

Abstract
Development of new vaccines, diagnostics and therapeutics for biodefense or other relatively rare infectious diseases is hindered by the lack of naturally occurring human disease on which to conduct clinical trials of efficacy. To overcome this experimental gap, the U.S. Food and Drug Administration established the Animal Rule, in which efficacy testing in two well-characterized animal models that closely resemble human disease may be accepted in lieu of large scale clinical trials for diseases with limited natural human incidence. In this report, we evaluated the Brown Norway rat as a model for pneumonic plague and describe the natural history of clinical disease following inhalation exposure to Yersinia pestis. In high-capacity, high-containment housing, we monitored temperature, activity, heart rate and rhythm by capturing electronic impulses transmitted from abdominal telemeter implants. Using this system, we show that reduced activity and development of fever are sensitive indications of disease progression. Furthermore, we identified heart arrhythmias as contributing factors to the rapid progression to lethality following the fever response. Together these data validate the Brown Norway rat as an experimental model for human pneumonic plague and provide new insight that may ultimately lead to novel approaches in post-exposure treatment of this devastating infection.
Keywords: infectious diseases, Yersinia pestis, animal models, telemetry
Introduction
Yersinia pestis causes plague, a disease that once killed 250 million people due to the bacteria’s extreme virulence and ability to be transmitted by respiratory droplets. This Gram-negative member of the Enterobacteriaceae family emerged as a highly virulent blood pathogen an estimated 3,000 years ago in China and is presently endemic in five continents (Cui, et al., 2013). Due to the ability and opportunity of Y. pestis to undergo horizontal gene exchange while residing in its flea reservoir, plague continually threatens to reemerge as a deadly multi-drug resistant pandemic (Hinnebusch, et al., 2002, Welch, et al., 2007). Y. pestis is maintained in rodent reservoirs, preventing its eradication as well as providing a source of bacteria for would-be malignant users (Gage & Kosoy, 2005). These factors, combined with its extreme virulence and respiratory transmission, resulted in its classification as a Tier 1 Select Agent, placing it as a top priority in the US government’s biodefense research agenda for the development of novel pre- and post-exposure treatments as well as vaccines (Inglesby, et al., 2000).
Pneumonic plague is the most deadly disease state caused by Yersina pestis, with a high degree of mortality and short window for initiating treatment (Inglesby, et al., 2000). Primary pneumonic plague begins after inhalation of Y. pestis aerosol and, shortly after infection, the bacteria establish an anti-inflammatory environment that permits colonization of the alveoli (Price, et al., 2012). Once this replicative niche is established, Y. pestis growth combined with a mounting inflammatory response leads to dissemination through the vasculature to seed distal organs such as the liver (Heine, et al., 2013). The resulting bacteremia is believed to cause a fever response, and in humans, this is often the first indication of infection (Pollitzer, 1954). High fever (>101.5°F), tachycardia, hypotension and leukocytosis are other symptoms of lethal plague in humans, and advanced cases of pneumonic plague can include severe sepsis and multi-organ failure (Wang, et al., 2011). Antibiotic treatment must be initiated shortly after the appearance of fever in order to be effective, often before a diagnosis can be made. The propensity of Y. pestis for genetic adaptation combined with the rapid progression and high transmissibility of plague have led to significant concern that public health could be threatened in the future by natural or intentional plague outbreaks.
Although naturally occurring human cases of plague occur every year in most parts of the world, 96% of these are bubonic plague (Butler, 2013). Human pneumonic plague outbreaks still occur, with nearly 1,000 pneumonic plague cases reported worldwide since 2000. Although modern diagnostics and medicine prevent escalation of the outbreak, the mortality rate remains high even when antibiotic therapy is administered (Wang, et al., 2011). In order to support licensure for new methods for prevention and treatment of pneumonic plague, large scale efficacy tests are needed that exceed the number of naturally occurring cases. To overcome this limitation, in 2012, the US Food and Drug Administration recently applied its “Animal Rule” to license levofloxacin as a pneumonic plague therapeutic. Efficacy data collected from rodent and non-human primate models were accepted in lieu of large scale phase III human clinical trials.
Y. pestis CO92 is a clinical isolate that has been used as the gold standard strain for efficacy testing in experimental models of pneumonic plague in rodents and non-human primates (Anderson, et al., 1996, Davis, et al., 1996, Cornelius, et al., 2008, Anderson, et al., 2009, Koster, et al., 2010, Heine, et al., 2013). Following inhalation of Y. pestis CO92, necrotizing bronchopneumonia and mortality occur in less than 7 days, with rodents undergoing a more rapid disease course than primates. Moribund animals often also develop high titer bacteremia, showing mild to moderate lesions in distal tissues such as the liver and spleen. The mortality rate approaches 100% at a relatively low challenge dose. Actual cause of death has not been well characterized, particularly in the rodent models.
Intranasal challenge of rodents leads to primary pneumonic plague, however this method of inoculation can lead to infection of the upper respiratory or gastro-intestinal tract which may have undesired impact on the host response to infection (Thomas, et al., 2008). This method is therefore unreliable for efficacy testing of candidate vaccines, diagnostics or therapeutics. We recently described an aerosol challenge model in Brown Norway rats, which uses a nose-only inhalation exposure system to deliver aerosolized Y. pestis CO92 in a controlled setting (Gater, et al., 2012). In this model, rats typically succumb to infection within 72 hours, often without showing signs of illness until just hours before death. For therapeutic development, it is essential to identify triggers, such as fever, from which to initiate treatment (FDA, 2004). Thus, for the rat to be a useful experimental model for plague therapeutics, it is essential to monitor for fever or other symptoms that indicate the onset of disease as a treatment trigger.
