

Abstract
In vitro models of ischemia have not historically recapitulated the cellular interactions and gradients of molecules that occur in a 3D tissue. This work demonstrates a paper-based 3D culture system that mimics some of the interactions that occur among populations of cells in the heart during ischemia. Multiple layers of paper containing cells, suspended in hydrogels, are stacked to form a layered 3D model of a tissue. Mass transport of oxygen and glucose into this 3D system can be modulated to induce an ischemic environment in the bottom layers of the stack. This ischemic stress induces cardiomyocytes at the bottom of the stack to secrete chemokines that subsequently trigger fibroblasts residing in adjacent layers to migrate toward the ischemic region. This work demonstrates the usefulness of patterned, stacked paper for performing in vitro mechanistic studies of cellular motility and viability within a model of the laminar ventricle tissue of the heart.
Keywords: cardiac ischemia, 3D cell culture, cardiomyocytes, co-culture, gradients
1. Introduction
Ischemia is a condition in which the cells within a tissue are deprived of the nutrients— in particular, oxygen and glucose—needed for normal metabolism. Ischemia commonly results in vivo from an occluded supply of blood, or from a defect in circulation.[1] In normal tissue, blood vessels supply oxygen and glucose to the cells surrounding the vessels, while also removing cellular byproducts. A radial gradient of nutrients forms around the blood vessels, and the concentration of these nutrients decreases as the radial distance from the blood vessel increases. This gradient results from two competing processes: i) the diffusive mass transport of these nutrients from the blood vessel into the tissue, and ii) the consumption of these nutrients by cellular metabolism. Most tissues in the body have capillaries every ~200 μm to ensure sufficient delivery of oxygen to all cells[2]. Cardiomyocytes are highly active metabolically, however, rely on aerobic respiration,[3] and therefore have capillaries spaced ~20 μm apart to maintain proper oxygenation.[4]
If a blood vessel occludes, (as, for example, in a myocardial infarction and stroke) the overall rate of mass transport can become insufficient to meet the rate of consumption of nutrients in the surrounding tissue, and the tissue becomes ischemic. In addition to a lack of nutrients needed for metabolism, the accumulation of cellular byproducts (e.g., carbon dioxide, carbonate ions, and lactate) during a period of prolonged ischemia results in acidosis,[5] which in turn damages the cellular membranes and mitochondria. Membrane damage leads to the release of proteolytic enzymes and contributes to the generation of toxic free radical species.[6]
Conventional in vitro models for tissues generate an ischemic environment in 2D cultures by using a hypoxia chamber and a nutrient-poor culture medium to define the amount of nutrients available to cells.[5, 7-17] These systems have provided a plethora of information about the responses of cells to ischemia (i.e., viability, changes in metabolism, release of free radicals), including the elucidation of many associated pathways.[7, 16-18] The culture conditions in these 2D models, however, provide information about the effects of global ischemia and not those resulting from physiological gradients formed in a tissue. Other models, such as 3D hydrogels,[4, 9, 19] enable physiological gradients to form in the culture, but these models are difficult to analyze and not amenable to creating precise co-cultures.[4, 19] Microfluidic devices can recapitulate the cellular environments needed to study ischemia better than conventional culture dishes because they can generate precise gradients of oxygen and glucose, induce physiologically relevant flows of liquids, and support complicated co-cultures of cells (both in 2D and 3D environments).[20-30] Despite the sophistication of microfluidic devices, and the generally accepted need for better in vitro models to study ischemia, the biology community has not adopted these technologies, since they are more difficult to establish, maintain, and analyze than 2D cultures.
Most in vitro model systems lack some, if not all, of the complex interactions that occur in an ischemic tissue.[1, 4, 10, 14, 25, 28] There is a need for an ischemia model that, at a minimum, allows for: i) co-cultures of multiple types of cells; ii) controlled temporal and spatial gradients of nutrients such as oxygen and glucose; and, iii) paracrine signaling between sub-populations of cells. Such a model system should also be easy to assemble, inexpensive enough for one-time use, compatible with the high-throughput tools already in use, and amenable to rapid analysis of data.
We previously described a 3D culture system that uses paper as a scaffold to support hydrogels containing cells; we refer to this system as “Cells-in-Gels-in-Paper” (or CiGiP).[31-33] The CiGiP culture system has six advantages over 2D culture and microfluidic-based devices. i) The hydrophilic surface and large (~70%) void-volume of the chromatography paper (i.e., a “layer”) wicks liquids, allowing easy seeding of cell-laden hydrogels into the scaffold with a micropipette. ii) The fibers of the paper provide strong mechanical support, and allow for the repeated handling of thin (~200 μm) slabs of cell-containing hydrogels. iii) Individual layers of paper are easily assembled to form thicker constructs, which we refer to as “stacks.” Assembly of a stack is modular, and allows us to control the thickness of the overall construct (i.e., the number of layers) and the composition of each layer (e.g., different densities of a single type of cell, or co-cultures of more than one type of cell). iv) Wax-printed patterns define an array of hydrophilic “zones” (i.e., regions not containing the hydrophobic wax of arbitrary size and shape). These zones allow us to perform multiple experiments, in parallel, on a single sheet of paper. v) Intercellular communication can occur between layers—via migration of cells or the diffusion of signaling molecules—because each layer of the stack is in conformal contact. vi) Diffusion of nutrients (e.g., oxygen and glucose) into the stack can be controlled by placing the stack in a holder composed of an impermeable material; diffusion of nutrients through the stack, and the shapes of gradients in the stacks, can be modulated by changing the density of cells in each layer.[33]
We demonstrate the versatility of the CiGiP culture system by modeling cardiac ischemia with two different experimental systems. In the first experimental system, we stacked layers containing primary neonatal rat cardiomyocytes, and limited the access of nutrients to these cells from a single direction (flux from the top of the stack). This experimental design is similar to the single cell-type cultures described previously[33] and mimics the region of cardiomyocytes between a healthy (top of the stack) and occluded blood vessel (bottom of the stack).
