

Abstract
Background
Factor XI (FXI) deficiency is a rare autosomal recessive disorder. Many patients with even very low FXI levels (<20 IU/dL) are asymptomatic or exhibit only mild bleeding, whereas others experience severe bleeding usually following trauma. Neither FXI antigen nor activity predicts bleeding risk in FXI-deficient patients.
Objectives
1) Characterize the formation, structure and stability of plasma clots from patients with severe FXI deficiency, and 2) Determine whether these assays can distinguish asymptomatic patients (“non-bleeders”) from those with a history of bleeding (“bleeders”).
Methods
Platelet-poor plasmas were prepared from 16 severe FXI-deficient patients who were divided into bleeders or non-bleeders, based on bleeding associated with at least two tooth extractions without prophylaxis. Clot formation was triggered by recalcification and addition of tissue factor and phospholipids in the absence or presence of tissue plasminogen activator and/or thrombomodulin. Clot formation and fibrinolysis were measured by turbidity, and fibrin network structure by laser scanning confocal microscopy.
Results
Non-bleeders and bleeders had similarly low FXI levels, normal prothrombin times, normal levels of fibrinogen, factor VIII, von Willebrand factor, factor XIII, and normal platelet number and function. Compared to non-bleeders, bleeders exhibited lower fibrin network density and lower clot stability in the presence of tissue plasminogen activator. In the presence of thrombomodulin, 7 of 8 bleeders failed to form a clot, whereas only 3 of 8 non-bleeders did not clot.
Conclusions
Plasma clot structure and stability assays distinguished non-bleeders from bleeders. These assays may reveal hemostatic mechanisms in FXI-deficient patients and have clinical utility for assessing bleeding risk.
Keywords: bleeding, Factor XI, Factor XI deficiency, plasma clot lysis time, fibrin clot lysis time
INTRODUCTION
Factor XI (FXI) deficiency is a rare, autosomal recessive disorder present in 1:1,000,000 individuals. Homozygous or compound heterozygous patients have low FXI levels (<20 IU/dL), whereas heterozygotes have moderately-reduced FXI levels (25–70 IU/dL) [1, 2]. Spontaneous bleeding is rare; however, patients with severe FXI deficiency can present with tissue-specific bleeding following surgery or injury, predominantly at sites with high fibrinolytic activity (mouth, nose, genitourinary tract), or menorrhagia [1, 2].
Previous studies have shown that when clotting is initiated with low tissue factor (TF) concentrations, the FXI level mediates thrombin generation, fibrin formation and inhibition of fibrinolysis. Reduced FXI levels lead to diminished thrombin generation and reduced rate of fibrin formation [3–6]. Subsequent studies extended these observations and showed that reduced thrombin generation results in reduced activation of the thrombin-activatable fibrinolysis inhibitor (TAFI) [7, 8] and consequently, reduced resistance of clots to fibrinolysis. These findings suggest an essential role for FXI in normal blood coagulation and clot stability, and provide a rationale for increased bleeding risk in FXI-deficient patients.
Interestingly, however, patients with similarly reduced FXI antigen and activity levels exhibit variable bleeding tendencies [9–11]. Some patients are asymptomatic even after trauma, while others display bleeding with trauma, or bleeding that begins several hours or even days following trauma. Neither FXI antigen nor activity correlate with clinical risk of bleeding, and activated partial thromboplastin time (APTT) assays do not predict bleeding risk.
Attempts to differentiate FXI-deficient patients with or without a bleeding tendency have focused on plasma thrombin generation characteristics. Rugeri et al. [12] isolated contact-inhibited platelet-rich plasma from healthy controls and patients with FXI deficiency divided into severe and mild/non-bleeder groups and measured thrombin generation triggered by addition of low TF. They showed that compared to controls, mild/non-bleeders have normal thrombin generation, but severe bleeders exhibit prolonged lag times and reduced rates and peaks of thrombin generation. In contrast, Guéguen et al. [13] did not detect significant differences in the thrombin generation peaks or endogenous thrombin potentials of either platelet-rich or platelet-poor plasma isolated from FXI-deficient patients categorized as bleeders or non-bleeders, although lag times in platelet-rich plasmas from bleeders were slightly (non-significantly, P=0.07) prolonged compared to those from non-bleeders. Neither of these studies evaluated plasma clot formation or stability. Consequently, the relationship between plasma clot quality and bleeding risk in these patients remains unknown.
Herein, we characterized plasma clot formation and quality in severe FXI-deficient patients and show that plasma clotting assays can identify patients with increased bleeding risk.
MATERIALS AND METHODS
Materials
Corn trypsin inhibitor and rabbit lung thrombomodulin were from Haematologic Technologies, Inc. (Essex Junction, VT, USA). Innovin (human TF) was from Siemens Healthcare Diagnostics (Newark, DE, USA). Tissue plasminogen activator (t-PA) was from American Diagnostica, Inc (Stamford, CT, USA). AlexaFluor488-conjugated fibrinogen (6 mol dye/mol fibrinogen) was prepared as described [14].
Human subjects
The FXI-deficient cohort consisted of 16 unrelated Israeli patients who were referred to the Sheba Medical Center for evaluation of a bleeding tendency or prolonged APTT, and whose FXI level was less than 9 IU/dL. A subgroup of 8 patients (5 males, 3 females) was defined as “bleeders” because they previously bled excessively following at least two separate sessions of tooth extractions performed under no prophylactic means. Excessive bleeding was determined when patients had oozing of blood for one hour or more after extraction, when bleeding reoccurred within 24 hours, or when the patient returned to the clinic or was hospitalized to achieve hemostasis. “Non-bleeders” were 8 patients (4 males, 4 females) who underwent at least two uneventful tooth extractions without prophylaxis (Table 1). Bleeding histories were obtained by two experienced clinicians and patients were classified as bleeders or non-bleeders prior to further laboratory testing. A cohort of healthy controls consisted of 10 unrelated individuals (4 males, 6 females) with normal levels of FXI. The mean age of patients (bleeders and non-bleeders) was 57±15 years, and of controls was 51±11 years (Table 1).
