

HSAF (1) was isolated from the biocontrol agent Lysobacter enzymogenes (Figure 1).[1-4] This bacterial metabolite belongs to polycyclic tetramate macrolactams (PTM) that are emerging as a new class of natural products with distinct structural features.[5, 6] HSAF exhibits a potent antifungal activity and shows a novel mode of action.[1-4] The HSAF biosynthetic gene cluster contains only a single-module hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS), although the PTM scaffold is apparently derived from two separate hexaketide chains and an ornithine residue.[1-4] This suggests that the same PKS module would act not only iteratively, but also separately, in order to link the two hexaketide chains with the NRPS-activated ornithine to form the characteristic PTM scaffold. Recently, the Gulder group reported heterologous expression of the ikarugamycin (4) biosynthetic gene cluster in E. coli,[7] and the Zhang group reported the enzymatic mechanism for formation of the inner 5-memebered ring and demonstrated the polyketide origin of the ikarugamycin skeleton.[8] Ikarugamycin is a Streptomyces-derived PTM which has a 5,6,5-tricyclic system (Figure 1). Both the Gulder and Zhang groups showed that a three-gene cluster is sufficient for ikarugamycin biosynthesis. Despite the progress, this iterative polyketide biosynthetic mechanism had not been demonstrated using purified PKS and NRPS. In addition, HSAF has a 5,5,6-tricyclic system, and its gene cluster contains at least six genes.[3] Finally, unlike most PTM compounds, HSAF is produced by a Gram-negative bacterium, L. enzymogenes. Here, we report the heterologous production of HSAF analogs in Gram-positive Streptomyces hosts, in which the native PKS have been deleted. We also obtained evidence for the formation of the polyene tetramate intermediate in Streptomyces when only the single-module hybrid PKS-NRPS gene was expressed. Finally, we showed the in vitro production of the polyene tetramate using the individually purified PKS and NRPS. The results provide direct evidence for this iterative polyketide biosynthetic mechanism that is likely general for the PTM-type hybrid polyketide-peptides.
Figure 1.

Chemical structures of HSAF and other PTM analogs and HPLC analysis of the HSAF analogs produced in Streptomyces. SR107, metabolites from non-transformed Streptomyces sp. LZ35 strain SR107, in which four native PKS gene clusters had been deleted; SR107HSAF, metabolites from strain SR107HSAF1 transformed with the PKS-NRPS biosynthetic gene cluster under the control of ermE* promoter. The first two HPLC profiles show the metabolites detected at 380 nm, and the bottom two show the metabolites detected at 318 nm. The asterisks indicate peaks that were absent in the control.
First, we isolated a cosmid clone, Cos4’-1, from the genomic library of L. enzymogenes C3, which contains the entire HSAF biosynthetic gene cluster.[2] The gene cluster was then transferred into vectors for expression in Streptomyces sp. However, the transformants failed to produce any detectable HSAF or analogs. Subsequently, we made two modifications in the experiments. One was to replace the putative promoter at the 5′-nontranslated region of the PKS-NRPS gene with the ermE* promoter.[9-11], generating pSETHSAF3 (Figure S1A). The other modification was to use an engineered host, strain SR107 derived from Streptomyces sp. LZ35 through deleting its four native PKS gene clusters.[12, 13] This host is expected to provide a relatively “clean” background for the heterologous production of HSAF. We introduced pSETHSAF3 into strain SR107 to generate strain SR107HSAF1 and analyzed the metabolites in the transformant using HPLC. Strain SR107HSAF1 produced approximately seven eminent peaks that were absent in the control strain SR107 (Figure 1). We first focused our attention on the main peak 2 at 18.8 min because it falls in the region that HSAF and analogs would appear.
