

Abstract
Multiple Sclerosis (MS) pathology is marked by the massive infiltration of myelin-specific T cells into the central nervous system (CNS). Hallmarks of T helper (Th) cells during active disease are pro-inflammatory Th1/Th17 cells that predominate over immunoregulatory Th2/Treg cells. Neurodegeneration, a major factor in progressive MS, is often overlooked when considering drug prescription. Here, we show that oral dosing with SNJ-1945, a novel water soluble calpain inhibitor, reduces experimental autoimmune encephalomyelitis (EAE) clinical scores in vivo and has a two pronged effect via anti-inflammation and protection against neurodegeneration. We also show that SNJ-1945 treatment downregulates Th1/Th17 inflammatory responses, and promotes regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) in vivo, which are known to have the capacity to suppress helper as well as cytotoxic T cell functions. Through analysis of spinal cord samples, we show a reduction of calpain expression, decreased infiltration of inflammatory cells and signs of inhibition of neurodegeneration. We also show a marked reduction of neuronal cell death in spinal cord (SC) sections. These results suggest that calpain inhibition attenuates EAE pathology by reducing both inflammation and neurodegeneration, and could be used in clinical settings to augment the efficacy of standard immunomodulatory agents used to treat MS.
Keywords: EAE, Calpain, Inflammation, Multiple Sclerosis
INTRODUCTION
Multiple Sclerosis (MS) is a multifaceted disease, involving interactions between the immune system and the central nervous system (CNS) leading to axonal and neuronal degeneration caused by pro-inflammatory attack on the CNS (Murray 2005, Poser & Brinar 2004). Current approved therapies for MS mainly focus on immunosuppression and immunomodulation while also having many side effects (Wipfler et al. 2011). However, MS is a heterogeneous disease involving both inflammation and neurodegeneration (Lassmann 2013). Another concern is that only two therapies on the market today are orally available, while most therapies are still invasive injections. Therefore, new therapeutic strategies must be developed that not only target the immune arm of the disease, but also the neurodegenerative arm, diminish unwanted side effects, and also be orally available.
While the etiology of MS remains unknown, the activation of myelin-specific T cells is thought to be a major event in the development and progression of MS and EAE. Thus, the immune system remains an active therapeutic target. Ca2+-activated neutral protease calpain is associated with increased disease in MS and EAE (Banik et al. 1987, Berlet 1987, Sato et al. 1982, Sato et al. 1984). Both ubiquitous and tissue-specific calpains have been identified which play multiple roles in physiological events including cell proliferation and differentiation, T cell activation, T cell migration, signal transduction, platelet activation, membrane fusion, necrosis, and apoptosis. Interestingly, STAT6, which is involved in Th2 cell survival and cytokine production, is a direct substrate degraded by calpain. It is widely accepted that decreasing Th1 and Th17 inflammatory cells while increasing Th2 and Treg cells can lead to decreased disease severity in EAE. (Kivisakk et al. 2003, Lassmann & Ransohoff 2004, Kennedy et al. 1992, Issazadeh et al. 1995, Harrington et al. 2005, Nishihara et al. 2007, Park et al. 2005). Inhibition of calpain has been shown by our laboratory and others to decrease EAE disease signs in rats and mice (Guyton et al. 2005, Guyton et al. 2010, Guyton et al. 2009, Guyton et al. 2006, Smith et al. 2011a, Hassen et al. 2008, Hassen et al. 2006). Enzymatic activity of calpain is increased in peripheral blood mononuclear cells (PBMCs) of MS relapse and remission patients compared to cells of healthy volunteers (Imam et al. 2007). Treatment of MS patient blood samples with calpain inhibitor decreased the levels of inflammatory T cells and cytokines (Smith et al. 2011b). The roles of calpain in T cell activation are multifaceted and include direct modulation of signaling proteins that lead to cytokine production (Hendry & John 2004, Schaecher et al. 2004). This shows that calpain inhibition can act to reduce inflammatory cytokines in vitro.
As stated before, inflammation is not the only mediator of disease; neurodegeneration plays a strong role in EAE and MS (Murray 2005, Poser & Brinar 2004, Lassmann 2013). It is well established that calpain expression and activity is upregulated in white matter, gray matter MS and EAE (Shields et al. 1999, Shields & Banik 1998a, Zheng & Bizzozero 2011). Studies have shown calpain is a mediator of degeneration of axons and neuronal death in EAE (Schaecher et al. 2001, Shields et al. 1999). Myelin proteins are substrates of calpain, and cleavage of myelin proteins directly damages the myelin sheath as well as creating antigenic peptides that activate myelin specific T cells (Schaecher et al. 2001, Deshpande et al. 1995, Medveczky et al. 2006). Previous studies demonstrated increased calpain activity and cell-specific overexpression in neural cells (astrocytes, microglia) in EAE and MS, implicating a pivotal role for calpain in myelin breakdown in these diseases (Shields & Banik 1998a, Shields & Banik 1999, Shields et al. 1999). Treatment of EAE animals with calpain inhibitor has been shown to decrease neuronal cell death and loss of myelin (Guyton et al. 2010), and is also considered as neuroprotective (Ryu et al. 2011). What remains to be studied is whether calpain inhibition by an orally available compound SNJ-1945 reduces immune arm (inflammatory Th1/Th17 cells) as well as neurodegeneration (neuronal death and axonal damage) in vivo. However, one drawback of calpain inhibitor treatment is the solubility of the previous inhibitors is very low, making it use as a treatment option unappealing as it has to be given I.P. While many calpain inhibitors are available, we have chosen SNJ-1945, which is more water soluble than other calpain inhibitors, and demonstrates a low toxicity (Shirasaki et al. 2006, Oka et al. 2006). The goal of this study is to investigate whether this calpain inhibitor given orally would reduce EAE disease in mice, by ameliorating inflammation and neurodegeneration. Here, we report that oral dosing with this new water soluble SNJ-1945 reduces EAE disease significantly. We also show a significant reduction in inflammatory cytokines and inflammatory Th cells, and an increase in anti-inflammatory Tregs and myeloid derived suppressor cells (MDCS) in the host. CNS inflammation is also markedly decreased with oral SNJ-1945 treatment. Importantly, we show that neurodegeneration is decreased by a reduction of gliosis, myelin loss and neuronal cell death. These findings support the notion that calpain inhibition is effective in both reducing inflammation and neurodegeneration in EAE leading to a possible more effective less toxic translational treatment for MS.
MATERIALS AND METHODS
EAE Induction
Male B10.PL mice 4-5 weeks old were purchased from Jackson Laboratories (Bar Harbor, ME). Mice (18-20 grams) were immunized with (1:1) CFA containing Mycobacterium tuberculosis H37Ra (10 mg/ml; Difco), phosphate-buffered saline (PBS), and guinea pig MBP (1 mg/ml). Each mouse received 4 subcutaneous (s.c.) injections, one over each haunch, totaling 400 μl of the emulsion which contains 400 μg of MBP. Control animals received PBS/CFA alone. At the time of the induction the mice also received an i.p. injection of pertussis toxin (Sigma) (200 ng/mouse) approximately 48 hours later. Mice were monitored daily for clinical scores of paralysis (0-no change; 1-limp tail; 2-hind limb weakness; 3-hind limbs plegic; 4-front limb weakness; 5-paraplegic). Half number scores were used if the animal was partway between clinical score criteria levels. Experiments were approved by the Medical University of South Carolina (MUSC).
