, Wu JW, Asano K, Hsu HP, Spector M, Yannas IV. Cells Tissues Organs. 2004;176:153. doi: 10.1159/000075035. [DOI] [PubMed] [Google Scholar]; Yannas IV, Lee E, Orgill DP, Skrabut EM, Murphy GF. Proc Natl Acad Sci USA. 1989;86:933. doi: 10.1073/pnas.86.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy CM, Matsiko A, Haugh MG, Gleeson JP, O'Brien FJ. J Mech Behav Biomed Mater. 2012;11:53. doi: 10.1016/j.jmbbm.2011.11.009. [DOI] [PubMed] [Google Scholar]; Farrell E, O'Brien FJ, Doyle P, Fischer J, Yannas I, Harley BA, O'Connell B, Prendergast PJ, Campbell VA. Tissue Eng. 2006;12:459. doi: 10.1089/ten.2006.12.459. [DOI] [PubMed] [Google Scholar]
- 20.Curtin CM, Cunniffe GM, Lyons FG, Bessho K, Dickson GR, Duffy GP, O'Brien FJ. Adv Mater. 2012;24:749. doi: 10.1002/adma.201103828. [DOI] [PubMed] [Google Scholar]
- 21.Lovati AB, Corradetti B, Cremonesi F, Bizzaro D, Consiglio AL. Int J Artif Organs. 2012;35:996. doi: 10.5301/ijao.5000129. [DOI] [PubMed] [Google Scholar]; Canseco JA, Kojima K, Penvose AR, Ross JD, Obokata H, Gomoll AH, Vacanti CA. Tissue Eng Part A. 2012;18:2549. doi: 10.1089/ten.tea.2012.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juncosa-Melvin N, Shearn JT, Boivin GP, Gooch C, Galloway MT, West JR, Nirmalanandhan VS, Bradica G, Butler DL. Tissue Eng. 2006;12:2291. doi: 10.1089/ten.2006.12.2291. [DOI] [PubMed] [Google Scholar]
- 23.Subramony SD, Dargis BR, Castillo M, Azeloglu EU, Tracey MS, Su A, Lu HH. Biomaterials. 2013;34:1942. doi: 10.1016/j.biomaterials.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapoor A, Caporali EH, Kenis PJ, Stewart MC. Acta Biomater. 2010;6:2580. doi: 10.1016/j.actbio.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 25.Cheng X, Gurkan UA, Dehen CJ, Tate MP, Hillhouse HW, Simpson GJ, Akkus O. Biomaterials. 2008;29:3278. doi: 10.1016/j.biomaterials.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 26.Harley BA, Lynn AK, Wissner-Gross Z, Bonfield W, Yannas IV, Gibson LJ. J Biomed Mater Res A. 2010;92:1078. doi: 10.1002/jbm.a.32387. [DOI] [PubMed] [Google Scholar]
- 27.Martin TA, Caliari SR, Williford PD, Harley BA, Bailey RC. Biomaterials. 2011;32:3949. doi: 10.1016/j.biomaterials.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caliari SR, Weisgerber DW, Ramirez MA, Kelkhoff DO, Harley BAC. J Mech Behav Biomed Mater. 2012;11:27. doi: 10.1016/j.jmbbm.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caliari SR, Harley BAC. Biomaterials. 2011;32:5330. doi: 10.1016/j.biomaterials.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harley BA, Lynn AK, Wissner-Gross Z, Bonfield W, Yannas IV, Gibson LJ. J Biomed Mater Res A. 2010;92:1066. doi: 10.1002/jbm.a.32361. [DOI] [PubMed] [Google Scholar]
- 31.Harley BA, Leung JH, Silva EC, Gibson LJ. Acta Biomater. 2007;3:463. doi: 10.1016/j.actbio.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 32.Vickers SM, Gotterbarm T, Spector M. J. Orthop. Res. 2010;28:1184. doi: 10.1002/jor.21113. [DOI] [PubMed] [Google Scholar]
- 33.Yin Z, Chen X, Chen JL, Shen WL, Hieu Nguyen TM, Gao L, Ouyang HW. Biomaterials. 2010;31:2163. doi: 10.1016/j.biomaterials.2009.11.083. [DOI] [PubMed] [Google Scholar]
- 34.Molloy T, Wang Y, Murrell G. Sports Med. 2003;33:381. doi: 10.2165/00007256-200333050-00004. [DOI] [PubMed] [Google Scholar]; Shen H, Gelberman RH, Silva MJ, Sakiyama-Elbert SE, Thomopoulos S. PLoS ONE. 2013;8:e77613. doi: 10.1371/journal.pone.0077613. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hagerty P, Lee A, Calve S, Lee CA, Vidal M, Baar K. Biomaterials. 2012;33:6355. doi: 10.1016/j.biomaterials.2012.05.045. [DOI] [PubMed] [Google Scholar]; Date H, Furumatsu T, Sakoma Y, Yoshida A, Hayashi Y, Abe N, Ozaki T. J Orthop Res. 2010;28:225. doi: 10.1002/jor.20981. [DOI] [PubMed] [Google Scholar]