Compound 2 was isolated (~1 mg/L titer) as yellow powder. HR-ESI-MS gave a quasi molecular ion at m/z 495.2837 for [M+H]+ (calculated 495.2853 for C29H38N2O5). The structure assignments were carried out by the analysis of 1D and 2D NMR data (HSQC, HMBC and 1H-1H COSY) (Table S1, Figures S14 – S20 in Supporting Information). The NMR comparison of 2 with alteramide A (5) indicated that the compounds are structurally similar,[14] except for the absence of the hydroxyl group at C3 and the Z-geometry for C10,C11 double bond in 2 (Figure 1 and Table S1). The relative configuration of 2 was established by proton couplings and NOE correlations. The large coupling constants (~ 15.0 Hz) between olefinic protons (H8/H9, H21/H22, and H23/H24) led to assignment of the E-configuration for the three double bonds, whereas the Z-configuration of C10,C11 double bond was deduced from the small coupling constant (11.2 Hz) between H10 and H11. The relative stereochemistry of the bicycle unit was determined from the NOESY experiment, which is identical to alteramide A (5).[14] Interestingly, the configuration at C12 in 2 and 5 is opposite to that in 1. Compounds 2 and 5 may result from an alternative stereospecific cycloaddition that leads to a “wrong” configuration at C12. This consequently may prevent the formation of the 6-membered ring in 1, as L. enzymogenes PKS-NRPS mutant was not able to convert 5 to 1.
The rest of the eminent peaks (indicated by asterisks in Figure 1) detected at 380 nm appeared unstable, and we were not able to obtain the NMR data. To see if any other isolable HSAF analog was produced in the transformant, we checked the metabolites under other wavelengths. At 318 nm, two peaks were detected at the HSAF region, one at 18.8 min (compound 2) and the other at 18.6 min (compound 3) (Figure 1). These two compounds were not produced by the control strain SR107 (at this wavelength, a main peak at 18.6 min was also detected in the control, but showed a different UV-Vis spectrum than compound 3). Compound 3 was then isolated (~0.4 mg/L titer) for structural determination. It appeared as white powder, with a quasi molecular ion at m/z 497.3021 for [M+H]+ (calculated 497.3015 for C29H40N2O5) as determined by HR-ESI-MS. Comparison of the 1H-NMR spectrum of 3 to that of the previously reported 3-deOH-HSAF readily established the structure of 3 as 3-deOH-HSAF (Figure 1 and S21-23).[15]
The production of compounds 2 and 3 in a Streptomyces strain supports the notion that the single-module hybrid PKS-NRPS in pSETHSAF3 is likely sufficient for the assembly of the PTM scaffold. To further prove this point, we transferred pSETHSAF3 into a second Streptomyces host that has a completely “clean” background. Strain ZM12 was derived from S. coelicolor, probably the best-studied model Streptomyces, through deleting all ten native PKS and NRPS gene clusters present in its genome.[16] HPLC showed that this strain produced very few metabolites (Figure S2A). However, upon introducing pSETHSAF3, both compounds 2 and 3 were produced as the main metabolites in strain ZM12, in addition to a number of minor peaks. Since no other PKS-NRPS is present in strain ZM12, our data clearly demonstrated that the biosynthesis of HSAF only requires a single-module hybrid PKS-NRPS. The results also imply that the five domains (KS-AT-DH-KR-ACP) of this PKS module act two separate times to assemble two separate hexaketide chains.
Compounds 2 and 3 lack the 3-hydroxyl group of HSAF (1), suggesting that the SD gene (see Figure S1 for the cluster) was not functional in the expression construct pSETHSAF3, where ermE* promoter was placed in front of the PKS-NRPS gene (Figure S1A). The SD gene encodes the 3-hydroxylase converting 3-deOH-HSAF (3) to 1.[15] We subsequently generated a second expression construct pSETHSAF4, where ermE* promoter was placed in front of the SD gene (Figure S1B). The construct was introduced into Streptomyces sp. SR107 to generate the transformant strain SR107HSAF2. However, a careful search of the metabolites in this strain did not find any HSAF-like compound (Figure S2B). The reason for this is unclear at this moment; one possibility is that the insertion of the ermE* cassette in front of SD gene might not lead to the transcription of the PKS-NRPS gene and downstream tailoring genes because SD gene and the rest genes in the cluster do not appear to share the same promoter (Figure S1).[15]
To further demonstrate that the single-module hybrid PKSNRPS is able to assemble two separate polyketide chains and then link them to ornithine, we generated the third Streptomyces expression construct pSETHSAF5 that contains only the PKS-NRPS gene under the control of ermE* promoter (Figure S1C). The construct was introduced into Streptomyces sp. SR107 to generate strain SR107PKS/NRPS. HPLC analysis showed that the strain produced three new peaks that were absent in the control strain. The peaks showed absorption λmax around 350-450 nm, suggesting the presence of conjugation systems in all compounds (Figure S3). The peaks were individually collected and analysed by LC-MS/MS. Among them, the peak (compound 6) with a retention time of 20.8 min gave a quasi molecular ion at m/z 475.26 (calculated 475.26 for [M+H]+ for the polyene tetramate 6) (Figure 2). MS/MS analysis showed fragments of 173.10 and 147.08 that are consistent with the polyene structure. The same polyene was also observed in the recent heterologous expression of ikarugamycin PKS/NRPS.[8] Together, the data support that single module hybrid PKS-NRPS is able to assemble two separate polyketide chains and then link them with ornithine residue to generate the polyene tetramate (6).