Administration of Calpain Inhibitor
SNJ-1945, a 3rd generation water-soluble calpain inhibitor was used (Oka et al. 2006). Mice received oral gavage of the calpain inhibitor SNJ-1945 (50mg/kg) or 0.5% carboxymethylcellulose vehicle twice daily, from days 9 post-induction until sacrifice. An effective dose was previously determined based on SNJ1945 IC50 (Shirasaki et al. 2006) and unpublished studies on dose-dependent data from our laboratory showing maximal biological effect at 50mg/kg dose from day 9 of disease, signs of inflammation and optometric response (OKR) sensitivity loss became apparent.
Isolation of PBMCs
Pooled blood samples (1mL/mouse) from EAE and control mice were collected. PBMCs were isolated from these blood samples and washed twice in Hanks Balanced Salt Solution (HBSS) as described (Imam et al., 2007). Briefly, anticoagulant-treated whole blood was mixed with equal volumes of HBSS and layered on top of Ficoll-Paque Plus™ (GE Healthcare, Piscataway, NJ), and centrifuged. The upper layer of plasma was carefully drawn off, leaving the lymphocyte layer undisturbed at the interface. This layer was transferred to a centrifuge tube and suspended in 6 ml of HBSS. After centrifugation, the supernatant was aspirated. The pellet was re-suspended in 6 ml of HBSS and centrifuged once more. PMBCs in the pellet were counted and diluted in RPMI 1640 medium containing 1% penicillin/streptomycin and 10% Fetal Bovine Serum to a concentration of 3 × 106 cells/ml.
Cell Proliferation
At time of sacrifice, the draining lymph nodes (LN) and spleen were dissected out and the tissue was ground into a single cell suspension. The LN cells were grown with a 10-fold excess of irradiated isogenic spleen cells and were aliquotted into 96-well plates. Triplicate wells were stimulated with 40μg/mL whole guinea pig myelin basic protein (MBP), 10μg/mL optic nerve homogenate (ONH), or left unstimulated. After 48h, 100 μl of supernatant was removed for cytokine profiling and 2 μCi of 3H-thymidine in media was added per well. After 24 more hours the cells were harvested with a semi-automated cell harvester then a scintillation analyzer was used to determine the amount of 3H-thymidine present in the cells as an indicator of DNA replication and proliferation.
ELISA
As described above, 100 μl of supernatant from duplicate wells containing LN cells was collected after 24h of culture. Single analyte ELISA kits for IL-17, IFNγ, and IL-10 (SABiosciences, Frederick, MD) were utilized to determine cytokine expression according to the manufacturer's protocol. Briefly, a serial dilution of the antigen standard (2000, 1000, 500, 250, 125, 61.5, 31.25, 0 pg/ml) was prepared for IL-17, IL-10 and IFNγ recombinant antigens. Assay buffer (50μL) was aliquotted into each well of an ELISA plate pre-coated with primary capture antibodies. Samples (50μL) were transferred in duplicate to the appropriate wells of the plate. The plate was then incubated for 2 hrs at room temperature (RT). The plate was washed three times with 350μL of wash buffer before 100μL of detection antibody was added to the wells. After an 1-hr incubation at RT, the plate was washed three times then 100μL of avidin-HRP was distributed to the wells. Following 30 minutes of incubation and four washes, 100μL of development solution was added and the plate incubated for 15 min in the dark. Stop solution was added (100μL) and the plate was immediately read at 450nm O.D on a spectrophotometer, using SoftMax Pro Software version 5.4 (Softmax Pro Sunnyvale, CA).
Flow Cytometry
PBMCs or lymph were isolated from EAE mice (5×106 cells per ml) were cultured in 6-well plates and stimulated for 3 days with 40μg/mL purified whole guinea pig MBP (Sigma-Aldrich, St. Louis, MO) in the presence or absence of 100μM calpeptin. Cells were stained using a Treg or Th17 multi-color flow cytometry kits or GR-1, Integrin α (R&D Systems, Minneapolis, MN) according to the manufacturer's protocol. Briefly, PBMCs were then harvested and washed twice in PBS. Approximately 5×105 cells/group, were re-suspended in 0.5 mL of Fixation/Permeabilization Buffer and incubated at 2 - 8° C for 30 minutes. After washing, the cell pellet was re-suspended in 200 μL of the Permeabilization/Wash Buffer. To each group of cells, 10 μL of primary antibody (CD4, CCR6, IL-17, IL-4, CD25, Gr-1, Integrin-α) or corresponding isotype control antibody was added. After incubating the mixture for 30-45 minutes at 2-8° C in the dark, the PBMCs were washed in 2 mL of Permeabilization/Wash Buffer and the final cell pellet was re-suspended in 300 μL of PBS for flow cytometric analysis using FACSDiva software 6.0 (BD Biosciences, San Jose, CA).
PCR
Total RNA was extracted from splenocytes using the RNEasy Mini Kit (Qiagen) according to the manufacturer's protocol. The reverse transcription reaction was performed using the iScript cDNA Synthesis Kit (Bio-Rad). Briefly, the following components were combined to form a 20 μL reaction volume: nuclease-free water, 5x iScript Select reaction mix, oligo (dT)20 primer, total RNA (2 μg), and Reverse Transcriptase (RT). The reaction tubes were incubated for 90 minutes at 42°C, then incubated for 5 minutes at 85°C (to inactivate RT). PCR was executed in a programmed thermal cycler (Biometra). β-actin mRNA was used as a control for each experiment using the primer sequence 5′- ATATCGCTGCGCTGGTCGTCGA -3′ (sense) and 5′- ACCCATTCCCACCATCACACCCTG -3′ (anti-sense). Cytokine and transcription factor primers were used to test for FoxP3 5′- TCCTTCCCAGAGTTCTTCCA -3′ (sense) 5’-GGCTAGGTTGGAACTAGGGG -3’(anti-sense); IL-17 5′- CTCCAGAAGGCCCTCAGACTA -3′ (sense) and 5′- AGCTTTCCCTCCGCATTGACACAG -3′ (anti-sense) Thermal cycling parameters were 95°C for 10 minutes followed by 40 cycles of amplifications at 96°C for 3 seconds, 55°C for 3 seconds, and 68°C for 5 seconds, followed by a final elongation step of 72°C for 10 minutes. PCR products were visualized using Geldoc software following electrophoresis in 1.5% agarose gel with ethidium bromide staining. Average relative density of 4 samples each were calculated, normalized to control and plotted.