In the second experimental system, we stacked layers containing either primary neonatal rat cardiomyocytes or primary cardiac fibroblasts to form a co-culture. With this arrangement, we demonstrated that increasing the thickness of the stack results in a greater amount of ischemic damage in the cardiomyocytes, and subsequently, a greater number of fibroblasts that migrate toward the ischemic cardiomyocytes (i.e., against the oxygen gradient). We also show migration of fibroblasts in this co-culture is due, at least partially, to the release of cytokines by the ischemic cardiomyocytes.
Both of these experimental systems serve as a proof-of-principle, and demonstrate that the modularity of the layer-by-layer construction of the CiGiP culture system can probe the complex interactions between cells that occur in a tissue-like construct. Although the proposed CiGiP models of cardiac ischemia do not perfectly mimic the in vivo microenvironment of native heart tissue, it does recapitulate certain aspects of the tissue (i.e., patterning of multiple types of cells, formation of gradients of cytokines and nutrients) that are not easily achieved with conventional 2D culture systems. We illustrate that cardiomyocytes in the CiGiP culture respond to ischemic conditions by undergoing changes in morphology, viability, and release of signaling molecules (ischemia-induced cytokines). These signaling molecules, and not just the gradient of nutrients, induce the migration of fibroblasts. We demonstrate the migration of the fibroblasts is partially due to signaling of transforming growth factor beta (TGF-β).
2. Results and Discussion
2.1. Cardiomyocytes cultured in a stack mimic the interface of living and dead tissue
To mimic the environment of a tissue surrounding an occluded coronary artery, we prepared a 3D construct of rat neonatal cardiomyocytes, and placed it in a holder that restricted the mass transport of nutrients into the stack. We patterned Whatman 114 chromatography paper (thickness 190 μm) with a commercial wax printer, as described previously,[32, 33] to have 20 circular hydrophilic zones (diameter 3 mm) surrounded by hydrophobic barriers (Fig. 1A). The 20 zones allowed us to perform 20 replicate cultures in parallel, and to obtain enough data to analyze a single experiment with statistical significance. We seeded each hydrophilic zone of six individual layers with primary cardiomyocytes (100,000 cells/zone) suspended in a 2 μL suspension of Matrigel (Fig. 1B). We cultured these layers for three days in media with 10% fetal bovine serum prior to stacking; this period allowed the cells to recover from the cell harvest procedure. The six-layer stack (~1.2 mm in total thickness) formed a tissue-like construct. We placed the stack in a custom-made Delrin holder (Fig. 1C); we chose Delrin because it is both non-toxic to cells and impermeable to gases and liquids.[34] This holder blocks diffusion of nutrients from the bulk medium to the stack, except for at the top of the stack (i.e., “layer 1”) where holes in the top plate of the holder are positioned above the zones of the stack of paper. As nutrients diffuse into the stack, they are metabolized by the cardiomyocytes and depleted; this depletion of nutrients results in an ischemic environment in the bottom layers.
Figure 1. Paper-based Model of Cardiac Ischemia.

A) Image of a layer of wax-patterned paper and the top and bottom portions of a custom-made Delrin holder in which the cell containing papers are cultured. The Delrin material is non-toxic to cells and fully insulates the cell-containing stack from mass transport of outside nutrients. Scale bar: 8mm. B) Schematic of a single layer of paper containing 20 hydrophilic zones surrounded by a hydrophobic wax-printed border. Cells suspended in a hydrogel are pipetted into the hydrophilic zones. C) Schematic of a stack of paper in the Delrin holder comprising a top piece containing holes for access to culture media and a solid bottom piece. D) Heat map of the average fluorescence intensities of calcein-stained cells in a stack of six layers of paper. A darker color indicates higher levels of calcein, and is proportional to the number of viable cells. Each layer (clustered together as a stack of 20 slices) contains 20 replicate zones initially containing 100,000 neonatal rat cardiomyocytes suspended in matrigel. The stack was cultured for seven days prior to staining. E) Total number of viable cells in each zone of the stack for three different time points. Error bars are the S.D. of 20 zones. Means with different letters are significantly different based on Holm-Sidak method, (P = <0.001). Detailed statistics of each value available in the SI.
To determine the effect of the ischemia on the cardiomyocytes after seven days of culture we peeled apart the layers of the stack, and determined the viability of the cells in each layer with calcein AM (a molecule that permeates the cell wall that becomes impermeable and fluorescent upon cleavage of the ester groups on the molecule by an esterase that is only present in living cells). We obtained images of each layer with a flatbed fluorescent scanner, and measured the average fluorescence intensity of each zone with custom-written script for the MatLab software.[33] The software generates heat maps representing a cross-sectional image of the fluorescence intensity (from which, we infer the viability) of cells in each layer of the stack (Fig. 1D). A thick black line demarcates the layers within the stack and each layer comprises the fluorescence intensity of 20 individual zones cultured in parallel.
Figure 1E illustrates the number of viable cardiomyocytes in each layer of three independently cultured stacks (n = 20 replicates) after culture intervals of 2-, 4-, and 7-days. The number of viable cardiomyocytes was determined by generating a calibration curve for the fluorescence intensity of calcein for different numbers of cardiomyocytes (Fig. S1). In each stack, the number of viable cells in the bottom-most layer (layer 6)—that is, the layer furthest from the oxygenated medium—was significantly lower than the number of viable cells in the top-most layer (layer 1). The greatest difference between these two layers occurred after 7 days of culture, and resulted in a 90% decrease in the viability of cells in layer 6 compared to those in layer 1. The difference in the number of viable cells between layer 1 and layer 6 increased by ~30% between day 2 and day 7, and suggests ischemia-induced injury is gradual under these experimental conditions and occurs over a period of days. These data suggest the gradient of nutrients formed within the stack mimics infarcted ventricular myocardium when the ischemic and non-ischemic cells are in direct contact. Culturing a stack of cardiomyocytes for different periods is potentially useful in comparing the effects of acute and chronic ischemia.