Table 1.
Demographic characteristics of subjects and clinical coagulation tests and clotting factor levels
| Controls (n=10) | Non-bleeders (n=8) | Bleeders (n=8) | P ANOVA | P* Controls vs Non-Bleeders | P* Controls vs Bleeders | P* Bleeders vs Non-Bleeders | |
|---|---|---|---|---|---|---|---|
| Age (years, mean ± SD) | 51 ± 11 | 58 ± 7 | 57 ± 21 | 0.41 | – | – | – |
| Male n (%) | 4 (40) | 4 (50) | 5 (63) | 0.67 | – | – | – |
| Prothrombin Time (%) | 107 ± 7 | 105 ± 13 | 104 ± 16 | 0.88 | – | – | – |
| APTT (seconds) | 31 ± 3 | 56 ± 6 | 68 ± 24 | <0.0001 | <0.01 | <0.001 | 0.11 |
| Thrombin Time (seconds) | 14.3 ± 1.6 | 14.9 ± 1.3 | 14.6 ± 0.5 | 0.65 | – | – | – |
| XI:Ac (%) | 114 ± 21.0 | 3 ± 1.4 | 3.34 ± 3.0 | <0.0001 | <0.001 | <0.001 | 0.40 |
| Fibrinogen (mg/dL) | 310 ± 51 | 318 ± 51 | 310 ± 35 | 0.93 | – | – | – |
| VIII:Ac (%) | 96 ± 27 | 104 ± 27 | 108 ± 24 | 0.63 | – | – | – |
| vWF:Ac (%) | 127 ± 38 | 161 ± 48 | 152 ± 49 | 0.27 | – | – | – |
| XIII:Ag (%) | 108±13$ | 116 ± 20 | 107 ± 25 | 0.60 | – | – | – |
| Antithrombin (%) | 93 ± 22 | 107 ± 17 | 102 ± 17 | 0.23 | – | – | – |
| TFPI (ng/mL) | 45.0 ± 19.8 | 54.0 ± 16.4 | 72.2 ± 32.8 | 0.07 | – | – | – |
| Protein C (%) | 103 ± 16 | 112 ± 18 | 107 ± 32 | 0.68 | – | – | – |
| Protein S:Ac (%) | 94 ± 18 | 100 ± 17 | 113 ± 28 | 0.20 | – | – | – |
| TAFI:Ag (%) | 62 ± 18 | 78 ± 31 | 87 ± 35 | 0.59 | – | – | – |
| PAI-1 (U/mL) | NA^ | 4.2 ± 3.4 | 4.2 ± 2.8 | 0.50& | – | – | – |
| Platelets/μL | 219700 ± 46162 | 207000 ± 71893 | 217000 ± 34297 | 0.87 | – | – | – |
| Platelet aggregation (%, collagen) | 88.0 ± 3 | 90.7 ± 3 | 89.5 ± 3 | 0.13 | – | – | – |
| Platelet aggregation (%, ADP) | 85.6 ± 6 | 90.4 ± 6 | 84.5 ± 5 | 0.11 | – | – | – |
| Platelet aggregation (%, epinephrine) | 85 ± 3 | 79 ± 20 | 77 ± 20 | 0.50 | – | – | – |
Mean±SD
ANOVA with Bonferroni post-hoc test,
t-test
FXIII:Ag only for 2 controls
NA, not assayed
Plasma preparation
Informed consent was obtained from each donor in accordance with the Declaration of Helsinki. Blood was collected by venipuncture through a 21-guage, butterfly needle into a syringe via a protocol approved by the Institutional Review Board of the Sheba Medical Center. The first 5 mL were discarded. The following 30 mL were drawn into a separate syringe containing sodium citrate/corn trypsin inhibitor (0.109 M/3.2% sodium citrate, pH 6.5, 18.3 μg/mL corn trypsin inhibitor) to minimize contact activation [15]. Platelet-poor plasma (PPP) was prepared by sequential centrifugation (150×g for 15 minutes, 1500×g for 15 minutes), aliquoted, and snap-frozen in −20 °C within 2 hours of blood collection. All analyses of plasma thrombin generation and clot formation, structure, and stability were performed in a blinded fashion.
Clinical coagulation testing
Prothrombin time, APTT, thrombin time, fibrinogen levels, plasma FXI and factor VIII activity levels were measured by ACL-TOP-500 (Instrumental Laboratories, Bedford, MA, USA), using RecombiPlasTin 2G, SynthASil, ThrombinTime, Fibrinogen C reagents, FXI- and FVIII-deficient plasma, respectively (HemosIL, Beckman Coulter Inc, Nyon, Switzerland). Von Willebrand factor (vWF) and protein S antigen levels were measured by ACL-TOP-500, using von-Willebrand antigen kit and free protein S antigen (HemosiL). PAI-1 activity levels were measured by Sysmex 1500 using Berichrom PAI kit (Siemens Healthcare Diagnostics, Marburg, Germany). Factor XIII, antithrombin and protein C activities were measured by chromogenic assays using Berichrom FXIII reagent, Liquid antithrombin, and Coamatic protein C chromagenix kit (Siemens). Platelet aggregation was evaluated by light transmission aggregometry (AggRAM, Helena Laboratories, Beaumont, TX, USA) using adenosine diphosphate (ADP, 10 μM, Diamed AG, Cressier, Switzerland), epinephrine (50 μM, Diamed AG) or collagen (9 μM/mL, Helena Laboratories) as platelet agonists. Changes in light transmission were recorded for 5 minutes and the aggregation maximal amplitude was measured. TAFI and tissue factor pathway inhibitor (TFPI) antigen levels were measured by ELISA (IMUCLONE TAFI ELISA kit, American Diagnostica Inc. CT, USA and Human TFPI ELISA, RayBiotech Inc, Norcross, USA, respectively).