Immunoblotting
Procedures for protein analyses have been previously described (Kupina et al. 2001, Das et al. 2005, Sharma & Rohrer 2007). Briefly, spinal cords were homogenized in 50 mM Tris buffer (pH 7.4) containing 5 mM ethylene glycol tetraacetic acid (EGTA) and 1 mM phenylmethylsulfonyl fluoride (PMSF). After determination of protein concentration, known amounts of all samples were separated by 4-20% linear gradient SDS-polyacrylamide gel electrophoresis (SDS-PAGE). After SDS-PAGE, proteins were transferred onto nitrocellulose membranes, which were probed with primary antibodies against m-calpain and β-actin. All antibodies for western blotting were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and diluted at a concentration of 1:200. Blots were incubated with horseradish peroxidase (HRP)-conjugated secondary anti-rabbit (1:2,000) antibody. Specific protein bands were detected using the FluorChem FC2 Chemiluminescent CCD detection system (Alpha Innotech, San Leandro, CA). Western blot data were analyzed using Image J software (National Institutes of Health, Bethesda, MD) to determine optical density (OD) of bands, average relative density of 5 samples each were calculated, normalized to control and plotted.
H&E Staining
Frozen spinal cord tissues were sliced into 8 μm sections. Immune cell infiltration into the spinal cord and perivascular cuffing were examined following H&E staining of the tissue sections, as we described previously (Shields et al. 1998). Photos taken at 400x magnification)
TUNEL Assay and Immunohistochemistry
Lumbar portions of the spinal cord (SC) from the EAE study were dissected. The tissue was then embedded in OCT, and cryosectioned (8 mm). In order to detect cell-specific death in spinal cord tissues, the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) assay was combined with cell-specific marker labeling, assay was performed according to a previously described protocol (Guyton et al. 2009). Briefly, spinal cord tissues were sectioned, fixed as described above, and then post-fixed in 4% methanol-free formaldehyde (in PBS) for 15 min. The slides were saturated with TdT buffer (50 μL/slide) for 5 min and then replaced with TUNEL reaction mixture (50 μL/slide) containing 10× polymerase chain reaction (PCR) mixture containing digoxigenin (DIG)-11-dUTP (2.5 μL) and terminal TdT (25 Units) in buffer (Promega, Madison, WI). Cell-specific immunohistochemical labeling was performed as previously described (Guyton et al. 2009). Briefly, prior to primary antibody staining, non-specific binding sites were blocked with the same serum as the secondary antibody for 1 hourr at room temperature then incubated with NeuN (1:100, clone A60; Chemicon, Temecula, CA, USA), monoclonal CD11b (1:100, clone OX42; Invitrogen, Carlsbad, CA, USA), monoclonal glial fibrillary acidic protein (GFAP; 1:400, clone GA5; Chemicon), or pan monoclonal dNFP (1:100, SMI-311; Covance Princeton, NJ, USA) overnight at 4°C. Sections were incubated with a (1:100) secondary antibody conjugated with Texas Red (Vector Laboratories, Burlingame, CA) or fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Vector Laboratories, Burlingame, CA, USA) a for 1 hour in the dark. The slides were mounted with 1 drop of Vectashield Mounting Medium (Vector Laboratories) and coverslipped. The sections were viewed under a fluorescence microscope at 200x magnification. CD11b and MBP/NFP stained sections were viewed at 400x magnification.
Statistical Analyses
All statistical tests were chosen on the basis of well-recognized recommendations (Fleming et al. 2005) for analyzing data from EAE studies and performed using the SAS statistics software (SAS Institute, Cary, NC, USA). Overall statistical significance for various data including median clinical score (CS) and day of onset were performed using the Kruskal–Wallis test followed by the Mann–Whitney U-tests for pairwise comparisons after area under the curve. Overall significant differences in protein expression, mRNA expression were analyzed using the non-parametric Kruskal–Wallis test followed by Mann–Whitney U-tests for pairwise comparisons. The null hypothesis for each analysis was rejected at p≤0.05.
RESULTS
The daily administration of calpain inhibitor SNJ-1945 decreases clinical scores in EAE mice
MS patients have few options when it comes to oral therapeutics, and the therapeutics that are on the market only focus on the immune arm of the disease. We speculate that calpain inhibition affects both the immune and neurodegenterative arms of MS and EAE, but until recently calpain inhibitors have not been viable human therapeutic because of its non-water solubility making it an injectable like previous MS therapies. While injectables are an option, they are less desired by the already suffering patients. SNJ-1945 is a new more water soluble calpain inhibitor that has shown therapeutic effects in other disease models such as retinal degeneration and lissencephaly (Azuma & Shearer 2008, Toba et al. 2013). Here we aimed to show the in vivo effect of oral treatment to directly asses the validity of this new calpain inhibitor SNJ-1945 as an oral therapeutic for EAE/MS. We performed daily oral dosing on B10-PL mice with EAE starting at day 9 post induction. The disease scores of Control-vehicle (Control), EAE-vehicle (EAE), and EAE SNJ-1945 treated mice (SNJ-1945) were recorded (Figure 1). EAE vehicle-treated mice were consistent with what has been described in the literature having severe paralysis symptoms (Papenfuss et al. 2004). Overall, there was a statistically significant difference in the paralysis scores between Control-Vehicle, EAE-Vehicle, and EAE-SNJ-1945-treated mice. Not only did mice treated with SNJ-1945 experience significantly milder manifestations of the disease, but also the monophasic peak of paralytic signs was delayed relative to EAE-vehicle mice. Both of these positive in vivo outcomes show that SNJ-1945 is a valid oral therapy for EAE mice.
Figure 1. Oral with SNJ 1945 improves clinical scores of paralysis in EAE.
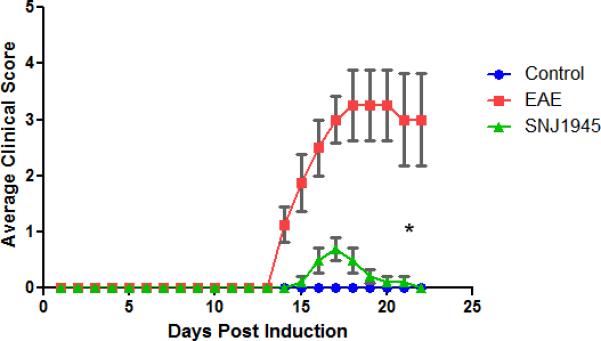
Average clinical scores are shown for mice treated with SNJ1945 by oral gavage twice daily starting at day 9 (arrow shows start of treatment), 50mg/kg. Scale: 0 (no clinical disease), 0.5 (piloerection), 1 (tail weakness), 1.5 (tail paralysis), 2 (hindlimb weakness), 3 (hindlimb paralysis), 3.5 (forelimb weakness), 4 (forelimb paralysis), 5 (moribund/death). Half numbers were used for animals in between scores to correctly reflect visible signs of the disease. Average clinical scores (0<5) are compared for control-vehicle, EAE-Vehicle, and EAE SNJ 1945 treated mice. N=5 for all groups, * p<0.05 compared to EAE.