To determine if the cardiomyocytes were dying from a lack of nutrients, and not from a change of pH in the stack, we measured the pH of each layer with a piece of pH paper immediately after destacking. The buffering capacity of the HEPES buffer in the culture media was sufficient to prevent the formation of a pH gradient in the stack (i.e., each of the layers, and the bulk media, were neutral). This result suggests that ischemic damage in our system is primarily due to a lack of nutrients; effects due to changes in pH could, however, be studied in the CiGiP system by lowering the buffering capacity of the HEPES buffer, or by eliminating the buffer altogether.
One unresolved result from these experiments is that a small sub-population (~10%) of cardiomyocytes in the bottom-most layer of the stack (Fig. 1E) survived after long-term ischemic culture. The survival of this sub-population could be due to the diffusion of nutrients through the edges of the wax-patterned zones, as described previously.[33] Another possible explanation for this result is that neonatal heart tissue primarily relies on glycolysis and lactate oxidation for the production of ATP,[35] and in newborn rats, the transition from glycolysis to fatty acid oxidation (aerobic metabolism) is incomplete until seven days after birth.[36] The cardiomyocytes isolated in our study were harvested from two-day-old rat pups, and are, therefore, less susceptible to ischemia than more mature cardiomyocytes.[37-39]
2.2 Morphology of the cardiomyocytes change in the ischemic layers of a stack
Although sub-populations of cardiomyocytes survive in each layer of the stack after prolonged culture, viability is not necessarily indicative of cellular health. The morphology of cardiomyocytes grown in 2D cultures change from an elongated to a rounded shape under ischemic conditions.[28, 40] To determine if the morphology of the cardiomyocytes cultured in the CiGiP system changed after prolonged periods of culture, we measured the morphology of a representative number of cardiomyocytes in each layer of a six-layer stack after a seven-day culture (Fig. 2A). We obtained confocal images of the cells from a single zone of each layer (Fig. 2B) after staining for sarcomeric α-actinin, which is a z-line protein expressed specifically in striated muscle and in cardiomyocytes. We measured the circularity of 9-12 representative cardiomyocytes in each layer with imageJ software.[41] The circularity, C, of a cell is defined by C = 4πAP–2, where A is the area and P is the perimeter of the projection of a cell. A cell that is perfectly round, and that exhibits no polarization or spreading, has C = 1.00; a cell that is polarized and elongated along a single axis has (with the limit being a single “line”) has C = 0.00.
Figure 2. Effect of Ischemia on Morphology.
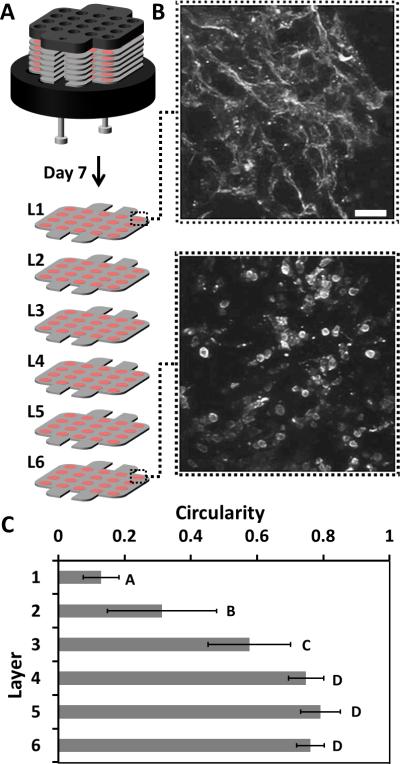
A-B) Confocal, immunofluorescence images of the sarcomeric α-actinin in cardiomyocytes within a single zone of layer 1 (the top layer) and layer 6 (the bottom layer) of the stack after 7 days of culture. Scale bar: 50 μm. Inset Scale Bar: 10 μm. C) Graph of the average circularity of a set of 9-12 cells chosen at random from a single zone in each layer of the stack. Error bars are a S.D. of 9–12 cells in each layer, averaged between two different stacks. Means with different letters are significantly different based on Holm-Sidak method, (P = <0.001). Detailed statistics of each value available in the SI.
Figure 2C shows a gradient in the circularity of the cells, with a trend similar to that seen in the viability of the cells (Fig. 1E) and suggests these cells are experiencing similar ischemic environments. The differences in circularity between layer 1 (C = 0.12 ± 0.06) and layer 6 (C = 0.76 ± 0.04) are both dramatic and statistically significant (p < 0.01), and suggest these populations of cells have different morphologies. The difference in circularity of the cells in layers 1 and 3 is also statistically significant (p < 0.01); the circularity of the cells in layers 4, 5, and 6 is not distinguishable (p > 0.05).