Phospholipid vesicles
Phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine were from Avanti Polar Lipids (Alabaster, AL). Large unilamellar vesicles (41% phosphatidylcholine/44% phosphatidylethanolamine/15% phosphatidylserine) were made as described [16]. Briefly, lipids were combined, dried under nitrogen gas, and resuspended in cyclohexanes. Resuspended lipids were lyophilized, resuspended in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) pH 7.4, 150 mM NaCl containing 1 mM ethylenediamine tetraacetic acid, and extruded through a 0.2 μm filter ten times.
Thrombin generation assays
Thrombin generation was measured by calibrated automated thrombography [17]. Briefly, TF/phospholipids were mixed with plasma in a 96-well round-bottom microtiter plate (Becton Dickinson, Falcon), inserted into a Fluoroskan Ascent fluorometer (ThermoLabsystem, Helsinki, Finland), and warmed to 37 °C for 10 minutes. Reactions were initiated by automatically dispensing fluorogenic substrate and CaCl2 to each well. Final TF, phospholipid, fluorogenic substrate, and CaCl2 concentrations were 1 pM, 4 μM, 416 μM and 16 mM, respectively. Thrombin parameters (lagtime, time to peak, peak, and endogenous thrombin potential [ETP]) were calculated using Thrombinoscope software version 3.0.0.29 (Thrombinoscope BV, Maastricht, Netherlands), as we have described [18].
Characterization of clot formation and lysis
Clotting was initiated by incubating recalcified (10 mM CaCl2, final) PPP with TF and phospholipids (1:30,000 dilution of Innovin [0.5 pM TF] and 4 μM, final, respectively) in the absence or presence of t-PA (0.5 μg/mL, final) and thrombomodulin (5 nM, final). Final reaction volumes were 100 μL (90% and 85% PPP, final, for clotting and fibrinolysis assays, respectively) in 96-well plates. Clot formation and lysis were monitored by turbidity at 405 nm in a SpectraMax 340Plus plate reader (Molecular Devices, Sunnyvale, CA) for 2 hours at room temperature. The onset of clot formation was the time to the inflection point prior to the turbidity increase. The maximum slope was the slope of a line fitted to the maximum rate of turbidity increase (“Vmax”) using 5–10 points to determine the line. The peak turbidity change was the maximum turbidity of the clot less the starting turbidity of the plasma sample. For lysis experiments, the time to peak turbidity was the time to the inflection point at peak turbidity, and the area under the curve was the sum of trapezoids formed by turbidity curves less a baseline established by the lowest measurement recorded (Softmax Pro 5.4, Molecular Devices, Sunnyvale, CA, USA).
Fibrin structure analysis
Clots were produced by incubating recalcified (10 mM CaCl2, final) PPP with TF and phospholipids (1:30,000 dilution of Innovin [0.5 pM TF] and 4 μM, final, respectively) in Labtek II glass chamber slides. Plasmas were spiked with trace AlexaFluor488-congujated fibrinogen (80 μg/mL, final, 2.6% of total fibrinogen) to visualize fibrin fibers, as described [14]. Clots were scanned with a Zeiss LSM700 confocal laser scanning microscope (Carl Zeiss, Inc, Thornwood, NY, USA) linked to a Zeiss inverted microscope equipped with a Zeiss 63x oil immersion plan apo-chromatic lens, as described [14]. Thirty optical sections (1024×1024 pixels) were collected at 0.36-μm intervals in the z-axis. Images were processed using 3D deconvolution algorithms in AutoQuant software X 3.0.1 (Media Cybernetics Inc, Bethesda, MD, USA) prior to image analysis. Fibrin density was determined using ImageJ 1.41o by summing individual sections to create Z-projections, as described [19]. Briefly, thresholding was performed on Z-projections using the ImageJ thresholding function to visualize fibers and minimize noise. Thresholds were set on a per experiment basis, to compensate for differences in gain settings. The area covered by pixels above the threshold cut-off was determined using the ImageJ Measure function. Because fiber diameter (~200–400 nm) is at the lower resolution limit of laser scanning confocal microscopy [20, 21], we did not quantify fibrin diameter.
Statistical analyses
All thrombin generation and clot formation, structure, and fibrinolysis experiments were carried out in blinded fashion. Descriptive statistics for clotting, fibrinolysis, and fibrin structure parameters were summarized using means and standard deviations (SD). Parameters were compared between groups using analysis of variance (ANOVA) in Kaleidagraph version 4.1.3 (Synergy Software). Parameters showing significant differences were then analyzed with Bonferroni post hoc testing. P<0.05 was considered significant.
RESULTS
Coagulation tests and clotting factor levels do not correlate with bleeding risk in severe FXI-deficient patients
Compared to healthy controls, both non-bleeders and bleeders had similarly reduced levels of FXI (3.0±1.4 versus 3.3±3.0%, respectively), and prolonged APTTs (56±6 and 68±24 seconds, respectively). Both non-bleeders and bleeders had normal prothrombin and thrombin times, and normal levels of fibrinogen, FVIII, vWF, factor XIII, antithrombin, proteins C and S, and TAFI, and normal platelet numbers and function (Table 1). Bleeders had higher levels of TFPI than controls or non-bleeders, although these differences did not reach statistical significance (Table 1).
Thrombin generation parameters do not differ between FXI-deficient patients and controls
We first measured thrombin generation in plasmas from control individuals and FXI-deficient patients using calibrated automated thrombography. Although both non-bleeders and bleeders showed slightly prolonged lagtimes and times to peak and decreased peak thrombin generation compared to controls, these differences did not reach significance (Table 2). There were no differences in thrombin generation parameters between non-bleeders and bleeders (Table 2).