Calpain inhibitor SNJ-1945 reduces inflammatory cells and cytokines while supports Tregs
It is widely accepted that autoreactive Th1 and Th17 cells predominate in the MS and EAE while Tregs and Th2 cells are more common in normal and remission patients. Here we investigated how daily oral dosing of SNJ-1945 affects the balance of Th cells. The responses of lymph node (LN) cells isolated from EAE mice to purified MBP antigenic components were assayed in a primary cell culture system. It was hypothesized that incubation of SNJ-1945-treated T cells with MBP antigens would evoke a relative immunosuppressive response; particularly from memory T cells, due to promotion of a Th2 bias. As expected, it was found that stimulation of LN cells from EAE vehicle-treated mice with MBP elicited significantly higher levels of proliferation than all groups of cells from control-vehicle and SNJ-1945-treated mice (Figure 2A). Within SNJ-1945-treated and untreated EAE T cells, there was a trend of increased proliferation of MBP-stimulated cells vs. unstimulated, which implies recognition of the immunizing antigen.
Figure 2. In vivo treatment with calpain inhibitor SNJ1945 decreases proliferation of primary lymph node cells, reduces secretion of Th cytokines, and supports Th2 cytokines and transcription factor STAT-6.
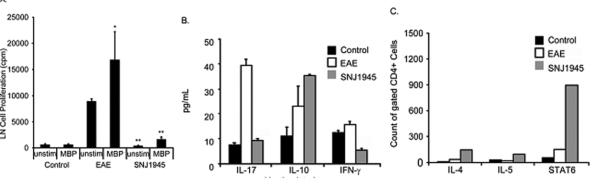
After immunization, animals were given SNJ1954 (50mg/kg, oral administration) daily on days 9-25 post-induction. Draining lymph nodes (LN) and spleen from control, EAE, and SNJ1945 treated mice were then analyzed. (A) Proliferation analysis of LN via scintillation counter for 3H-thymidine present in the cells as an indicator of DNA replication in the presence or absence of MBP stimulation. (B) The LN cells were grown with a 10-fold excess of irradiated isogenic spleen cells and were stimulated with 40μg/mL whole guinea pig myelin basic protein (MBP). After 48h, 100 μl of supernatant was removed per well for analysis of cytokines by ELISA. (C) Cells stimulated with 40μg/mL MBP were analyzed by FACS for cytokines IL-4, IL-5 and the transcription factor STAT6. N=3, animals in each group. Cells were pooled and one experiment was run for each. *p≤0.05 compared to control; **p≤0.05 compared to EAE.
Looking deeper in Th cell response upon stimulation, LN cells were again isolated from control, EAE, and SNJ-1945 treated mice. Cytokine profiling revealed that EAE LN cells secreted significantly more IL-17 and less IL-10 compared with control LN cells (Figure 2B). Conversely, treatment with SNJ-1945 showed that cells expressed less inflammatory IL-17 and more anti-inflammatory IL-10 compared with vehicle-treated EAE mouse cells (Figure 2B). Therefore, cells from SNJ-1945-treated animals were strongly biased away from inflammation (IL-17), towards higher levels of IL-10, this may serve to decrease antigen presentation and differentially regulate circulating pro-inflammatory cells.
To further investigate Th helper cell bias with SNJ-1945 treatment, PBMCs were isolated from treated mice to perform intracellular FACS. Focusing on cells gated for CD4 expression, we graphed the absolute number of cells expressing IL-4, IL-5 and STAT6. Compared to control and EAE, many more SNJ-1945-treated PBMCs expressed IL-4, IL-5, and STAT6. The number of IL-5-expressing cells was relatively quite low. As expected, Stat-6 levels significantly increased when mice were treated with SNJ-1945, indicating that it is working through a STAT mediated mechanism (Figure 2C).
T-regulatory cells can help diminish inflammation; we looked at CD25 expressing cells in the lymph nodes of mice treated with SNJ-1945. We saw a trend toward increasing CD25 positive cells with treatment as compared to vehicle treated EAE mice (Figure 3A). Conversely, when we looked at CCR6 expression (a marker for inflammatory Th17 cells) from mice treated with SNJ-1945, we saw a significant decrease in these CCR6-expressing cells as compared to vehicle treated EAE animals. To confirm this finding, we analyzed mRNA of the splenic cells taken from these same mice. We observed an inhibition of Th17 SNJ-1945 treated mice by a significant decrease in IL-17 mRNA as compared to vehicle treated mice (Figure 3B). FoxP3 (a marker of T-regulatory cells) mRNA expression was also significantly increased in the SNJ-1945 mice as compared to the vehicle treated EAE mice (Figure 3B). These findings further confirm the T cell bias in EAE mice, a change after calpain inhibition treatment to more of a regulatory or anti-inflammatory type from inflammatory. It is also believed that MDSCs serve as regulatory cells and are mainly anti-inflammatory. Interestingly, the number of MDSCs increased upon in vivo treatment of EAE mice with SNJ-1945. This data suggests that they may have rendered an important anti-inflammatory effect in the progression of the disease (Figure 3A). Overall, here we show that inflammatory Th cells are reduced upon treatment with SNJ-1945 while regulatory/anti-inflammatory factors and cells are increased in mice. Taken together, these data confirm that SNJ-1945 treatment aids in suppressing the inflammatory immune arm of the disease.
Figure 3. SNJ1945 treatment effects on myeloid suppressor cells, Th17 cells, and Tregs.
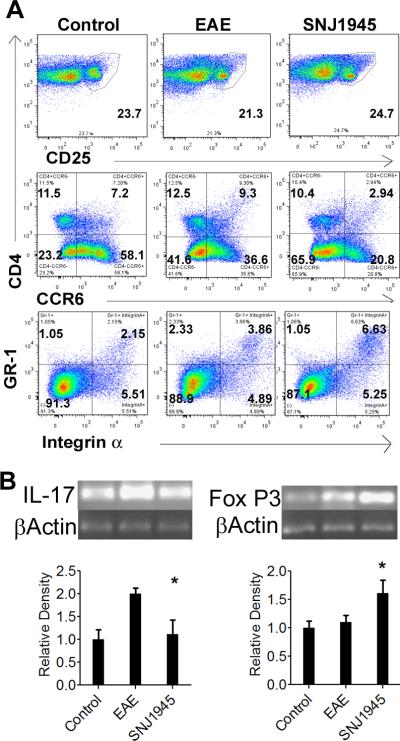
After immunization with MBP, animals were treated with 50mg/kg orally twice daily starting on 9 days post induction and continuing every day until sacrifice. (A) Primary lymph node cells from Control, EAE Vehicle (untreated), and SNJ1945 treated mice were analyzed via flow cytometry for expression of Tregs cells (CD4 + CD25+), Th17 cells (CD4+CCR6+) and MDSCs (GR1+Integrin-α). For each data point, cells were pooled from 4 animals. (B) Spleen cells from the same animals were analyzed by semi-quantiative RT-PCR for inflammatory cytokine IL-17 and anti-inflammatory FoxP3 gene expression for Control, EAE and SNJ1945 groups. The relative density shown is normalized to control. N=4, *p≤0.05.