Figure 2.
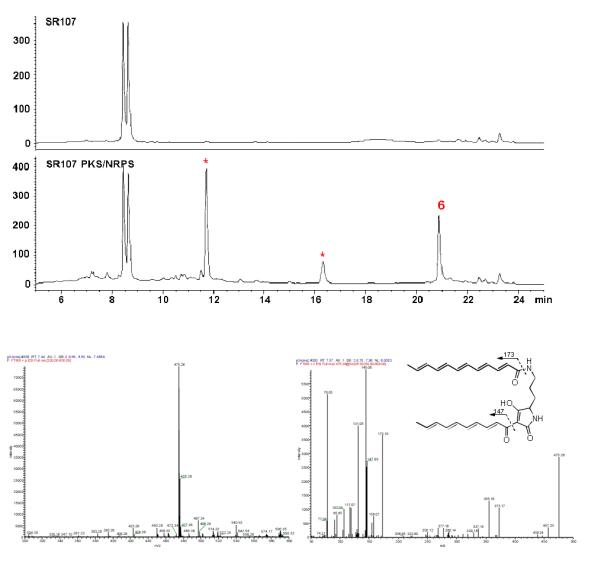
Production of the polyene tetramate (6) in Streptomyces transformed with the PKS-NRPS only. Top figure, HPLC analysis; bottom figure, MS analysis of 6, with the full ESI-MS at the left and MS/MS analysis at the right. SR107, metabolites from non-transformed Streptomyces sp. LZ35 strain SR107; SR107 PKS/NRPS, metabolites from the strain transformed with only the PKS-NRPS gene under the control of ermE* promoter. The metabolites were detected at 380 nm. The asterisks indicate peaks that were absent in the control.
To obtain direct evidence for this iterative single-module PKS, we expressed the PKS in E. coli and purified the 199.8 kDa protein (Figure S4-6). To test its activity, we converted the PKS to its holo form by incubating it with CoA and Svp, a promiscuous 4′-phosphopantetheinyl transferase (PPTase).[17] Due to the huge size of this protein, we treated the PKS with trypsin after the reaction and followed the mass change of the tryptic fragment within the ACP domain of the PKS, to which the PPT moiety and biosynthetic intermediates are covalently linked.[18, 19] Specifically, the trypsin digestion[20] is predicted to release a 26-residue fragment, VKPEQIDADASLNALGLDSLLAMELR (the active site serine residue underlined), within the ACP domain.
First, we wanted to confirm that this 26-residue fragment was indeed released from the PKS that was heterologously expressed in E. coli. Q-TOF-MS showed that the tryptic fragment of the apo-PKS had m/z 928.1374 for [M+3H]3+ (calculated 928.1604) (Figure 3), and tandem MS-MS showed that this tryptic fragment had the predicted amino acid sequence (Figure S7). After the correct tryptic PKS fragment was identified, we analyzed the holo-PKS and detected a tryptic fragment of m/z 1041.5487 for [M+3H]3+ (calculated 1041.5221) and m/z 781.4270 for [M+4H]4+ (calculated 781.3936) (Figure 3, S8; Table S2). The holo-PKS was also confirmed using phosphopantetheine ejection assay,[21] which detected m/z 357.2021 (calculated 357.0891) for the predicted PPT-ejected product (Figure S8). The data showed that the E. coli produced PKS was as expected and the enzyme was active.