2.3 Fibroblasts within a stack migrate toward ischemic layers
Although cardiomyocytes occupy the majority of the volume of the myocardium, cardiac fibroblasts contribute up to 66% of the total number of cells in cardiac muscle.[36] Under ischemic conditions, metabolically stressed cardiomyocytes produce cytokines that induce fibroblasts to both proliferate and migrate into the injured regions—by chemotaxis toward the increased concentrations of cytokines—to remodel and repair the area of damage.[36]
To investigate the migratory response of fibroblasts toward cardiomyocytes in the CiGiP culture system, we co-cultured stacks composed of layers containing primary cardiomyocytes, primary cardiac fibroblasts, or an immortalized line of (3T3) fibroblasts (Fig. 3A). Each stack contained two layers of cardiomyocytes situated below a layer of primary cardiac fibroblasts and an increasing number of layers of 3T3 fibroblasts. The two types of fibroblasts were always seeded into separate layers. We labeled the primary cardiac fibroblasts with a fluorescent marker (Cell Tracker Orange) to distinguish them from the cardiomyocytes and 3T3 fibroblasts when invading adjacent layers. Cell Tracker Orange, like calcein AM, is a cell-permeable probe that becomes impermeable and fluorescent when cleaved by esterases in the cells; the Cell Tracker Orange also reacts with thiols to provide long-term stability of the fluorophore over days of culture.[22, 42] We used 3T3 fibroblasts in these experiments because there were an insufficient number of primary fibroblasts available.
Figure 3. Migration of Cardiac Fibroblasts in Response to Varying Levels of Ischemia.

A) Schematic of three stacks used to induce an environment of low ischemia (left), medium ischemia (middle), and high ischemia (right) for two layers containing cardiomyocytes (100,000 cells/zone) at the bottom of the stack. Each stack contains a different number of layers of immortalized 3T3 fibroblast (50,000 cells/zone) placed on top of the stack (the cells in these upper layers deplete nutrients diffusing toward the cardiomyocytes). Each stack contained a single layer of primary cardiac fibroblasts (10,000 cells/zone) between the 3T3 fibroblasts and cardiomyocytes. Cardiac fibroblasts were labeled with Cell Tracker Orange prior to seeding. Each stack was cultured for three days. B) Viability (determined with calcein AM) of the top layer of cardiomyocytes (e.g., cardiomyocytes in the second from bottom layer of the stack shown in A). C) Relative amount of migration of the cardiac fibroblasts into the top layer of cardiomyocytes, determined by the fluorescence intensity of Cell Tracker Orange in the second from bottom layer that contains either cardiomyocytes (in matrigel) or only matrigel. Error bars are the standard deviation of 10 replicates. Means with different letters are significantly different based on Holm-Sidak method, (P = <0.001). Detailed statistics of each value available in the SI.
Increasing the number of layers of fibroblasts in a stack formed three different cellular environments, which we call: i) low ischemia, ii) medium ischemia, and iii) high ischemia (Fig. 3A). We hypothesized that increased ischemic damage of the cardiomyocytes would result in an increased number of migrating cardiac fibroblasts. We measured the fluorescence intensity of the cardiomyocyte-containing layers after a 7-day culture, and determined the relative amount of migration of the cardiac fibroblasts toward the cardiomyocytes in the three different ischemia environments. In each stack, we found that only the top layer of cardiomyocytes contained primary fibroblasts.
We observed two results that suggest the cardiomyocytes in the “high ischemia” stacks (containing seven layers of fibroblasts) were under a higher nutrient/oxygen stress than the cardiomyocytes in the “low ischemia” stacks (containing one layer of fibroblasts): i) the cardiomyocytes in the high ischemia stacks are ~75% less viable, as determined with calcein AM stain, than cardiomyocytes in the low ischemia stacks, and ii) a larger number of fibroblasts (~ 2.5-fold) migrated to the layer of cardiomyocytes in the high ischemia stack than in the low ischemia stack.
Relying on the fluorescence intensities of the viability stains alone is misleading because the calcein dye does not distinguish between the cardiomyocytes and fibroblasts cells. Although the medium and high conditions of ischemia appear to contain similar numbers of viable cardiomyocytes (Fig. 3B), the number of fibroblasts in the cardiomyocyte layer of the high ischemia condition is 30% greater than the cardiomyocyte layer of the low ischemia condition. The cardiomyocytes in the high ischemia condition, therefore, are of a lower viability than the medium ischemia condition. These results suggest the level of ischemia imposed on the cardiomyocytes directly affected the migration of the cardiac fibroblasts.
2.4 Signaling from Cardiomyocytes induce the migration of fibroblasts to ischemic regions
In a separate set of experiments we compared the migration of cardiac fibroblasts in the low ischemia, medium ischemia, and high ischemia stacks under identical experimental conditions, but replaced the cardiomyocyte-containing layers in one stack with hydrogel-only containing layers. This comparison assessed whether the cardiac fibroblasts were migrating toward the cardiomyocytes, or migrating either away from either the immortalized fibroblasts or against the oxygen gradient. Figure 3C illustrates that the number of cardiac fibroblasts that migrated to the adjacent layer below reduces dramatically in the absence of cardiomyocytes: greater than 20-fold for the low ischemia condition, 12-fold for medium ischemia condition, and 3-fold for high ischemia condition. This reduction in migration of the fibroblasts suggests signaling molecules expressed by the cardiomyocytes are chemoattractants and that migration is not due to unexpected interactions of the two types of fibroblasts, random movement (i.e., chemokinesis), or the oxygen gradient.
2.5 TGF-β neutralizing antibodies decrease the migration of fibroblasts
The accumulation of signaling molecules produced under ischemic conditions is another factor that complicates our ability to predict the response of cardiac cells in 2D cultures. Increased levels of transforming growth factor-beta (TGF-β), for example, induce hypertrophy in cardiomyocytes, cause the chemotaxis of fibroblasts, and activate the elevated secretion of ECM by fibroblasts.[43, 44] The addition of a pan-specific antibody (i.e., an antibody that binds to many epitopes of the protein) to neutralize TGF-β suppresses chemotaxis of fibroblasts and reduces the progression of wound healing in vivo.[45, 46] Treatment of cardiac ischemia with antibodies that antagonize TGF-β signaling to reduce the chemotaxis of fibroblasts is not routinely used in clinical treatments because the effect(s) of these antibodies on wound healing is not understood.[45, 46] The reduced blood flow to ischemic tissues makes it difficult to predict the efficacy of directly delivered therapeutics; it is difficult to predict the optimal amount of anti-TGF-β to administer to a patient because the molecule must diffuse to its target site from a distant blood vessel.[47] We added pan-specific TGF-β neutralizing antibodies to the culture medium to determine if they could diffuse through the stack and alter the behavior of cells. Although our system is not a perfect model of a tissue, such diffusive transport is reminiscent of a drug transport to a region of ischemia.