The daily administration of calpain inhibitor SNJ-1945 reduces EAE disease in the CNS
MS has many effects in the periphery of patients, however, the CNS is where long lasting neurodegeneration is executed. Previous studies have indicated that SNJ-1945 can enter the BBB (Toba et al. 2013). Here, we show that in EAE animals, SNJ-1945 given orally was able to inhibit calpain expression in the CNS. We looked at calpain protein levels in SCs of EAE mice with and without SNJ-1945 oral treatment. We observed a significant reduction of calpain expression in animals treated with SNJ-1945 as compared to EAE animals treated with vehicle (Figure 4). This is a good indication that SNJ-1945 given orally was able to cross into the CNS and render its suppressive effects.
Figure 4. Calpain is inhibited in the CNS.
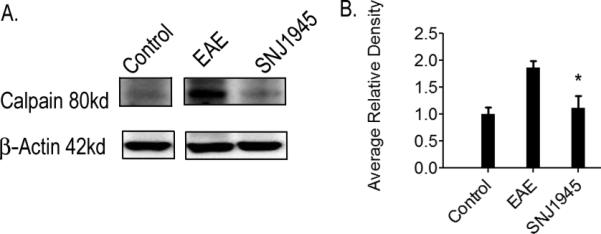
After induction of EAE, mice received twice-daily oral doses of SNJ-1945 or vehicle, and were sacrificed on day 25 post-induction for tissue analysis. Treatment groups consist of Control, EAE, EAE-50μg/kg SNJ-145 (SNJ1945). Representative blots and densitometric analysis were shown. (A) Calpain expression. (B) Quantification of densitometry represented as average relative density normalized to control. N=5
Inflammation in the CNS is one of the hallmarks of MS and EAE. Treating EAE animals with IP calpain inhibitor is thought to dampen T cell activation and migration into the CNS but weather oral administration of SNJ-1945 also has similar effects remains unknown. To address this question, we looked at inflammatory cell inflammation in SC sections. EAE animals treated with vehicle alone demonstrated a marked increase in perivascular cuffing of immune cells (Figure 5A). Animals treated with SNJ-1945, in contrast, demonstrated fewer immune cells in the spinal cord tissue and less perivascular cuffing. We also assessed gliosis in EAE samples. Gliosis is a proliferation process of resident glial cells (astrocytes, microglia), which is increased during neuron damage and CNS inflammation. We investigated these cells by staining for glial fibrillary acidic protein (GFAP). We stained lumbar sections of SC from animals treated orally with SNJ-1945 and showed a significant decrease in GFAP expression as compared to vehicle treated EAE animals (Figure 5B). To further examine the effects of SNJ-1945 therapy on inflammatory cells during EAE, spinal cord sections were stained with antibody against CD11b (Fig. 5C). CD11b is expressed on the surface of many leukocytes involved in the immune system, including monocytes, granulocytes, macrophages, and natural killer cells, as well as on CNS resident macrophages known as microglia. Treatment of EAE animals with vehicle alone exhibited prominent increases in microgliosis compared with control animals. In contrast, staining for CD11b spinal cord tissues from animals treated with SNJ-1945 were reduced. These data suggest that treating EAE animals with SNJ-1945 can reduce clinical signs of the disease, in part by reducing infiltration of immune cells and or proliferation of resident inflammatory cells.
Figure 5. Treatment with SNJ 1945 reduces CNS inflammation in EAE mice.

Frozen spinal cord sections were taken from Control, EAE and SNJ-1945 treated (SNJ) mice and stained for (A) Inflammatory cell infiltration as shown by H&E staining (arrows), 400x magnification. (B) Gliosis is shown by GFAP single staining (red), 200x magnification. (C) Microglial populations were shown by CD11b single staining (green), 400x magnification.
SNJ-1945 calpain inhibitor is different from current FDA approved therapies for MS because it has the potential to be neuroprotective in the CNS itself and not just be anti-inflammatory as well as being orally bioavailable. To explore this, we examined signs of neuroprotection in the CNS. Part of the pathology of neurodegeneration in MS and EAE is a marked increase in axonal degeneration. This occurs when the myelin sheath is damaged and the axon is exposed. We stained lumbar SC sections with an antibody against dephosphorylated neurofilament protein (dNFP), which is known to be increased after axonal injury. A significant decrease in dNFP was seen in mice treated with SNJ-1945 as compared to vehicle treated EAE animals (Figure 6A).
Figure 6. Treatment with SNJ 1945 reduces neurodegeneration in the CNS.

Frozen spinal cord sections were taken from Control, EAE and SNJ-1945 treated (SNJ) mice, and stained for (A) Axonal degeneration marker dNFP using single staining, 200x magnification. (B) Myelin loss around axons MBP (red) & NFP (green) were shown by double staining, 400x magnification. (C) Neuronal cell death (arrows), TUNEL (red), NeuN (green), and TUNEL-NeuN double staining with DAPI (overlay); 200x magnification.
In order for axon exposure and degradation to occur myelin itself has to be damaged or degraded. No oral calpain therapy has been shown to protect against myelin degradation. To investigate whether oral treatment of EAE with SNJ-1945 calpain inhibitor can reduce degradation of myelin around axons, we looked at myelin loss via double staining of MBP (a myelin marker) and NFP (an axonal marker). We show a marked decrease in myelin surrounding axons in EAE vehicle treated animals (Figure 6B). Treatment with SNJ-1945 maintains myelin protection around axons. Apoptotic cell death of neurons is seen during the disease states of EAE. We investigated the effect of SNJ-1945 treatment on neuron cell death in the CNS. We show significantly higher levels of TUNEL-positive neurons in lumbar sections of the SC taken from vehicle treated EAE as compared with SC sections from EAE animals treated with SNJ-1945 and controls (Figure 5C). This indicates a neuroprotective effect by SNJ-1945 treatment in EAE. These results further confirm the reduction of inflammation and neurodegeneration provided by an oral administration of calpain inhibitor SNJ-1945 in the CNS.
DISCUSSION
MS is a multifaceted disease and not only involves a severe autoimmune arm but also a prolonged progressive neurodegenerative component. Current therapeutics for MS are mainly focused on immune modulation and have significant drawbacks while only reducing relapses by 30% (Hassan-Smith & Douglas 2011). None of the current therapeutics on the market aim to help protect against neurodegeneration. Also, there are limited treatments that are orally available. The present study revealed that treatment of animals orally with calpain inhibitor SNJ-1945 was sufficient to decrease clinical disease scores. Moreover, we found that calpain inhibition significantly decreased Th inflammatory cells in the periphery of EAE mice while increasing immune regulatory cells. Inflammation in the CNS was shown to be reduced via loss of infiltrating immune cells, loss of gliosis and CD11b positive inflammatory cells. We also determined that SNJ-1945 provided neuroprotection in the CNS through decreased axonal damage and neuron death. Furthermore, myelin protection was found to be retained with treatment of calpain inhibitor. Thus, calpain inhibition shows effectiveness in reducing EAE both by being anti-inflammatory and neuroprotective.