Table 2.
Thrombin generation parameters
| Controls (n=9) | Non-bleeders (n=8) | Bleeders (n=8) | P ANOVA | P Controls vs Non-Bleeders | P Controls vs Bleeders | P Bleeders vs Non-Bleeders | |
|---|---|---|---|---|---|---|---|
| Lagtime (minutes) | 4.2 ± 1.2 | 5.1 ± 1.7 | 4.9 ± 1.0 | 0.37 | – | – | – |
| Time to peak (minutes) | 9.5 ± 2.4 | 11.4 ± 2.9 | 10.8 ± 2.4 | 0.32 | – | – | – |
| Thrombin Peak (nM) | 86.1 ± 59.7 | 57.1 ± 26.3 | 57.6 ± 41.5 | 0.33 | – | – | – |
| ETP (nM*minutes) | 809 ± 387 | 789 ± 319 | 684 ± 394 | 0.76 | – | – | – |
Mean±SD
Reduced plasma clot formation rate reflects FXI deficiency
We then triggered clotting in plasmas from control individuals and FXI-deficient patients by recalcification and addition of TF and phospholipids, and monitored clot formation by turbidity (Fig. 1A). The onset times of fibrin formation for both bleeder and non-bleeder clots were prolonged compared to that seen in control clots, although these differences did not reach statistical significance (Table 3). Compared to controls, both bleeders and non-bleeders had significantly slower rate (Vmax) of clot formation (Fig. 1B, Table 3), suggesting the FXI level modulates the clot formation rate. Both bleeders and non-bleeders also had a prolonged time to turbidity plateau (maximal fibrin formation), though only bleeders were significantly different from controls (P<0.003, Fig. 1C, Table 3). These data suggest that the FXI level is a major determinant of the clot formation rate.
Figure 1. Plasma clot formation correlates with bleeding risk in FXI-deficient patients.
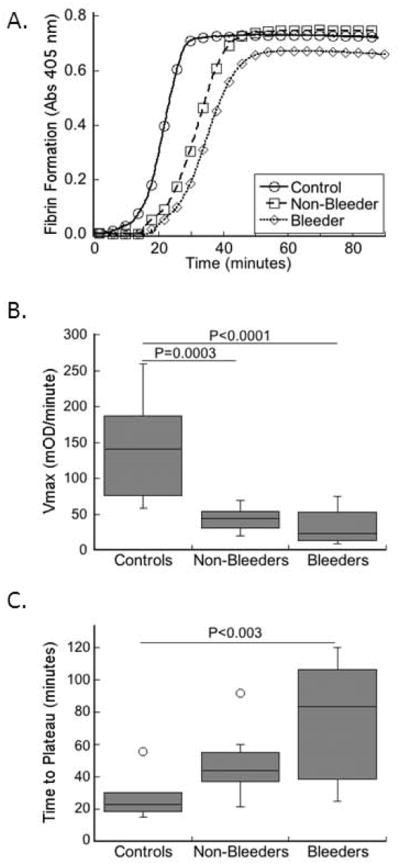
Clotting was triggered in plasmas from healthy individuals and FXI-deficient patients in a blinded fashion by recalcification and addition of TF and phospholipids. Clot formation was monitored by turbidity. A) Representative clot formation curves. B) Rate (Vmax) of clot formation in controls and FXI-deficient patients (non-bleeders and bleeders, as indicated). C) Time to plateau of turbidity in controls and FXI-deficient patients (non-bleeders and bleeders, as indicated). The boxes enclose 50% of the data with the median value displayed as a horizontal line, and lines enclose the interquartile distance (IQD). Open symbols represent points whose value falls more than 1.5-fold outside the IQD.
Table 3.
Plasma clot formation and fibrinolysis parameters
| Controls (n=10) | Non-bleeders (n=8) | Bleeders (n=8) | P ANOVA | P* Controls vs Non-Bleeders | P* Controls vs Bleeders | P* Bleeders vs Non-Bleeders | |
|---|---|---|---|---|---|---|---|
| Clotting | |||||||
| Onset Time (minutes) | 13.4 ± 7.8 | 16.0 ± 7.4 | 23.8 ± 16.9 | 0.16 | – | – | – |
| Vmax (mOD/minute) | 143.4 ± 67.5 | 43.3 ± 16.9 | 32.4 ± 25.5 | <0.0001 | 0.0003 | <0.0001 | 1 |
| Time to Plateau (minutes) | 28.4 ± 15.3 | 47.4 ± 17.4 | 74.5 ± 37.4 | <0.003 | 0.34 | <0.003 | 0.14 |
| Peak Turbidity Change | 0.69 ± 0.14 | 0.68 ± 0.13 | 0.56 ± 0.11 | 0.07 | – | – | – |
| Fibrin density (A.U.) | 222786 ± 37171 | 234574 ± 42621 | 177387 ± 22610 | <0.008 | 1 | <0.05 | <0.02 |
| Fibrinolysis | |||||||
| Onset Time (minutes) | 15.8 ± 8.8 | 11.6 ± 4.1 | 17.0 ± 6.1 | 0.28 | – | – | – |
| Vmax (mOD/minute) | 166.0 ± 50.0 | 49.5 ± 12.5 | 39.5 ± 16.6 | <0.0001 | <0.0001 | <0.0001 | 1 |
| Time to Peak (minutes) | 27.0 ± 11.3 | 32.2 ± 7.4 | 43.2 ± 17.3 | <0.05 | 1 | <0.05 | 0.28 |
| Lysis Time$ (minutes) | 36.3 ± 11.4 | 29.9 ± 5.6 | 23.7 ± 3.5 | <0.02 | 0.32 | <0.01 | 0.39 |
| Peak Turbidity Change | 0.64 ± 0.11 | 0.58 ± 0.14 | 0.40 ± 0.12 | <0.002 | 0.83 | <0.002 | <0.05 |
| Area under the Curve | 1482 ± 530 | 1176 ± 386 | 673 ± 210 | <0.002 | 0.39 | <0.002 | <0.07 |
Mean±SD
ANOVA with Bonferroni post-hoc test
Length of time between the midpoints of the increase and decrease in turbidity
Fibrin network structure correlates with bleeding risk in severe FXI-deficient patients
Given the differences in the clot formation parameters between control and FXI-deficient plasmas, we measured the fibrin structure of clots produced from these plasmas. Plasmas were re-calcified, and clot formation was triggered by addition of TF and phospholipids. Fluorescently-labeled fibrinogen was included as a tracer in these reactions to visualize fibrin network structure by confocal microscopy, as described [14]. Whereas fibrin structure in plasma clots from non-bleeders did not differ from controls, fibrin network density in plasma clots from bleeders was reduced ~20–25% compared to both controls (P<0.05) and non-bleeders (P<0.02) (Figs. 2A–B, Table 3). These findings demonstrate a unique feature of clots formed in bleeders. Specifically, FXI-deficient patients with increased bleeding produce abnormal fibrin network structure following TF-initiated clotting.