To test the effect of TGF-β in our system, we prepared stacks composed of a single layer of primary rat cardiomyocytes (100,000 cells/zone) situated below three layers of primary rat cardiac fibroblasts (100,000 cells/zone) (Fig. 4A), which were pre-labeled with a fluorescent dye (Cell Tracker Orange). We added different concentrations (0, 50, or 100 μg/mL) of a pan-specific TGF-β neutralizing antibody (polyclonal rabbit IgG) to the medium of three separate stacks (the antibody therefore needed to diffuse into the stacks), cultured the stacks for three days, and determined the extent of fibroblast movement from fluorescence images of each layer. We found that the number of fibroblasts that migrated into the cardiomyocyte-containing layer decreased by 35% when the highest concentration (100 μg/mL) of the TGF-β neutralizing antibody was added to the media (Fig. 4B). These results suggest this system could screen biopharmaceuticals (specifically therapeutic antibodies) for their ability to alter cellular phenotypes and to diffuse through tissue-like constructs.
Figure 4. Role of TGF-β Signaling in Invasion of Cardiac Fibroblasts.
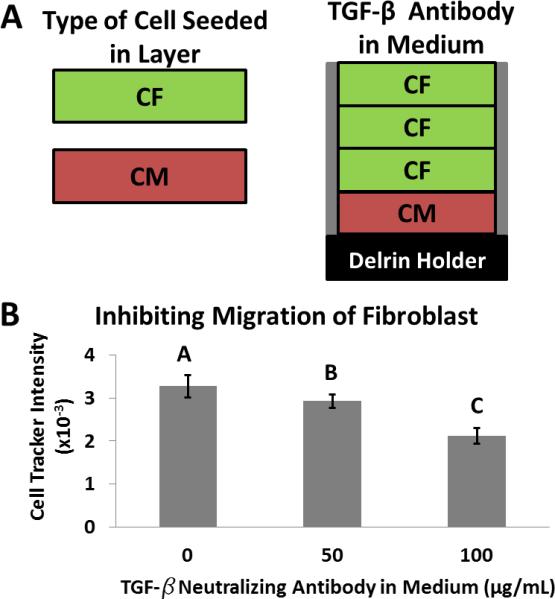
A) Schematic of a stack consisting of layers of cardiomyocytes and cardiac fibroblasts. B) Values of fluorescence intensity of fibroblasts, labeled with Cell Tracker Orange, that migrated into the adjacent layer of cardiomyocytes. Each condition is in the presence of a different concentration of TGF-β neutralizing antibody, for three days in a stacked culture. Error bars are the standard deviations for 20 replicates. Means with different letters are significantly different based on Holm-Sidak method, (P = <0.001). Detailed statistics of each value available in the SI.
3. Conclusion
This paper demonstrates that the CiGiP culture system supports the co-culture of multiple types of cells—primary neonatal rat cardiomyocytes, primary rat cardiac fibroblasts, and 3T3 fibroblasts—and 3D stacks of cells mimic certain aspects of the pathology of cardiac tissue undergoing an ischemic event. The modularity of the system allowed us to: i) control the level of ischemia imposed on the cardiomyocytes by changing the thickness of the stack (i.e., the number of fibroblast-containing layers), ii) measure the migratory response of fibroblasts caused by cardiomyocytes under ischemic distress, and iii) test the effect of different concentrations of a protein on the migratory response of the fibroblasts.
We evaluated the cells in each layer of paper a flatbed gel scanner, which is a high-throughput technique, but provides low-resolution data, and a confocal microscope, which is a low-throughput technique, but provides high-resolution data. The fluorescent images obtained from the gel scanner allowed us to determine the overall viability of the cardiomyocytes in each layer and the relative amount of migration the fibroblasts in the stack. The confocal microscope allowed us to visualize the morphology of individual cardiomyocytes within each layer. The ability to quantify quickly and easily the migration of fibroblasts provides an opportunity to screen new biopharmaceutical candidates in an environment that is more physiologically relevant than other commonly used 2D and 3D culture systems.
There are a number of drawbacks of the current system which require further development of the paper-based scaffolds and the analytical techniques used to analyze the cells in each layer of paper: i) The wax-printed, hydrophobic borders of the chromatography paper are permeable to liquid when immersed (for hours) and allow diffusion of nutrients laterally into the periphery of each zone, as described previously.[33] This unwanted diffusion through the paper causes orthogonal gradients of nutrients to form in the stack and contributes to the heterogeneity of cells within a given zone. ii) The fibers of the chromatography paper scatter light, and limit the imaging capabilities to fluorescence-based readouts. iii) It is currently difficult to measure the concentration of small molecules (e.g., oxygen, glucose, or cytokines) in situ, and destacking the layers prior to analysis perturbs these gradients and (possibly) the metabolic state of the cells. iv) This system is not easily modified to measure the contractility of cardiomyocytes without complex optics (i.e., confocal microscopy); contractility is an important indicator of their physiological activity.
We recently developed an alternative scaffold fabricated from paraffin-embossed polymer meshes.[48] The new mesh-based scaffolds allow light to pass through its open pores without significant scattering and the pores can be filled more easily to make barriers that block the lateral diffusion of nutrients within the stack. Further work with this new scaffold will enable more precise gradients to be formed and measured; it is, however, more difficult to use technically than the paper-based system described here.