Calpain inhibition as a therapy is a novel approach. Here we have shown that it offers great potential in reducing EAE through anti-inflammatory and neuroprotective mechanisms. Before recently, calpain inhibition as a therapy has been hindered by lack of solubility. SNJ-1945 was engineered to be more water soluble and thus more bioavailable. We have previously shown that calpain expression is increased in CNS diseases, however the exact mechanism is not known but post transcriptional regulation is thought to play a major role (Shields & Banik 1998b, Shields & Banik 1999). In order to verify that calpain was inhibited in SC tissues from EAE animals and those treated with SNJ-1945 were measured for calpain expression and showed a significant reduction.
In MS, Th1/Th17 cells predominate in relapses while Th2/Treg cells are shown in remission patients and balanced in control patients (Smith et al. 2011b, Shields & Banik 1999). Here, we show a relative increase in Treg cells and a decrease in Th17 cells after oral administration of SNJ-1945, which correlates with the lower disease scores found in the treated animals. Recently, Treg cells were shown to be only slightly decreased in EAE but have a significant reduction of function (Venken et al. 2010, Venken et al. 2008). This might explain our slight trend in reduction of Treg cells but not completely diminishing in EAE vehicle treated animals. MDSCs also serve as regulatory cells to help decrease inflammation. We found that these types of cells were increased upon treatment with SNJ-1945 indicating its anti-inflammatory effect. Furthermore, infiltration of immune cells into the CNS was reduced when SNJ-1945 was given. This may have been due to inhibition of activated peripheral immune cells which have been shown to enter the CNS even before the onset of disease (Shields & Banik 1999). Importantly, calpain has been found to be involved in immune cell activation and migration, and thus, its inhibition will likely reduce CNS inflammatory response (Schaecher et al. 2001, Deshpande et al. 1995, Guyton et al. 2010). CD11b is expressed on the surface of many leukocytes involved in the immune system as well as in resident microglia, the macrophages of the CNS. CD11b functions include regulating leukocyte adhesion and migration as well as other processes such as phagocytosis, cell-mediated cytotoxicity, chemotaxis and cellular activation. We have shown that CD11b is decreased in the CNS with SNJ-1945 oral administration to EAE animals. These outcomes show that SNJ-1945 like traditional therapies for MS can reduce and regulate inflammation during disease but with the added benefit of being orally deliverable.
While other therapeutics often ignore neuroprotection as a part of treatment, axonal damage is a key factor in neurological disability in MS patients (Trapp et al. 1999, Bjartmar & Trapp 2001). Myelin and other cytoskeletal proteins are known to be degraded by calpain and their degeneration has been detected in EAE as well as MS (Schaecher et al. 2001). Further support for calpain's role in axonal degeneration are provided by studies from MS patients post mortem tissue and have indicated that calpain could be co-localized with damaged axons (Diaz-Sanchez et al. 2006). In vitro studies have indicated that endogenous calpain inhibitor calpastatin partially blocked cleavage and degradation of myelin into antigenic fragments (Deshpande et al. 1995). Also studies show that when purified MBP was incubated with purified calpain, calpain cleaved MBP and this degradation was inhibited by calpeptin (Banik et al. 1997). Thus, therapies that reduce calpain activity may attenuate the destruction of myelin directly by reducing cleavage of axonal proteins and MBP. Protecting MBP degradation can also impact the immune arm of the disease by diminishing epitope spreading (Goebels et al. 2000). In the current study, we show a reduction of axonal damage through a reduction of dNFP and GFAP staining in spinal cords from animals treated with calpain inhibitor SNJ-1945. We also show an increase of myelin protection on the axons via MBP and NFP double staining.
MS inflammation involves both peripheral and CNS components, creating a global inflammation in the host. A number of studies suggest that calpain expression and activity is critically involved in the disease process and inhibition of calpain decreases inflammation (Cuzzocrea et al. 2000, Smith et al. 2011b, Guyton et al. 2009). Since one of the common factors involved in neurodegeneration is inflammation, it is expected that inhibition of global inflammation by calpain inhibitor will attenuate neurodegenerative process in EAE. However, whether inhibition of peripheral inflammation in EAE by calpain inhibition will completely block the neurodegeneration is not clear, but it may partially ameliorate the outcome. Nonetheless, this concept may be tested in a chemically induced demyelination model of EAE where peripheral inflammation is absent. Studies in our laboratory also suggest that calpain inhibition reduces neurodegeneration (Guyton et al. 2010, Ryu et al. 2011). EAE and MS are such multifaceted diseases, that it is hard to parse out whether the reduction of inflammation itself lead to the reduction of neurodegeneration. It has been shown that a reduction of inflammation in MS and EAE coincides with a reduction of CNS neuronal signs (Peterson et al. 2001, Guyton et al. 2006, Guyton et al. 2010). These published studies suggest that attenuation of only inflammation may not completely lead to beneficial reduction of neurodegeneration. However, since calpain has many direct substrates (e.g., cytoskeletal proteins, axon and myelin proteins) degradation of which will lead to neurodegeneration itself, a lot of the neuroprotective effects can be provided directly by SNJ-1945 as previously published (Ryu et al. 2011).
Previously, we demonstrated for the first time that calpain inhibition retained both sensory and motor neurons as well as protective myelin forming oligodendrocytes (Guyton et al. 2010). The prevention of MBP degradation and myelin by SNJ-1945 as shown in the present study also suggests that treatment increased protection of myelin forming oligodendroglia cells. It is also known that calpain inhibition can protect many neuronal cell types in vitro (Das et al. 2005). In the current study, SNJ-1945 therapy reduced the loss of neurons in spinal cord in EAE animals. These findings are significant since patients with MS suffer from many symptomatic outcomes based on loss of neurons, and therapies that protect multiple neuronal cell types can be advantageous in treating heterogeneous neurological diseases such as MS (Tekok-Kilic et al. 2006)
In conclusion, the present study demonstrates that calpain is a promising target for treating the inflammatory and neurodegenerative events associated with disability in EAE and MS. In the current investigation, EAE mice were treated twice daily with the calpain inhibitor SNJ-1945 and showed a marked amelioration in clinical scores of the disease. This treatment was two pronged, 1) targeted inflammatory arm of EAE by reducing inflammatory immune cells in the periphery to potentially block T cell activity and immune cell migration; and 2) neurodegenerative arm where we also showed a reduction of gliosis, axonal damage, and cell death directly in the CNS by inhibiting calpain. Studies to address both the immune and neurodegenerative arm are needed to help improve therapeutics for MS.
Acknowledgements
These authors have no financial or other conflict of interest.
Funding sources: This study is supported by NIH R01 NS041088 and NIH 5R01NS056176. We thank the Flow Cytometry and Cell Sorting Core of Medical University of South Carolina.