- 35.Caliari SR, Harley BA. Tissue Eng Part A. 2013;19:1100. doi: 10.1089/ten.tea.2012.0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomopoulos S, Harwood FL, Silva MJ, Amiel D, Gelberman RH. J Hand Surg Am. 2005;30A:441. doi: 10.1016/j.jhsa.2004.12.006. [DOI] [PubMed] [Google Scholar]; Costa MA, Wu C, Pham BV, Chong AKS, Pham HM, Chang J. Tissue Eng. 2006;12:1937. doi: 10.1089/ten.2006.12.1937. [DOI] [PubMed] [Google Scholar]
- 37.Park A, Hogan MV, Kesturu GS, James R, Balian G, Chhabra AB. Tissue Eng Part A. 2010;16:2941. doi: 10.1089/ten.tea.2009.0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harley BA, Kim HD, Zaman MH, Yannas IV, Lauffenburger DA, Gibson LJ. Biophys J. 2008;95:4013. doi: 10.1529/biophysj.107.122598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu B, Song G, Ju Y, Li X, Song Y, Watanabe S. Journal of Cellular Physiology. 2012;227:2722. doi: 10.1002/jcp.23016. [DOI] [PubMed] [Google Scholar]
- 40.Andalib MN, Lee JS, Ha L, Dzenis Y, Lim JY. Acta Biomater. 2013;9:7737. doi: 10.1016/j.actbio.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 41.Paxton JZ, Hagerty P, Andrick JJ, Baar K. Tissue Eng Pt A. 2012;18:277. doi: 10.1089/ten.tea.2011.0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Day TF, Guo XZ, Garrett-Beal L, Yang YZ. Developmental Cell. 2005;8:739. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 43.Hartmann C, Tabin CJ. Cell. 2001;104:341. doi: 10.1016/s0092-8674(01)00222-7. [DOI] [PubMed] [Google Scholar]; Guo XZ, Day TF, Jiang XY, Garrett-Beal L, Topol L, Yang YZ. Gene Dev. 2004;18:2404. doi: 10.1101/gad.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berendsen AD, Fisher LW, Kilts TM, Owens RT, Robey PG, Gutkind JS, Young MF. Proc Natl Acad Sci U S A. 2011;108:17022. doi: 10.1073/pnas.1110629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuo CK, Tuan RS. Tissue Eng Part A. 2008;14:1615. doi: 10.1089/ten.tea.2006.0415. [DOI] [PubMed] [Google Scholar]
- 46.Weisgerber DW, Kelkhoff DO, Caliari SR, Harley BA. J Mech Behav Biomed Mater. 2013;28C:26. doi: 10.1016/j.jmbbm.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang Y, Kim S, Khademhosseini A, Yang Y. Biomaterials. 2011;32:6119. doi: 10.1016/j.biomaterials.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muller P, Bulnheim U, Diener A, Luthen F, Teller M, Klinkenberg ED, Neumann HG, Nebe B, Liebold A, Steinhoff G, Rychly J. J Cell Mol Med. 2008;12:281. doi: 10.1111/j.1582-4934.2007.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Beck GR, Zerler B, Moran E. P Natl Acad Sci USA. 2000;97:8352. doi: 10.1073/pnas.140021997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torres DS, Freyman TM, Yannas IV, Spector M. Biomaterials. 2000;21:1607. doi: 10.1016/s0142-9612(00)00051-x. [DOI] [PubMed] [Google Scholar]; Rowland CR, Lennon DP, Caplan AI, Guilak F. Biomaterials. 2013;34:5802. doi: 10.1016/j.biomaterials.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomopoulos S, Williams GR, Gimbel JA, Favata M, Soslowsky LJ. J Orthop Res. 2003;21:413. doi: 10.1016/S0736-0266(03)00057-3. [DOI] [PubMed] [Google Scholar]
- 51.O'Brien FJ, Harley BA, Yannas IV, Gibson L. Biomaterials. 2004;25:1077. doi: 10.1016/s0142-9612(03)00630-6. [DOI] [PubMed] [Google Scholar]
- 52.Olde Damink LH, Dijkstra PJ, van Luyn MJ, van Wachem PB, Nieuwenhuis P, Feijen J. Biomaterials. 1996;17:765. doi: 10.1016/0142-9612(96)81413-x. [DOI] [PubMed] [Google Scholar]
- 53.Tierney CM, Jaasma MJ, O'Brien FJ. J Biomed Mater Res A. 2009;91:92. doi: 10.1002/jbm.a.32207. [DOI] [PubMed] [Google Scholar]
- 54.Choi JS, Harley BA. Biomaterials. 2012;33:4460. doi: 10.1016/j.biomaterials.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 55.Duffy GP, McFadden TM, Byrne EM, Gill SL, Farrell E, O'Brien FJ. Eur Cell Mater. 2011;21:15. doi: 10.22203/ecm.v021a02. [DOI] [PubMed] [Google Scholar]
- 56.Zhou J, Xu C, Wu G, Cao X, Zhang L, Zhai Z, Zheng Z, Chen X, Wang Y. Acta Biomater. 2011;7:3999. doi: 10.1016/j.actbio.2011.06.040. [DOI] [PubMed] [Google Scholar]
- 57.Pauly S, Klatte F, Strobel C, Schmidmaier G, Greiner S, Scheibel M, Wildemann B. Eur Cell Mater. 2010;20:84. doi: 10.22203/ecm.v020a08. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.