Figure 3.
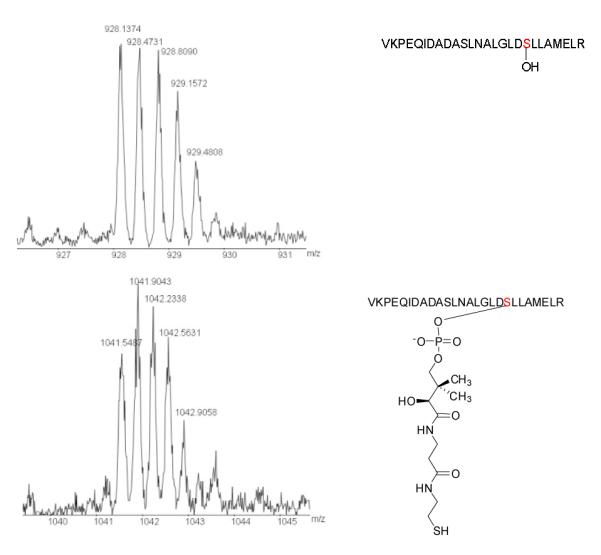
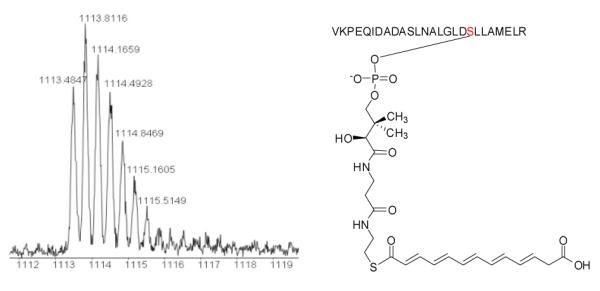
Q-TOF MS detection of the 26-residue tryptic PKS fragment that harbors the serine active site of the ACP domain. Top panel, the tryptic fragment from the PKS produced in E. coli, with m/z 928.1374 for [M+3H]3+ (calculated 928.1604); mid panel, the tryptic fragment from the 4′-phosphopantetheinylated PKS, with m/z 1041.5487 for [M+3H]3+ (calculated 1041.5221); bottom panel, the tryptic fragment from acylated PKS, with m/z 1113.4847 for [M+3H]3+ (calculated 1113.5533), showing the hexaketide polyene synthesized by the single-module PKS.
Next, we sought for potential biosynthetic intermediates that were covalently attached to the ACP domain of the PKS. Upon incubating the holo-PKS with malonyl-CoA, acetyl-CoA and NADPH, the tryptic fragment released from the PKS showed m/z 1113.4847 for [M+3H]3+ (calculated 1113.5533) and m/z 835.4001 for [M+4H]4+ (calculated 835.4170) (Figure 3, S9; Table S2). This mass change of the ACP fragment is coincident with a hexaketide polyene intermediate attached to the PPT of the PKS (Figure 3). We also varied reaction conditions and searched for other potential biosynthetic intermediates with a varied carbon chain. However, the hexaketide polyene was the only one detected. The results are consistent with that obtained from the heterologous production of HSAF analogs/intermediate in Streptomyces. Interestingly, the in vitro data suggest that the PKS used malonyl-CoA as both the starter and the extender in the polyketide chain synthesis because a carboxylate appeared present in the intermediate (Figure 3 and S9). To verify this, we generated another PKS expression construct, in which the active site cysteine in the KS domain of this PKS was mutated to alanine (C176A, protein accession number ABL86391) (Figure S10). The point-mutated PKS (mPKS) gene was heterologously expressed in E. coli, and the enzyme was purified and confirmed to contain the C176A mutation by Q-TOF-MS detection of the tryptic fragment containing the KS active site, GPSLSIDTAASSSLVAVHLACHSLRR (underlined residue to indicate the C176A mutation), with m/z 883.16 for [M+3H]3+ (calculated 883.45) (Figure S11). Because all the domains, except the KS domain, are still active in this PKS, the synthase is expected to transfer an acyl-CoA (malonyl- or acetyl-CoA) to the ACP but unable to elongate the polyketide chain. So the acyl group from the starter acyl-CoA will be “stuck” on the ACP domain. Indeed the ACP domain of the mPKS was 4′-phosphopantetheinylated by Svp, as evident by the tryptic fragment of m/z 1041.21 for [M+3H]3+ (calculated 1041.52) (Figure S11). Upon incubating the holo-mPKS