Abbreviations
- MS
Multiple Sclerosis
- CNS
central nervous system
- Th
T helper cells
- MDSC
myeloid-derived suppressor cells
- SC
Spinal cord
- EAE
experimental autoimmune encephalomyelitis
- STAT
Signal Transducer and Activator of Transcription
- PBMC
peripheral blood mononuclear cells
- CFA
complete freuds adjuvant
- MBP
Myelin Basic Protein
- PBS
Phosphate buffered saline
- HBSS
Hanks balanced salt solution
- LN
lymph node
- RT
room temperature
- PMSF
phenylmethylsulfonyl fluoride
- EGTA
ethylene glycol tetraacetic
- GFAP
Glial fibrillary acidic protein
- BBB
blood brain barrier
- dNFP
dephosphorylated neurofilament protein
- NFP
neurofilament protein
References
- Azuma M, Shearer TR. The role of calcium-activated protease calpain in experimental retinal pathology. Survey of ophthalmology. 2008;53:150–163. doi: 10.1016/j.survophthal.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik NL, Chakrabarti AK, Hogan EL. Distribution of calcium activated neutral proteinase (mM CANP) in myelin and cytosolic fractions in bovine brain white matter. Life Sci. 1987;41:1089–1095. doi: 10.1016/0024-3205(87)90626-6. [DOI] [PubMed] [Google Scholar]
- Banik NL, Matzelle DC, Gantt-Wilford G, Osborne A, Hogan EL. Increased calpain content and progressive degradation of neurofilament protein in spinal cord injury. Brain research. 1997;752:301–306. doi: 10.1016/s0006-8993(96)01488-6. [DOI] [PubMed] [Google Scholar]
- Berlet HH. Calcium-dependent neutral protease activity of myelin from bovine spinal cord: evidence for soluble cleavage products of myelin proteins. Neurosci Lett. 1987;73:266–270. doi: 10.1016/0304-3940(87)90256-4. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Current opinion in neurology. 2001;14:271–278. doi: 10.1097/00019052-200106000-00003. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, McDonald MC, Mazzon E, et al. Calpain inhibitor I reduces the development of acute and chronic inflammation. Am J Pathol. 2000;157:2065–2079. doi: 10.1016/S0002-9440(10)64845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Sribnick EA, Wingrave JM, Del Re AM, Woodward JJ, Appel SH, Banik NL, Ray SK. Calpain activation in apoptosis of ventral spinal cord 4.1 (VSC4.1) motoneurons exposed to glutamate: calpain inhibition provides functional neuroprotection. J Neurosci Res. 2005;81:551–562. doi: 10.1002/jnr.20581. [DOI] [PubMed] [Google Scholar]
- Deshpande RV, Goust JM, Hogan EL, Banik NL. Calpain secreted by activated human lymphoid cells degrades myelin. J Neurosci Res. 1995;42:259–265. doi: 10.1002/jnr.490420214. [DOI] [PubMed] [Google Scholar]
- Diaz-Sanchez M, Williams K, DeLuca GC, Esiri MM. Protein co-expression with axonal injury in multiple sclerosis plaques. Acta neuropathologica. 2006;111:289–299. doi: 10.1007/s00401-006-0045-0. [DOI] [PubMed] [Google Scholar]
- Fleming KK, Bovaird JA, Mosier MC, Emerson MR, LeVine SM, Marquis JG. Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;170:71–84. doi: 10.1016/j.jneuroim.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Goebels N, Hofstetter H, Schmidt S, Brunner C, Wekerle H, Hohlfeld R. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain : a journal of neurology. 2000;123(Pt 3):508–518. doi: 10.1093/brain/123.3.508. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Brahmachari S, Das A, Samantaray S, Inoue J, Azuma M, Ray SK, Banik NL. Inhibition of calpain attenuates encephalitogenicity of MBPspecific T cells. J Neurochem. 2009;110:1895–1907. doi: 10.1111/j.1471-4159.2009.06287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton MK, Das A, Matzelle DD, Samantaray S, Azuma M, Inoue J, Ray S, Banik NL. SJA6017 Attenuates Immune Cell Infiltration and Neurodegneration in EAE. 8th International Congress of Neuroimmunology. 2006:107–112. [Google Scholar]
- Guyton MK, Das A, Samantaray S, Wallace G. C. t., Butler JT, Ray SK, Banik NL. Calpeptin attenuated inflammation, cell death, and axonal damage in animal model of multiple sclerosis. J Neurosci Res. 2010;88:2398–2408. doi: 10.1002/jnr.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Sribnick EA, Matzelle DD, Tyor WR, Ray SK, Banik NL. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res. 2005;81:53–61. doi: 10.1002/jnr.20470. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature immunology. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Hassan-Smith G, Douglas MR. Management and prognosis of multiple sclerosis. Br J Hosp Med (Lond) 2011;72:M174–176. doi: 10.12968/hmed.2011.72.sup11.m174. [DOI] [PubMed] [Google Scholar]
- Hassen GW, Feliberti J, Kesner L, Stracher A, Mokhtarian F. A novel calpain inhibitor for the treatment of acute experimental autoimmune encephalomyelitis. J Neuroimmunol. 2006;180:135–146. doi: 10.1016/j.jneuroim.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Hassen GW, Feliberti J, Kesner L, Stracher A, Mokhtarian F. Prevention of axonal injury using calpain inhibitor in chronic progressive experimental autoimmune encephalomyelitis. Brain Res. 2008;1236:206–215. doi: 10.1016/j.brainres.2008.07.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry L, John S. Regulation of STAT signalling by proteolytic processing. Eur J Biochem. 2004;271:4613–4620. doi: 10.1111/j.1432-1033.2004.04424.x. [DOI] [PubMed] [Google Scholar]
- Imam SZ, Guyton MK, Haque A, Vandenbark A, Tyor WR, Ray SK, Banik NL. Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J Neuroimmunol. 2007;190:139–145. doi: 10.1016/j.jneuroim.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issazadeh S, Ljungdahl A, Hojeberg B, Mustafa M, Olsson T. Cytokine production in the central nervous system of Lewis rats with experimental autoimmune encephalomyelitis: dynamics of mRNA expression for interleukin-10, interleukin-12, cytolysin, tumor necrosis factor alpha and tumor necrosis factor beta. J Neuroimmunol. 1995;61:205–212. doi: 10.1016/0165-5728(95)00100-g. [DOI] [PubMed] [Google Scholar]
- Kennedy MK, Torrance DS, Picha KS, Mohler KM. Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol. 1992;149:2496–2505. [PubMed] [Google Scholar]
- Kivisakk P, Mahad DJ, Callahan MK, et al. Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A. 2003;100:8389–8394. doi: 10.1073/pnas.1433000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupina NC, Nath R, Bernath EE, Inoue J, Mitsuyoshi A, Yuen PW, Wang KK, Hall ED. The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. J Neurotrauma. 2001;18:1229–1240. doi: 10.1089/089771501317095269. [DOI] [PubMed] [Google Scholar]
- Lassmann H. Multiple sclerosis: Lessons from molecular neuropathology. Experimental neurology. 2013 doi: 10.1016/j.expneurol.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Ransohoff RM. The CD4-Th1 model for multiple sclerosis: a critical [correction of crucial] re-appraisal. Trends in immunology. 2004;25:132–137. doi: 10.1016/j.it.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Medveczky P, Antal J, Patthy A, Kekesi K, Juhasz G, Szilagyi L, Graf L. Myelin basic protein, an autoantigen in multiple sclerosis, is selectively processed by human trypsin 4. FEBS letters. 2006;580:545–552. doi: 10.1016/j.febslet.2005.12.067. [DOI] [PubMed] [Google Scholar]