Figure 2. Fibrin network structure correlates with bleeding risk in FXI-deficient patients.

Clots were formed in a blinded fashion by recalcification and addition of TF and phospholipids in the presence of AlexFluor488-conjugated fibrinogen. Laser scanning confocal microscopy was performed as described in Methods. A) Representative confocal micrographs (z-projections of 30 individual slices) of clots formed in plasma from control individuals and FXI-deficient patients (non-bleeder and bleeder, as indicated). B) Fibrin network density (arbitrary units, A.U.) for controls and FXI-deficient patients (non-bleeders and bleeders, as indicated). The boxes enclose 50% of the data with the median value displayed as a horizontal line, and lines enclose the IQD. Open symbols represent points whose value falls more than 1.5-fold outside the IQD.
Plasma clot stability correlates with bleeding risk in severe FXI-deficient patients
The fibrin formation rate and fibrin network structure are major determinants of the ability of clots to withstand fibrinolysis; both reduced clot formation rate and reduced fibrin density are associated with increased susceptibility to fibrinolysis [19, 22]. To determine the ability of control and FXI-deficient plasma clots to resist fibrinolysis, clotting reactions were initiated by recalcification and addition of TF and phospholipids in the presence of t-PA. In this assay, clot formation competes with clot lysis, which is detected as an increase and subsequent decrease in turbidity (Fig. 3A) [23–25]. Similar to that seen in the absence of t-PA, both bleeders and non-bleeders plasmas exhibited a significantly (P<0.0001) slower rate of clot formation (Fig. 3B, Table 3). Other parameters (lag time, time to peak, peak turbidity change, lysis time, and area under the curve) were not significantly different between controls and non-bleeders. In contrast, compared to controls, FXI-deficient bleeder patient plasmas exhibited a significantly prolonged time to peak turbidity (Fig. 3C, P<0.05), shortened lysis time (Fig. 3D, P<0.01), and reduced peak turbidity change and area under the curve (Figs. 3E–F, P<0.002). Moreover, compared to plasmas from non-bleeders, plasmas from bleeders exhibited significantly lower peak turbidity change and a trend to lower area under the curve (Figs. 3E–F). These data suggest FXI-deficient patients who bleed have reduced resistance of clots to fibrinolysis.
Figure 3. Plasma clot stability correlates with bleeding risk in FXI-deficient patients.

Clotting was triggered by recalcification and addition of TF and phospholipids in the presence of t-PA in a blinded fashion. Clot formation and lysis were monitored by turbidity. A) Representative clot formation and lysis curves. B–F) Values for: B) rate (Vmax), C) time to peak turbidity, D) lysis time, E) peak turbidity change, and F) area under the clotting curve for plasmas from controls and FXI-deficient patients (non-bleeders and bleeders, as indicated). The boxes enclose 50% of the data with the median value displayed as a horizontal line, and lines enclose the IQD. Open symbols represent points whose value falls more than 1.5-fold outside the IQD.
Addition of thrombomodulin to plasma clot formation assays differentiates non-bleeders and bleeders
Finally, since the protein C/S anticoagulant system decreases procoagulant (fibrin-generating) activity, we tested the effect of this pathway on clot formation in plasmas from non-bleeders and bleeders. For these experiments, we triggered clotting by recalcification and addition of TF and phospholipids in the presence of thrombomodulin (5 nM, final). This concentration of thrombomodulin decreases thrombin generation in normal plasma by ~90% [18]. Under these conditions, 5 of 8 non-bleeders were still able to form a clot, whereas only 1 of 8 bleeders was able to form a clot. These data suggest addition of thrombomodulin to clot formation assays improves the sensitivity and ability to distinguish plasmas from non-bleeders and bleeders.
Together, these data suggest that bleeding in FXI-deficient patients results from the formation of abnormally-structured clots that have decreased resistance to fibrinolysis.
DISCUSSION
Coagulation factor deficiencies typically manifest with a high correlation between bleeding severity and clotting factor levels in the patients’ plasma. For most clotting factors, plasma levels above 5% of the normal level are usually sufficient to prevent bleeding. However, FXI deficiency is unique. Patients with FXI levels above 5% can develop significant bleeding symptoms, while patients having less than 1% FXI can be asymptomatic [26, 27]. Currently there is no explanation for this phenomenon. In this study, we present an assay that distinguishes bleeders from non-bleeders among patients with severe FXI deficiency.