The study of non-human cells is necessary because primary human cardiomyocytes are difficult to obtain, and cardiomyocytes derived from embryonic- or induced pluripotent stem cells do not provide all of the phenotypes expected for mature cardiomyocytes.[49] A limitation of the current set of experiments is the use of rat neonatal cardiac cells; general conclusions about ischemic tissue cannot be drawn since there are significant differences between rat and human cardiomyocytes.[50] We chose neonatal cells because they are more viable and amenable to in vitro culture conditions than adult cardiomyocytes. The use of primary adult rat cardiomyocytes, however, might provide more relevant data than the use of neonatal cells. Our current model of ischemic tissue—the co-culture of fibroblasts and cardiomyocytes—also omitted immune cells, which are known to respond to ischemic injury.[46] This particular limitation is addressable with the current system by incorporating other layers of paper containing immune cells of interest.
We believe that CiGiP, despite the limitations listed above, due to the simplicity of the system with which it allows the assembly of tissue-like constructs, and the dis-assembly of these constructs to analyze sub-populations of cells after culture will open the door to experimental tests of complex co-cultures in 3D environments.
4. Experimental
Fabrication of layers of paper
Layers of paper were prepared using the procedure described by Derda et al.[33] We printed wax directly onto sheets of Whatman 114 chromatography paper using a solid-ink printer (Xerox Phaser 8560). The wax-patterned paper was placed in a 150 °C oven for 2 minutes; this step ensures the wax melted through the thickness of the paper. We cut the paper into individual layers with a laser cutter (Versa Laser). Prior to usage, we soaked the layers in ethanol to remove any particulates remaining after laser cutting, autoclaved them in water (121 °C for min 15 min), and reheated the sheets in air to render the wax hydrophobic (the wax becomes hydrophilic when autoclaved in water).
Harvest and culture of cardiomyocytes and cardiac fibroblasts
All procedures were conducted according to the guidelines of the Harvard University Animal Care and Use Committee. Primary neonatal rat cardiomyocytes and cardiac fibroblasts were isolated from two-day old Sprague Dawley rats, as described previously.[51, 52] Left ventricular tissue was isolated from the neonatal rat, trypsinized overnight (1mg/mL) on ice. The cardiac tissue was further digested with collagenase (1mg/mL) at 37°C for four 15-minute cycles. After each cycle, the supernatant containing cardiomyocytes was collected; after the cycles of digestion were completed, the cellular mixture was pre-plated in a tissue culture flask for 45 minutes to select for cardiomyocytes. All cardiomyocytes and cardiac fibroblasts were cultured in M199 culture media supplemented with 10% heat-inactivated fetal bovine serum (FBS, Invitrogen, Carlsbad, CA), 10 mM HEPES, 0.1 mM MEM non-essential amino acids, 20 mM glucose, 2mM L-glutamine, 1.5 μm vitamin B-12, and 50 U ml-1 penicillin.
Seeding cells into layers of paper
All cells were suspended in ice-cold Matrigel (diluted 1:1 with 10% FBS culture media). Each zone of a layer was spotted with 2 μL of solution using a micropipette. Cardiomyocytes were seeded at a concentration of either 40,000 cells/zone or 100,000 cells/zone, as indicated. Cardiac fibroblasts were seeded at a concentration of either 10,000 cells/zone or 100,000 cells/zone, as indicated. The freshly harvested cardiomyocytes and cardiac fibroblasts were seeded in the paper layers and cultured for three days prior to any experimentation to allow for recovery. NIH 3T3 fibroblasts (ATCC) were seeded at 50,000 cells/zone the day prior to an experiment, and cultured in DMEM with 10% calf serum. After seeding of cells, all layers of paper were immediately submerged into pre-warmed (37 °C) culture media and incubated at 37 °C and 5% CO2.
Immunostaining, imaging, and analysis
For sarcomeric α-actinin staining, the cardiomyocytes were first rinsed 3x with 37 °C 1X PBS before being fixed and permeabilized (15 min) in a solution of 1X PBS containing 4% paraformaldehyde and 0.05% Triton X-100. The layers were incubated with a blocking solution (5% BSA in 1X PBS) for 30 minutes at room temperature before incubation (1 h) with a primary mouse anti-sarcomeric α-actinin antibody (Invitrogen) at a dilution of 1:200 in 1X PBS. Following a three times rinse in 1x PBS, the cardiomyocytes were incubated with Alexa Fluor 635 goat anti-mouse secondary antibody (Invitrogen) at a dilution of 1:200 for 90 minutes. Samples were then rinsed three times in 1x PBS before mounting on slides with ProLong Gold antifade reagent (Invitrogen). Images were taken with a Zeiss LSM 5LIVE confocal microscope.
Cardiac fibroblasts or NIH 3T3 fibroblasts were stained with the fluorescent marker (Cell Tracker Orange, Invitrogen) according to the manufacturer's protocol prior to seeding into the layers of paper. Viability of the cells was determined using calcein AM (Invitrogen; 0.4 μg/mL in culture media); the cells were incubated in the calcein-containing media for 20 minutes at 37 °C and 5% CO2. After staining with calcein-AM, the cells were washed with DPBS three times and imaged a Typhoon 9000 FLA scanner (GE Healthcare).
Supplementary Material
Acknowledgements
We acknowledge funding from the Wyss Institute for Biologically Inspired Engineering, National Institutes of Health (1 UH2 TR000522-01), Vertex Pharmaceuticals Incorporated, and MRSEC. We also thank Megan McCain for harvesting some of the primary rat cardiomyocytes used in this work.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Bobak Mosadegh, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA; Wyss Institute for Biologically Inspired Engineering, Harvard University, 60 Oxford Street, Cambridge, MA 02138, USA.