with both acetyl-CoA and malonyl-CoA in the presence of NADPH, we analyzed the acylated ACP fragment. The tryptic fragment showed m/z 1069.89 for [M+3H]3+ (calculated 1070.19 for malonyl-S-ACP, 1055.52 for acetyl-S-ACP) and m/z 802.69 for [M+4H]4+ (calculated 802.87 for malonyl-S-ACP, 791.86 for acetyl-S-ACP) (Figure S12). The results showed that the C176A mutated PKS was active in transferring the malonyl group from malonyl-CoA, but not the acetyl group from acetyl-CoA, to the ACP domain (Figure S12). The result demonstrated that the PKS prefers malonyl-CoA over acetyl-CoA as the starter. It also explains why a carboxylate was observed in the ACP-bound polyketide intermediate (Figure 3).
Finally, we attempted to reconstitute the activity of HSAF PKSNRPS in vitro using the individually purified enzymes. The NRPS module (C-A-PCP-TE, 148.6 kDa) was separately expressed and purified.[3] Prior to reconstituting the activity, the purified NRPS was converted to the holo form by incubating with CoA and Svp, followed by incubating with L-ornithine and ATP to form L-ornithine-S-NRPS.[3] Since a thioesterase (TE) domain is present in this module, we expect the product(s) be released into the reaction medium.[3, 4, 22] We incubated the polyene-S-PKS with L-ornithine-S-NRPS and used LC-MS to search for released product from the reconstitution reaction (Figure 4). Total ion chromatography of selected mass range detected a distinct peak (15.48 min under this LC-MS condition, see Supporting Information) that was absent in the control reaction. In HR-ESI-MS analysis, this peak gave m/z 475.2572 that is coincident with the expected [M+H]+ for the polyene tetramate 6 (Figure S13). The result is also in agreement with the in vivo data where the production of compound 6 was observed in Streptomyces containing the PKS-NRPS gene alone (Figure 2). The product of the reconstituted PKS and NRPS did not contain the carboxylate from the starter malonyl-CoA, suggesting that a decarboxylation must have taken place after the PKS-linked hexaketide polyene was transferred to the NRPS. Together, the in vivo heterologous production data (with whole gene cluster and PKS-NRPS only) and the in vitro enzyme assays (with PKS alone and PKS-NRPS reconstitution) support that the PKS module acts iteratively and, together the NPRS module, are sufficient for the synthesis of the framework of PTM.
Figure 4.
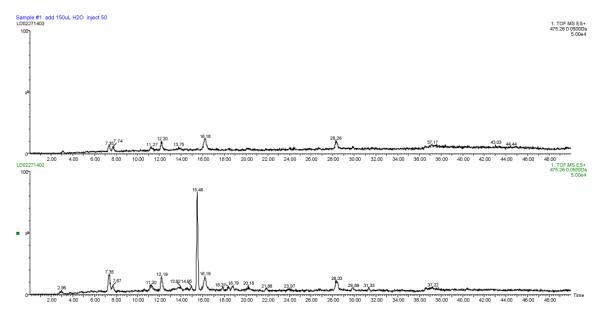
Analysis of the in vitro reconstituted PKS-NRPS. Total ion chromatography of the mass (475.2597) for compound 6 is shown for the control reaction (top panel) and the PKS-NRPS reaction (bottom). The peak at 15.48 min in the PKS-NRPS reaction gave m/z 475.2572 in HR-ESI-MS.