Our findings show that FXI deficiency (both in bleeders and non-bleeders) correlated with a lower rate of fibrin formation, suggesting that the plasma FXI level is an important determinant of thrombin generation and accordingly, the rate of conversion of fibrinogen to fibrin. Interestingly, these data imply that a reduced fibrin formation rate, alone, does not predict bleeding in FXI-deficient patients ipso facto. In contrast, compared to both controls and non-bleeders, bleeders exhibited significantly reduced fibrin network density, a parameter that has been previously associated with decreased clot stability and bleeding disorders (reviewed in [22]). Accordingly, as expected, bleeders exhibited reduced clot stability, with reduced peak turbidity and area under the curve, compared to both controls and non-bleeders. These data suggest fibrin clot structure and stability are major determinants of the risk of bleeding in FXI-deficient patients, but are not wholly dependent on the plasma FXI level. This finding suggests that in the setting of reduced FXI levels, additional, as yet undetermined, modifiers influence fibrin network structure and stability, and mediate the risk of bleeding.
Previously, Guéguen et al. investigated biological determinants of bleeding in a cohort of patients with FXI deficiency [13] and found that compared to non-bleeders, bleeders had normal levels of fibrinogen and factors II, V, VIII, IX, X, and XII, and anticoagulant and profibrinolytic proteins (proteins C and S, antithrombin, plasminogen, α2-plasmin inhibitor, and TAFI) that were not different than the levels in non-bleeders. Compared to non-bleeders, bleeders in the Guéguen et al. study had lower levels of thrombomodulin and vWF antigen; however, the difference in thrombomodulin levels was diminished in subsequent analysis. In contrast to that study, our cohort of bleeders had normal levels of vWF, suggesting plasma levels of this protein do not modify bleeding risk in our patient population. Other studies have shown that thrombin-mediated activation of FXI results in TAFI-dependent inhibition of fibrinolysis [7], and that reduced plasma procoagulant activity results in less TAFI activation and increased fibrinolysis. Since plasma clot structure also influences clot stability (reviewed in [28, 29]), and since we found that in the absence of fibrinolysis, bleeders and non-bleeders exhibited significant differences in fibrin network density, our data suggest that decreased clot stability in the bleeder group may also result from the failure of these plasmas to produce adequate clot structure. We previously showed that the presence of factor V Leiden or prothrombin G20210A did not affect post partum bleeding in patients with severe FXI deficiency [30], suggesting the risk of bleeding is also not modified by co-existence of the common thrombophilias. Although Ruggeri et al. [12] was able to distinguish bleeding risk in FXI-deficient patients using thrombin generation analysis of platelet-rich plasma, Guéguen et al. [13] were not able to reproduce these findings in either platelet-rich or platelet-poor plasma. Like Guéguen et al., we also did not detect significant differences in thrombin generation parameters in platelet-poor plasma, suggesting that differences in clot quality do not result from differences in plasma procoagulant (thrombin-generating) activity. Thus, at the present time, it remains unclear whether any single activity determines clot structure and stability in FXI-deficient patients, or whether the phenotype reflects the cumulative activity of the complete coagulation proteome. These findings support the use of global assays to assess hemostasis.
Bleeding typically occurs in tissues with high fibrinolytic activity, suggesting that in addition to differences in plasma clot formation, local fibrinolytic activity decreases clot stability. Consequently, inter-individual differences in the expression of endothelial procoagulant or fibrinolytic activity may also influence bleeding risk in FXI-deficient patients. To our knowledge, data on inter-individual differences in tissue-specific endothelial activity (e.g., t-PA or thrombomodulin expression) have not been reported. These measurements, though challenging, may reveal additional information on the mechanism in future studies.
The development of an assay that can identify patients with severe FXI deficiency who are likely to bleed has important implications. First, the ability to prospectively identify patients with high bleeding risk is likely to improve the standard of care of FXI-deficient patients facing surgery. Since antifibrinolytic agents such as tranexamic acid can prevent surgery-associated bleeding in FXI-deficient patients [31, 32], prophylactic use in at-risk patients prior to surgical procedures would be expected to reduce bleeding complications in these patients. Although confocal microscopy is not commonly used by clinical diagnostic laboratories, turbidity analysis of clot formation can be performed on widely-available microplate readers and provides information on clot stability in under 2 hours. Moreover, in contrast to thrombin generation tests, clot formation and lysis assays do not require a fluorescent plate reader or fluorescent substrates and are sensitive to fibrinolytic pathways. Consequently, plasma clot assays may be used in future studies to assess the efficacy of antifibrinolytic agents ex vivo. Second, recent work has suggested that FXI is a potential clinical target for anticoagulation and prevention of thrombosis [33]; however, experience with individuals who are congenitally deficient in FXI suggests this approach may lead to bleeding in some patients. As efforts to develop FXI antagonists move forward, our assay may provide a means to prospectively identify patients at risk for bleeding. Information provided by this assay may provide a method of individualizing anticoagulation therapy based on clot structure and stability characteristics.
Our study has several limitations. First, the number of individuals analyzed was small. However, even in this small cohort, several parameters emerged as significantly different between bleeders and non-bleeders, suggesting profound differences in clot structure and stability determine bleeding risk in these patients. Second, the characterization of patients into bleeder and non-bleeder categories is challenging across the clinical spectrum. Most patients with severe FXI deficiency do not exhibit spontaneous bleeding, but may bleed only following trauma to tissues with high fibrinolytic activity, e.g., oral mucosa, nasal mucosa, and urinary tract. We defined the bleeder population according to strict criteria that required a history of at least two tooth extractions carried out without prophylaxis. This standard hemostatic challenge is a common clinical procedure in which 49% of FXI-deficient patients bleed in the absence of prophylactic hemostatic measures [32], and provided an objective distinction between patient groups. Third, although TAFI contributes to clot stability in in vitro assays of hemostasis [34], we were not able to explicitly test the role of TAFI activity in these samples. Findings of similar thrombin generation and similar TAFI antigen levels in plasmas from bleeders and non-bleeders suggest TAFI activation would not differ substantially between these two groups. However, the TAFI ELISA has higher affinity for the Thr325Thr isoform than for the more active Thr325Ile isoform [35]. Thus, over-representation of the Thr325Thr isoform in bleeders could both inflate measured TAFI antigen and contribute to the observed decreased fibrinolytic stability. Additional studies are warranted to delineate any effect of TAFI polymorphism in FXI-deficient patients. Finally, our clot analysis was performed in the absence of platelets. Since platelets stabilize clots by several mechanisms, including increased thrombin generation, platelet-mediated clot retraction, and release of polyphosphates, inter-individual differences in platelet function may also contribute to the bleeder versus non-bleeder phenotype in FXI-deficient patients. We did not detect significant differences in standard measures of platelet function among groups in our study, although this finding does not preclude roles for functions that are not assessed by these clinical assays. Importantly, our data indicate that differences in fibrin network formation, structure, and stability between bleeder and non-bleeder patients arise independently of, though perhaps in addition to, any differences in platelet function.