- Murray TJ. Multiple sclerosis: the history of a disease. Demo; New York: 2005. [Google Scholar]
- Nishihara M, Ogura H, Ueda N, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. International immunology. 2007;19:695–702. doi: 10.1093/intimm/dxm045. [DOI] [PubMed] [Google Scholar]
- Oka T, Walkup RD, Tamada Y, Nakajima E, Tochigi A, Shearer TR, Azuma M. Amelioration of retinal degeneration and proteolysis in acute ocular hypertensive rats by calpain inhibitor ((1S)-1-((((1S)-1-benzyl-3-cyclopropylamino-2,3-di-oxopropyl)amino)carbonyl)-3-me thylbutyl)carbamic acid 5-methoxy-3-oxapentyl ester. Neuroscience. 2006;141:2139–2145. doi: 10.1016/j.neuroscience.2006.05.060. [DOI] [PubMed] [Google Scholar]
- Papenfuss TL, Rogers CJ, Gienapp I, Yurrita M, McClain M, Damico N, Valo J, Song F, Whitacre CC. Sex differences in experimental autoimmune encephalomyelitis in multiple murine strains. J Neuroimmunol. 2004;150:59–69. doi: 10.1016/j.jneuroim.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature immunology. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Annals of neurology. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Poser CM, Brinar VV. The nature of multiple sclerosis. Clinical neurology and neurosurgery. 2004;106:159–171. doi: 10.1016/j.clineuro.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Ryu M, Yasuda M, Shi D, et al. Critical role of calpain in axonal damage-induced retinal ganglion cell death. J Neurosci Res. 2011 doi: 10.1002/jnr.22800. [DOI] [PubMed] [Google Scholar]
- Sato S, Quarles RH, Brady RO. Susceptibility of the myelin-associated glycoprotein and basic protein to a neutral protease in highly purified myelin from human and rat brain. J Neurochem. 1982;39:97–105. doi: 10.1111/j.1471-4159.1982.tb04706.x. [DOI] [PubMed] [Google Scholar]
- Sato S, Quarles RH, Brady RO, Tourtellotte WW. Elevated neutral protease activity in myelin from brains of patients with multiple sclerosis. Ann Neurol. 1984;15:264–267. doi: 10.1002/ana.410150310. [DOI] [PubMed] [Google Scholar]
- Schaecher K, Goust JM, Banik NL. The effects of calpain inhibition on IkB alpha degradation after activation of PBMCs: identification of the calpain cleavage sites. Neurochem Res. 2004;29:1443–1451. doi: 10.1023/b:nere.0000026410.56000.dd. [DOI] [PubMed] [Google Scholar]
- Schaecher KE, Shields DC, Banik NL. Mechanism of myelin breakdown in experimental demyelination: a putative role for calpain. Neurochem Res. 2001;26:731–737. doi: 10.1023/a:1010903823668. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Rohrer B. Sustained elevation of intracellular cGMP causes oxidative stress triggering calpain-mediated apoptosis in photoreceptor degeneration. Curr Eye Res. 2007;32:259–269. doi: 10.1080/02713680601161238. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Putative role of calpain in the pathophysiology of experimental optic neuritis. Exp Eye Res. 1998a;67:403–410. doi: 10.1006/exer.1998.0537. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Upregulation of calpain activity and expression in experimental allergic encephalomyelitis: a putative role for calpain in demyelination. Brain Res. 1998b;794:68–74. doi: 10.1016/s0006-8993(98)00193-0. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Pathophysiological role of calpain in experimental demyelination. J Neurosci Res. 1999;55:533–541. doi: 10.1002/(SICI)1097-4547(19990301)55:5<533::AID-JNR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proceedings of the National Academy of Sciences. 1999;96:11486–11491. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Tyor WR, Deibler GE, Hogan EL, Banik NL. Increased calpain expression in activated glial and inflammatory cells in experimental allergic encephalomyelitis. Proc Natl Acad Sci U S A. 1998;95:5768–5772. doi: 10.1073/pnas.95.10.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki Y, Yamaguchi M, Miyashita H. Retinal penetration of calpain inhibitors in rats after oral administration. Journal of ocular pharmacology and therapeutics : the official journal of the Association for Ocular Pharmacology and Therapeutics. 2006;22:417–424. doi: 10.1089/jop.2006.22.417. [DOI] [PubMed] [Google Scholar]
- Smith AW, Das A, Guyton MK, Ray SK, Rohrer B, Banik NL. Calpain inhibition attenuates apoptosis of retinal ganglion cells in acute optic neuritis. Invest Ophthalmol Vis Sci. 2011a;52:4935–4941. doi: 10.1167/iovs.10-7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AW, Doonan BP, Tyor WR, Abou-Fayssal N, Haque A, Banik NL. Regulation of Th1/Th17 cytokines and IDO gene expression by inhibition of calpain in PBMCs from MS patients. J Neuroimmunol. 2011b;232:179–185. doi: 10.1016/j.jneuroim.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekok-Kilic A, Benedict RH, Zivadinov R. Update on the relationships between neuropsychological dysfunction and structural MRI in multiple sclerosis. Expert review of neurotherapeutics. 2006;6:323–331. doi: 10.1586/14737175.6.3.323. [DOI] [PubMed] [Google Scholar]
- Toba S, Tamura Y, Kumamoto K, et al. Post-natal treatment by a blood-brain-barrier permeable calpain inhibitor, SNJ1945 rescued defective function in lissencephaly. Scientific reports. 2013;3:1224. doi: 10.1038/srep01224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Current opinion in neurology. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Venken K, Hellings N, Broekmans T, Hensen K, Rummens JL, Stinissen P. Natural naive CD4+CD25+CD127low regulatory T cell (Treg) development and function are disturbed in multiple sclerosis patients: recovery of memory Treg homeostasis during disease progression. J Immunol. 2008;180:6411–6420. doi: 10.4049/jimmunol.180.9.6411. [DOI] [PubMed] [Google Scholar]
- Venken K, Hellings N, Liblau R, Stinissen P. Disturbed regulatory T cell homeostasis in multiple sclerosis. Trends in molecular medicine. 2010;16:58–68. doi: 10.1016/j.molmed.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Wipfler P, Harrer A, Pilz G, Oppermann K, Trinka E, Kraus J. Recent developments in approved and oral multiple sclerosis treatment and an update on future treatment options. Drug Discov Today. 2011;16:8–21. doi: 10.1016/j.drudis.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Zheng J, Bizzozero OA. Decreased activity of the 20S proteasome in the brain white matter and gray matter of patients with multiple sclerosis. Journal of neurochemistry. 2011;117:143–153. doi: 10.1111/j.1471-4159.2011.07182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]