Based on the results, we proposed a mechanism for the formation of PTM scaffold (Figure 5). First, the five-domain PKS module iteratively catalyzes the formation of a polyene hexaketide. The geometry of the double bonds is unknown, but most likely to be trans. This hexaketide is transferred to the four-domain NRPS module, which activates L-orinithine and catalyzes the first amide bond formation, probably between the delta-amino group of L-ornithine and thioester carbonyl group of the hexaketide. This leads to an NRPS-bound polyene-ornithine intermediate. Meantime, the PKS module continues to assemble the second hexaketide, which is subsequently transferred to the NRPS through forming the second amide bond, probably between the alpha-amino group of L-ornithine and the second hexaketide chain. The timing of this transfer is right after the fifth cycle of polyketide chain elongation, in which the newly formed beta-keto group of the second hexaketide chain has not been processed by the KR domain and DH domain. All PTM natural products contain this keto group in the final structure (C25 in HSAF structure, Figure 1).[3, 5, 6, 23] The determining factor for this timing is not known, but probably related to the redox enzymes that are proposed to cooperate with the PKS-NRPS in forming the PTM scaffold.[7, 8, 15, 23] The transfer of the second hexaketide chain leads to a NRPS-bound polyene-ornithine-polyene intermediate. Finally, the intermediate is released from the NRPS by formation of the tetramate moiety, which is through the attack of the nucleophilic alpha carbon of the second hexaketide at the thioester carbonyl carbon of L-ornithine. The presence of the beta-keto group on the second hexaketide makes the alpha-carbon a good nucleophile, which promotes the tetramate formation and product release. This perhaps contributes to the timing of the transfer of this hexaketide to NRPS. In case the beta-keto was fully processed, the second polyene chain would not undergo tetramate formation, which results in an “unproductive process”. In vivo, the polyene tetramate 6 is ultimately converted to the PTM scaffold by tailoring enzymes.[8, 23] Our previous study showed that this NRPS prefers a 12-carbon chain when forming tetramate acyl-ornithine-acyl products in vitro.[3] Thus, the specificity of the domain (most likely the C domain) responsible for transferring the polyketide chain from the PKS to the NRPS is the key determinant for the macrolactam ring size observed in the characteristic PTM framework.
Figure 5.
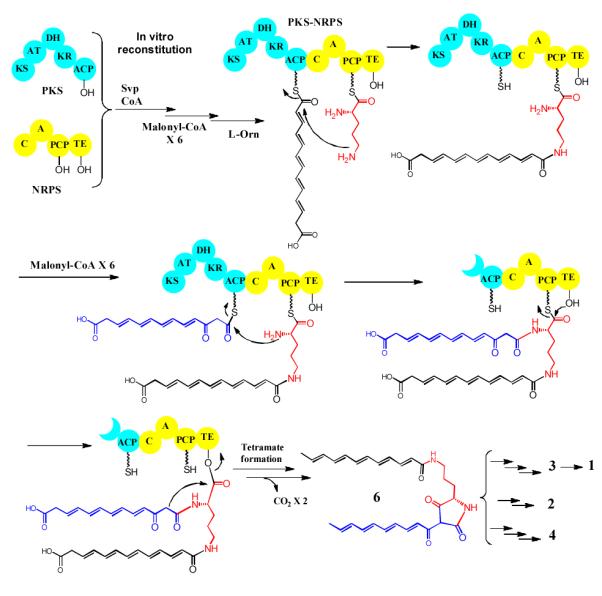
A proposed biosynthetic mechanism for the iterative bacterial PKS-NRPS in the assembly of the scaffold of HSAF and analogs.