In sum, plasma clot formation assays have clinical utility in predicting bleeding risk in FXI-deficient patients. These assays may reveal pathways that differentiate hemostatic mechanisms in these patients.
Acknowledgments
The authors acknowledge Maria M. Aleman, Bethany L. Walton, and James R. Byrnes for reading the manuscript, and Laura D. Gray and Lori A. Holle for technical assistance.
FUNDING
This study was supported by funding from the National Institutes of Health (R01HL094740 to ASW).
Footnotes
AUTHORSHIP CONTRIBUTIONS
M. Zucker designed and performed experiments, analyzed results and wrote the manuscript. U. Seligsohn supervised experiments and reviewed the manuscript. O. Salomon designed the clinical protocol and recruited the patients. A. S. Wolberg designed and supervised experiments, analyzed results and wrote the manuscript.
DISCLOSURE OF CONFLICT OF INTERESTS
The authors report no relevant conflict of interest.
References
- 1.Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost. 2009;7 (Suppl 1):84–7. doi: 10.1111/j.1538-7836.2009.03395.x. [DOI] [PubMed] [Google Scholar]
- 2.Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991;325:153–8. doi: 10.1056/NEJM199107183250303. [DOI] [PubMed] [Google Scholar]
- 3.von dem Borne PA, Meijers JC, Bouma BN. Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood. 1995;86:3035–42. [PubMed] [Google Scholar]
- 4.Cawthern KM, van’t Veer C, Lock JB, Di Lorenzo ME, Branda RF, Mann KG. Blood coagulation in hemophilia A and hemophilia C. Blood. 1998;91:4581–92. [PubMed] [Google Scholar]
- 5.Oliver JA, Monroe DM, Roberts HR, Hoffman M. Thrombin activates factor XI in activated platelets in the absence of factor XII. Arterio Thromb Vasc Biol. 1999;19:170–7. doi: 10.1161/01.atv.19.1.170. [DOI] [PubMed] [Google Scholar]
- 6.Allen GA, Wolberg AS, Oliver JA, Hoffman M, Roberts HR, Monroe DM. Impact of procoagulant concentration on rate, peak and total thrombin generation in a model system. J Thromb Haemost. 2004;2:402–13. doi: 10.1111/j.1538-7933.2003.00617.x. [DOI] [PubMed] [Google Scholar]
- 7.Von dem Borne PA, Bajzar L, Meijers JC, Nesheim ME, Bouma BN. Thrombin-mediated activation of factor XI results in a thrombin-activatable fibrinolysis inhibitor-dependent inhibition of fibrinolysis. J Clin Invest. 1997;99:2323–7. doi: 10.1172/JCI119412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bajzar L, Manuel R, Nesheim ME. Purification and characterization of TAFI, a thrombin-activatable fibrinolysis inhibitor. J Biol Chem. 1995;270:14477–84. doi: 10.1074/jbc.270.24.14477. [DOI] [PubMed] [Google Scholar]
- 9.Seligsohn U. High gene frequency of factor XI (PTA) deficiency in Ashkenazi Jews. Blood. 1978;51:1223–8. [PubMed] [Google Scholar]
- 10.Bolton-Maggs PH, Young Wan-Yin B, McCraw AH, Slack J, Kernoff PB. Inheritance and bleeding in factor XI deficiency. Br J Haematol. 1988;69:521–8. doi: 10.1111/j.1365-2141.1988.tb02409.x. [DOI] [PubMed] [Google Scholar]
- 11.Ragni MV, Sinha D, Seaman F, Lewis JH, Spero JA, Walsh PN. Comparison of bleeding tendency, factor XI coagulant activity, and factor XI antigen in 25 factor XI-deficient kindreds. Blood. 1985;65:719–24. [PubMed] [Google Scholar]
- 12.Rugeri L, Quelin F, Chatard B, De Mazancourt P, Negrier C, Dargaud Y. Thrombin generation in patients with factor XI deficiency and clinical bleeding risk. Haemophilia. 2010;16:771–7. doi: 10.1111/j.1365-2516.2010.02246.x. [DOI] [PubMed] [Google Scholar]
- 13.Guéguen P, Galinat H, Blouch MT, Bridey F, Duchemin J, Le Gal G, Abgrall JF, Pan-Petesch B. Biological determinants of bleeding in patients with heterozygous factor XI deficiency. Br J Haematol. 2012;156:245–51. doi: 10.1111/j.1365-2141.2011.08945.x. [DOI] [PubMed] [Google Scholar]
- 14.Campbell RA, Overmyer KA, Bagnell CR, Wolberg AS. Cellular procoagulant activities dictate clot structure and stability as a function of distance from the cell surface. Arterio Thromb Vasc Biol. 2008;28:2247–54. doi: 10.1161/ATVBAHA.108.176008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dargaud Y, Luddington R, Baglin TP. Elimination of contact factor activation improves measurement of platelet-dependent thrombin generation by calibrated automated thrombography at low-concentration tissue factor. J Thromb Haemost. 2006;4:1160–1. doi: 10.1111/j.1538-7836.2006.01905.x. [DOI] [PubMed] [Google Scholar]
- 16.Hope MJ, Bally MB, Webb G, Cullis PR. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochem Biophys Acta. 1985;812:55–65. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- 17.Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, Lecompte T, Beguin S. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4–15. doi: 10.1159/000071636. [DOI] [PubMed] [Google Scholar]