Borna E. Dabiri, Wyss Institute for Biologically Inspired Engineering, Harvard University, 60 Oxford Street, Cambridge, MA 02138, USA Disease Biophysics Group, Harvard School of Engineering and Applied Sciences, Cambridge, MA 02138, USA.
Matthew R. Lockett, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
Ratmir Derda, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA; Wyss Institute for Biologically Inspired Engineering, Harvard University, 60 Oxford Street, Cambridge, MA 02138, USA.
Patrick Campbell, Disease Biophysics Group, Harvard School of Engineering and Applied Sciences, Cambridge, MA 02138, USA.
Kevin Kit Parker, Wyss Institute for Biologically Inspired Engineering, Harvard University, 60 Oxford Street, Cambridge, MA 02138, USA; Disease Biophysics Group, Harvard School of Engineering and Applied Sciences, Cambridge, MA 02138, USA.
George M. Whitesides, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA; Wyss Institute for Biologically Inspired Engineering, Harvard University, 60 Oxford Street, Cambridge, MA 02138, USA.
References
- 1.Beanes SR, Dang C, Soo C, Ting K. Expert. Rev. Mol. Med. 2003;5:1. doi: 10.1017/S1462399403005817. [DOI] [PubMed] [Google Scholar]
- 2.Curtis MW, Russell B. J. Cardiovasc. Nurs. 2009;24:87. doi: 10.1097/01.JCN.0000343562.06614.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuznetsov AV, Hermann M, Saks V, Hengster P, Margreiter R. Intern. J. Biochem. Cell Bio. 2009;41:1928. doi: 10.1016/j.biocel.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Radisic M, Malda J, Epping E, Geng WL, Langer R, Vunjak-Novakovic G. Biotechnol. Bioeng. 2006;93:332. doi: 10.1002/bit.20722. [DOI] [PubMed] [Google Scholar]
- 5.Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Am. J. Physiol. 1996;270:H1334. doi: 10.1152/ajpheart.1996.270.4.H1334. [DOI] [PubMed] [Google Scholar]
- 6.Prasad A, Stone GW, Holmes DR, Gersh B. Circ. 2009;120:2105. doi: 10.1161/CIRCULATIONAHA.108.814640. [DOI] [PubMed] [Google Scholar]
- 7.Sauvant C, Schneider R, Holzinger H, Renker S, Wanner C, Gekle M. Cell Physiol. Biochem. 2009;24:567. doi: 10.1159/000257513. [DOI] [PubMed] [Google Scholar]
- 8.Rohr S. J. Cardiovasc. Pharmacol. 2011;57:389. doi: 10.1097/FJC.0b013e3182137e17. [DOI] [PubMed] [Google Scholar]
- 9.Chen CZ, Peng YX, Wang ZB, Fish PV, Kaar JL, Koepsel RR, Russell AJ, Lareu RR, Raghunath M. Br. J. Pharmacol. 2009;158:1196. doi: 10.1111/j.1476-5381.2009.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Q, Norman JT, Shrivastav S, Lucio-Cazana J, Kopp JB. Am. J. Physiol. Renal Physiol. 2007;293:F631. doi: 10.1152/ajprenal.00379.2006. [DOI] [PubMed] [Google Scholar]
- 11.Thampatty BP, Wang JH. Cell Motil. Cytoskeleton. 2007;64:1. doi: 10.1002/cm.20166. [DOI] [PubMed] [Google Scholar]
- 12.Yarrow JC, Perlman ZE, Westwood NJ, Mitchison TJ. BMC Biotechnol. 2004:4. doi: 10.1186/1472-6750-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belete HA, Godin LM, Stroetz RW, Hubmayr RD. Cell. Physiol. Biochem. 2010;25:71. doi: 10.1159/000272052. [DOI] [PubMed] [Google Scholar]
- 14.Liang CC, Park AY, Guan JL. Nat. Prot. 2007;2:329. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 15.Hu HW, Li XK, Zheng RY, Xiao J, Zeng JQ, Hou ST. Biochem. Biophys. Res. Comm. 2009;390:115. doi: 10.1016/j.bbrc.2009.09.077. [DOI] [PubMed] [Google Scholar]
- 16.Clarke TC, Williams OJS, Martin PEM, Evans WH. Eur. J. Pharm. 2009;605:9. doi: 10.1016/j.ejphar.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Kang PM, Haunstetter A, Aoki H, Usheva A, Izumo S. Circ. Res. 2000;87:118. doi: 10.1161/01.res.87.2.118. [DOI] [PubMed] [Google Scholar]
- 18.Lee WH, Kang S, Vlachos PP, Lee YW. Arch. Pharm. Res. 2009;32:421. doi: 10.1007/s12272-009-1316-9. [DOI] [PubMed] [Google Scholar]
- 19.Brown DA, MacLellan WR, Laks H, Dunn JCY, Wu BM, Beygui RE. Biotech. Bioeng. 2007;97:962. doi: 10.1002/bit.21295. [DOI] [PubMed] [Google Scholar]
- 20.Mosadegh B, Huang C, Park JW, Shin HS, Chung BG, Hwang SK, Lee KH, Kim HJ, Brody J, Jeon NL. Langmuir. 2007;23:10910. doi: 10.1021/la7026835. [DOI] [PubMed] [Google Scholar]
- 21.Mosadegh B, Saadi W, Wang SJ, Jeon NL. Biotechnol. Bioeng. 2008;100:1205. doi: 10.1002/bit.21851. [DOI] [PubMed] [Google Scholar]
- 22.Mosadegh B, Kuo C-H, Tung Y-C, Torisawa Y, Bersano-Begey T, Takayama S. Nat. Phys. 2010;6:433. doi: 10.1038/nphys1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosadegh B, Agarwal M, Tavana H, Bersano-Begey T, Torisawa YS, Morell M, Wyatt MJ, O'Shea KS, Barald KF, Takayama S. Lab Chip. 2010;10:2959. doi: 10.1039/c0lc00086h. [DOI] [PubMed] [Google Scholar]