HSAF is the main antifungal factor that the biocontrol agent Lysobacter enzymogenes uses to fight against fungal diseases. Lysobacter are ubiquitous in the environment, but largely remain untapped for bioactive natural products.[23] HSAF has a potent activity against a broad spectrum of fungi, using a new mode of action. Its chemical structure is distinct from any existing fungicide or antifungal drug. Most interestingly, its biosynthesis involves an iterative PKS mechanism. The hybrid PKS-NRPS has a typical modular organization, including KS-AT-DH-KR-ACP for the PKS module and C-A-PCP-TE for the NRPS module. There is no obvious remnants of an inactive enoylreductase (ER°) domain or a methyltransferase (CMeT) domain, as seen in several iterative fungal PKS-NRPS with the similar organization, such as PKS for tenellin, lovastain, and compactin.[24-27]. Although this type of iterative PKS-NRPS is commonly seen in fungi, it is not common in bacteria until recent years. Furthermore, PTM-type gene clusters have been identified from numerous bacterial genomes,[5, 6] and the biochemical investigation of this system has only started recently.[3, 7, 8, 28] Here, our studies of HSAF provide direct evidence for this iterative biosynthetic mechanism for bacterial polyketide-peptide natural products. The evidence comes from both the in vivo approach — heterologous production of HSAF analogs and the polyene tetramate intermediate in the Gram-positive bacterial hosts with a clean background, and the in vitro approach — expression of the ~200 kDa PKS and the ~150 kDa NRPS separately in E. coli, purification of the giant enzymes, and reconstitution of the biosynthetic activity. In light of the huge number of uninvestigated PTM-type gene clusters in databases, our studies present here will facilitate the future exploitation of new PTM products.
Experimental Section
Details of experimental procedures, construction of expression vectors, production of HSAF in heterologous hosts, PKS and NRPS gene expression in E. coli, protein purification, enzyme reactions and activity assay, and spectroscopic data are included in the Supporting Information.
Supplementary Material
Footnotes
We thank Dr. Zhongjun Qin for providing S. coelicolor strain ZM12. This work was supported in part by NSFC (31329005 and 3120032), the NIH (R01AI097260), Nebraska Research Initiatives, and Program for Changjiang Scholars and Innovative Research
Contributor Information
Dr. Yaoyao Li, Key Laboratory of Chemical Biology, School of Pharmaceutical Sciences, Shandong University, Jinan 250100, China
Haotong Chen, Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
Yanjiao Ding, Key Laboratory of Chemical Biology, School of Pharmaceutical Sciences, Shandong University, Jinan 250100, China.
Yunxuan Xie, Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
Dr. Haoxin Wang, Key Laboratory of Chemical Biology, School of Pharmaceutical Sciences, Shandong University, Jinan 250100, China; State Key Laboratory of Microbial Technology, School of Life Science, Shandong University, Jinan 250100, China
Dr. Ronald L. Cerny, Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA
Dr. Yuemao Shen, Key Laboratory of Chemical Biology, School of Pharmaceutical Sciences, Shandong University, Jinan 250100, China; State Key Laboratory of Microbial Technology, School of Life Science, Shandong University, Jinan 250100, China.
Dr. Liangcheng Du, Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
References