- 18.Machlus KR, Colby EA, Wu JR, Koch GG, Key NS, Wolberg AS. Effect of tissue factor, thrombomodulin, and elevated clotting factor levels on thrombin generation in the calibrated automated thrombogram. Thromb Haemost. 2009;102:936–44. doi: 10.1160/TH09-03-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gray LD, Hussey MA, Larson BM, Machlus KR, Campbell RA, Koch G, Ezban M, Hedner U, Wolberg AS. Recombinant factor VIIa analog NN1731 (V158D/E296V/M298Q-FVIIa) enhances fibrin formation, structure and stability in lipidated hemophilic plasma. Thromb Res. 2011;128:570–6. doi: 10.1016/j.thromres.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blomback B, Carlsson K, Hessel B, Liljeborg A, Procyk R, Aslund N. Native fibrin gel networks observed by 3D microscopy, permeation and turbidity. Biochim Biophys Acta. 1989;997:96–110. doi: 10.1016/0167-4838(89)90140-4. [DOI] [PubMed] [Google Scholar]
- 21.Collet JP, Moen JL, Veklich YI, Gorkun OV, Lord ST, Montalescot G, Weisel JW. The alphaC domains of fibrinogen affect the structure of the fibrin clot, its physical properties, and its susceptibility to fibrinolysis. Blood. 2005;106:3824–30. doi: 10.1182/blood-2005-05-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21:131–42. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Wolberg AS, Gabriel DA, Hoffman M. Analyzing fibrin clot structure using a microplate reader. Blood Coagul Fibrinolysis. 2002;13:533–9. doi: 10.1097/00001721-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Wolberg AS, Allen GA, Monroe DM, Hedner U, Roberts HR, Hoffman M. High dose factor VIIa enhances clot stability in a model of hemophilia B. Brit J Haematol. 2005;131:645–55. doi: 10.1111/j.1365-2141.2005.05820.x. [DOI] [PubMed] [Google Scholar]
- 25.Allen GA, Persson E, Campbell RA, Ezban M, Hedner U, Wolberg AS. A variant of recombinant factor VIIa with enhanced procoagulant and antifibrinolytic activities in an in vitro model of hemophilia. Arterioscler Thromb Vasc Biol. 2007;27:683–9. doi: 10.1161/01.ATV.0000257204.82396.2b. [DOI] [PubMed] [Google Scholar]
- 26.Bolton-Maggs PH. Factor XI deficiency--resolving the enigma? Hematology Am Soc Hematol Educ Program. 2009:97–105. doi: 10.1182/asheducation-2009.1.97. [DOI] [PubMed] [Google Scholar]
- 27.Peyvandi F, Di Michele D, Bolton-Maggs PH, Lee CA, Tripodi A, Srivastava A Project on Consensus Definitions in Rare Bleeeding Disorders of the Factor VFIXS, Standardisation Committee of the International Society on T Haemostasis. Classification of rare bleeding disorders (RBDs) based on the association between coagulant factor activity and clinical bleeding severity. J Thromb Haemost. 2012;10:1938–43. doi: 10.1111/j.1538-7836.2012.04844.x. [DOI] [PubMed] [Google Scholar]
- 28.Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haemost. 2007;5 (Suppl 1):116–24. doi: 10.1111/j.1538-7836.2007.02504.x. [DOI] [PubMed] [Google Scholar]
- 29.Lord ST. Molecular mechanisms affecting fibrin structure and stability. Arterioscler Thromb Vasc Biol. 2011;31:494–9. doi: 10.1161/ATVBAHA.110.213389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salomon O, Steinberg DM, Tamarin I, Zivelin A, Seligsohn U. Plasma replacement therapy during labor is not mandatory for women with severe factor XI deficiency. Blood Coagul Fibrinolysis. 2005;16:37–41. doi: 10.1097/00001721-200501000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Berliner S, Horowitz I, Martinowitz U, Brenner B, Seligsohn U. Dental surgery in patients with severe factor XI deficiency without plasma replacement. Blood Coagul Fibrinolysis. 1992;3:465–8. [PubMed] [Google Scholar]
- 32.Salomon O, Steinberg DM, Seligshon U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia. 2006;12:490–3. doi: 10.1111/j.1365-2516.2006.01304.x. [DOI] [PubMed] [Google Scholar]
- 33.Crosby JR, Marzec U, Revenko AS, Zhao C, Gao D, Matafonov A, Gailani D, Macleod AR, Tucker EI, Gruber A, Hanson SR, Monia BP. Antithrombotic effect of antisense factor XI oligonucleotide treatment in primates. Arterioscler Thromb Vasc Biol. 2013;33:1670–8. doi: 10.1161/ATVBAHA.113.301282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mosnier LO, Lisman T, van den Berg HM, Nieuwenhuis HK, Meijers JC, Bouma BN. The defective down regulation of fibrinolysis in haemophilia A can be restored by increasing the TAFI plasma concentration. Thromb Haemost. 2001;86:1035–9. [PubMed] [Google Scholar]
- 35.Schneider M, Boffa M, Stewart R, Rahman M, Koschinsky M, Nesheim M. Two naturally occurring variants of TAFI (Thr-325 and Ile-325) differ substantially with respect to thermal stability and antifibrinolytic activity of the enzyme. J Biol Chem. 2002;277:1021–30. doi: 10.1074/jbc.M104444200. [DOI] [PubMed] [Google Scholar]