- 24.Keenan TM, Folch A. Lab Chip. 2008;8:34. doi: 10.1039/b711887b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Jeon N, Baskaran H, Dertinger SK, Whitesides GM, Van de Water L, Toner M. Nat. Biotechnol. 2002;20:826. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- 26.Oppegard SC, Blake AJ, Williams JC, Eddington DT. Lab Chip. 2010;10:2366. doi: 10.1039/c004856a. [DOI] [PubMed] [Google Scholar]
- 27.Chen YA, King AD, Shih HC, Peng CC, Wu CY, Liao WH, Tung YC. Lab Chip. 2011;11:3626. doi: 10.1039/c1lc20325h. [DOI] [PubMed] [Google Scholar]
- 28.Khanal G, Chung K, Solis-Wever X, Johnson B, Pappas D. Analyst. 2011;136:3519. doi: 10.1039/c0an00845a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen JW, Bhatia SN. Biotechnol. Bioeng. 2003;82:253. doi: 10.1002/bit.10569. [DOI] [PubMed] [Google Scholar]
- 30.Mehta G, Mehta K, Sud D, Song JW, Bersano-Begey T, Futai N, Heo YS, Mycek MA, Linderman JJ, Takayama S. Biomed. Microdevices. 2007;9:123. doi: 10.1007/s10544-006-9005-7. [DOI] [PubMed] [Google Scholar]
- 31.Deiss F, Mazzeo A, Hong E, Ingber DE, Derda R, Whitesides GM. Anal. Chem. 2013;85:8085. doi: 10.1021/ac400161j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Derda R, Laromaine A, Mammoto A, Tang SK, Mammoto T, Ingber DE, Whitesides GM. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18457. doi: 10.1073/pnas.0910666106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Derda R, Tang SK, Laromaine A, Mosadegh B, Hong E, Mwangi M, Mammoto A, Ingber DE, Whitesides GM. PLoS One. 2011;6:e18940. doi: 10.1371/journal.pone.0018940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fister JS, Memoli VA, Galante JO, Rostoker W, Urban RM. J. Biomed. Mater. Res. 1985;19:519. doi: 10.1002/jbm.820190505. [DOI] [PubMed] [Google Scholar]
- 35.Ascuitto RJ, Ross-Ascuitto NT. Sem. Perinatol. 1996;20:542. doi: 10.1016/s0146-0005(96)80068-1. [DOI] [PubMed] [Google Scholar]
- 36.Hoerter JA, Opie LH. Biol. Neonate. 1978;33:144. doi: 10.1159/000241064. [DOI] [PubMed] [Google Scholar]
- 37.Rowland RT, Meng XZ, Ao LH, Terada LS, Harken AH, Brown JM. Surgery. 1995;118:446. doi: 10.1016/s0039-6060(05)80357-5. [DOI] [PubMed] [Google Scholar]
- 38.Singer D. Compar. Biochem. Physiol. a. 1999;123:221. doi: 10.1016/s1095-6433(99)00057-4. [DOI] [PubMed] [Google Scholar]
- 39.Yano Y, Braimbridge MV, Hearse DJ. J. Thor. Cardiovasc. Surg. 1987;94:887. [PubMed] [Google Scholar]
- 40.Zhu H, McElwee-Witmer S, Perrone M, Clark KL, Zilberstein A. Cell Death Diff. 2000;7:773. doi: 10.1038/sj.cdd.4400721. [DOI] [PubMed] [Google Scholar]
- 41.Chi NC, Bussen M, Brand-Arzamendi K, Ding C, Olgin JE, Shaw RM, Martin GR, Stainier DYR. Proc. Natl. Acad. Sci. U.S.A. 2010;107:14662. doi: 10.1073/pnas.0909432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torisawa YS, Mosadegh B, Luker GD, Morell M, O'Shea KS, Takayama S. Integr. Biol. 2009;1:649. doi: 10.1039/b915965g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bujak M, Frangogiannis NG. Cardiovasc. Res. 2007;74:184. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Postlethwaite AE, Keski-Oja J, Moses HL, Kang AH. J. Exp. Med. 1987;165:251. doi: 10.1084/jem.165.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Circ. 2002;106:130. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 46.Arslan F, de Kleijn DP, Pasterkamp G. Nat. Rev. Cardiol. 2011;8:292. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 47.Vidi V, Waxman S. Naghavi M, editor. Humana Press. 2010:671. [Google Scholar]
- 48.Simon K, Park KM, Mosadegh B, Subramaniam AB, Mazzeo A, Ngo PM, Whitesides GM. Biomat. 2014;1:259. doi: 10.1016/j.biomaterials.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iglesias-Garcia O, Pelacho B, Prosper F. J. Molec. and Cell. Cardiol. 2013;62:43. doi: 10.1016/j.yjmcc.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 50.Schaaf S, Shibamiya A, Mewe M, Eder A, Stohr A, Hirt MN, Rau T, Zimmermann WH, Conradi L, Eschenhagen T, Hansen A. PLoS One. 2011;6:e26397. doi: 10.1371/journal.pone.0026397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams WJ, Pong T, Geisse NA, Sheehy SP, Diop-Frimpong B, Parker KK. J. Computer-Aided Mater. Des. 2007;14:19. [Google Scholar]
- 52.Geisse NA, Sheehy SP, Parker KK. Vitro Cellul. Develop. Biol.-Animal. 2009;45:343. doi: 10.1007/s11626-009-9182-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.