- [1].Li S, Du L, Yuen G, Harris SD. Mol Biol Cell. 2006;17:1218–1227. doi: 10.1091/mbc.E05-06-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yu F, Zaleta-Rivera K, Zhu X, Huffman J, Millet JC, Harris SD, Yuen G, Li XC, Du L. Antimicrob Agents Chemother. 2007;51:64–72. doi: 10.1128/AAC.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lou L, Qian G, Xie Y, Hang J, Chen H, Zaleta-Rivera K, Li Y, Shen Y, Dussault PH, Liu F, Du L. J Am Chem Soc. 2011;133:643–645. doi: 10.1021/ja105732c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lou L, Chen H, Cerny RL, Li Y, Shen Y, Du L. Biochemistry. 2012;51:4–6. doi: 10.1021/bi2015025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Blodgett JA, Oh DC, Cao S, Currie CR, Kolter R, Clardy J. Proc Natl Acad Sci U S A. 2010;107:11692–11697. doi: 10.1073/pnas.1001513107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cao S, Blodgett JA, Clardy J. Organic Letters. 2010;12:4652–4654. doi: 10.1021/ol1020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Antosch J, Schaefers F, Gulder TAM. Angew Chem Int Ed Engl. 2014;53:3011–3014. doi: 10.1002/anie.201310641. [DOI] [PubMed] [Google Scholar]
- [8].Zhang G, Zhang W, Zhang Q, Shi T, Ma L, Zhu Y, Li S, Zhang H, Zhao Y, Shi R, Zhang C. Angew Chem Int Ed Engl. 2014;53 doi: 10.1002/anie.201402078. in press. [DOI] [PubMed] [Google Scholar]
- [9].Bibb MJ, Janssen GR, Ward JM. Gene. 1985;38:215–216. doi: 10.1016/0378-1119(85)90220-3. [DOI] [PubMed] [Google Scholar]
- [10].Wilkinson CJ, Hughes-Thomas ZA, Martin CJ, Bohm I, Mironenko T, Deacon M, Wheatcroft M, Wirtz G, Staunton J, Leadlay PF. J Mol Microbiol Biotechnol. 2002;4:417–426. [PubMed] [Google Scholar]
- [11].Datsenko KA, Wanner BL. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jiang Y, Wang H, Lu C, Ding Y, Li Y, Shen Y. ChemBiochem. 2013;14:1468–1475. doi: 10.1002/cbic.201300316. [DOI] [PubMed] [Google Scholar]
- [13].Zhao GS, Li SR, Wang YY, Hao HL, Shen YM, Lu CH. Drug Discov Ther. 2013;7:185–188. [PubMed] [Google Scholar]
- [14].Shigemori H, Bae MA, Yazawa K, Sasaki T, Kobayashi J. Journal of Organic Chemistry. 1992;57:4317–4320. [Google Scholar]
- [15].Li Y, Huffman J, Li Y, Du L, Shen Y. MedChemComm. 2012;9:982–986. [Google Scholar]
- [16].Zhou M, Jing X, Xie P, Chen W, Wang T, Xia H, Qin Z. FEMS Microbiology Letters. 2012;333:169–179. doi: 10.1111/j.1574-6968.2012.02609.x. [DOI] [PubMed] [Google Scholar]
- [17].Sanchez C, Du L, Edwards DJ, Toney MD, Shen B. Chemistry & Biology. 2001;8:725–738. doi: 10.1016/s1074-5521(01)00047-3. [DOI] [PubMed] [Google Scholar]
- [18].Hitchman TS, Crosby J, Byrom KJ, Cox RJ, Simpson TJ. Chem Biol. 1998;5:35–47. doi: 10.1016/s1074-5521(98)90085-0. [DOI] [PubMed] [Google Scholar]
- [19].Crosby J, Byrom KJ, Hitchman TS, Cox RJ, Crump MP, Findlow IS, Bibb MJ, Simpson TJ. FEBS Lett. 1998;433:132–138. doi: 10.1016/s0014-5793(98)00840-0. [DOI] [PubMed] [Google Scholar]
- [20].Kayser JP, Vallet JL, Cerny RL. J Biomol Tech. 2004;15:285–295. [PMC free article] [PubMed] [Google Scholar]
- [21].Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Biochem. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Du L, Lou L. Nat Prod Rep. 2010;27:255–278. doi: 10.1039/b912037h. [DOI] [PubMed] [Google Scholar]
- [23].Xie Y, Wright S, Shen Y, Du L. Nat Prod Rep. 2012;19:1277–1287. doi: 10.1039/c2np20064c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yakasai AA, Davison J, Wasil Z, Halo LM, Butts CP, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. Journal of the American Chemical Society. 2011;133:10990–10998. doi: 10.1021/ja204200x. [DOI] [PubMed] [Google Scholar]
- [25].Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Science. 1999;284:1368–1372. doi: 10.1126/science.284.5418.1368. [DOI] [PubMed] [Google Scholar]
- [26].Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Science. 2009;326:589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Abe Y, Suzuki T, Ono C, Iwamoto K, Hosobuchi M, Yoshikawa H. Mol Genet Genomics. 2002;267:636–646. doi: 10.1007/s00438-002-0697-y. [DOI] [PubMed] [Google Scholar]
- [28].Luo Y, Huang H, Liang J, Wang M, Lu L, Shao Z, Cobb RE, Zhao H. Nat Commun. 2014;4 doi: 10.1038/ncomms3894. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.