Recently, the African green non-human primate model was used to test efficacy of levofloxacin as a post-exposure therapeutic to prevent pneumonic plague. In this model, fever was shown to be a reliable indication of the onset of disease and was used as a trigger to initiate treatment (Layton, et al., 2011). Heart arrhythmias and increased respiratory rate accompanied the fever response. In this work, our goal was to evaluate these parameters in the rat model. Rodents are a preferred experimental model for early large scale pre-clinical trials where statistically reliable data can be obtained and mouse and rat plague models have been commonly used (Byrne, et al., 1998, Williamson, et al., 2001, Lathem, et al., 2005, Agar, et al., 2008, Agar, et al., 2009, Anderson, et al., 2009, Gater, et al., 2012). Plague in rats is more similar to humans compared to mice, though both species rapidly develop lethal disease following respiratory or dermal inoculation of Y. pestis (Sebbane, et al., 2005). In addition, more clinical and postmortem analyses are possible in the rat model, given the substantially larger size of the rat compared to the mouse. Yet, high capacity housing causes challenges in signal interference when using monitoring systems that depend on radio transmission, especially in the high containment laboratory. Our results not only demonstrate feasibility but with remote biomonitoring of symptoms, we present a more clearly defined progression of disease.
Materials and Methods
Animals
These experiments were conducted in accordance with the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals and the study protocol was approved by the University of Missouri Animal Care and Use Committee.
Telemetry study protocol
Ten male (>230g) and 18 female (>170g) aged-matched Brown Norway (BN) rats (Rattus norvegicus) were obtained from specific pathogen free breeding colonies at Charles River Laboratories (Wilmington, MA). All rats were in good health, with no evidence of congenital malformations, and exhibited no visible signs of disease. CTA-F40 telemetry units (Data Sciences International, St Paul, MN) were implanted in the abdomen of each of 27 rats 7 days prior to the start of the study. One animal (male) did not undergo implant surgery and was included in the challenge and survival data. The unit was placed into the abdominal cavity along the sagittal plane ventral to the digestive organs and secured to the ventral abdominal wall musculature. The positive lead for electrocardiography (ECG) was attached to the thoracic musculature and the negative lead was attached to the pectoralis superficialis. None of the animals developed visible complications from the implant.
For data collection and analysis, body temperature, heart rate, rhythm, and locomotor activity were recorded for one minute every fifteen minutes during the course of the study. Locomotor activity was measured by telemetric detection of changes in the distance between the transmitter and receiver, indicating the animal was moving around its cage. An initial baseline period of 22 hours was recorded in each animal (Barrientos, et al., 2009). Animals were not monitored during the exposure procedure.
Timecourse study protocol
Groups of 35 male Brown Norway rats, weighing >350g, were challenged in 2 cohorts by inhalation exposure. Animals were numbered consecutively and euthanized every 24 hours in numerical order for post-mortem analysis.
System design
Animals were housed in a high containment rodent housing system in HEPA-filtered isolator caging (Tecniplast, Italy). Double sided housing units hold cages approximately 6″ apart and 12″ above and below. The CTA-F40 units transmit using radio frequency. Individual cages were shielded from others and were placed in staggered configuration with an aluminum shield inserted in the middle of the rack to block interference from back-back cages. Receivers were mounted to the bottom of each cage. Two matrix boxes were used to transmit the signal outside of the high containment laboratory. Data collection and real-time monitoring of telemetry transmissions were accomplished outside of containment. The transmitters, receivers, consolidation matrices, cabling, and computers with Dataquest A.R.T. data acquisition system were components of the Physiotel Telemetry System (Data Science International, St Paul, MN). Dataquest A.R.T. telemetry software was used to capture and analyze the data.
Aerosol Challenge
Yersinia pestis CO92 is a clinical isolate of the Orientalis biovar (Welkos, et al., 1997, DeBord, et al., 2006). Bacteria were streaked from frozen stock on heart infusion agar (HIA) containing congo red to verify retention of the pigmentation locus (Surgalla & Beesley, 1969). Single, isolated, pigmented colonies were used to inoculate 6 cultures (50 ml each) of heart infusion broth (HIB) containing 2.5mM CaCl2 and cultures were grown for 24 hours at 37°C with aeration (120 rpm). Bacteria were concentrated by centrifugation and suspended in 10ml sterile PBS (final culture OD600=1.0). Y. pestis was aerosolized in a Sparging Liquid Aerosol Generator as previously described (Gater, et al., 2012). Aerosol samples were collected using a Teflon impinger (SKC, Inc, Eighty Four, PA, USA) at 5 slpm. Aerosol concentration (CAero) for a determined impinger sample concentration (CImp), impinger sample volume (V), sampling flow rate (QImp), and time (t) was calculated in CFU/mL as follows: CAero = (CImp × V)/(QImp × t). The presented dose is defined as the maximum aerosolized dose that might be inhaled by the subject, calculated as CAero × MV × t (M=mass of animal). All laboratories containing live Y. pestis were approved for Tier 1 Select Agent research by the US Centers of Disease Control and Prevention and MU Institutional Biosafety Committee.
Rats were moved into biocontainment and single-housed in cages that were each fitted with a receiver for 1 day prior to challenge. Rats were exposed to a light/dark cycle of 12 hours per condition each day. Temperature and humidity were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Rats were given rodent chow and water ad libitum throughout the study. Each rat was given a plastic tube for environmental enrichment.
On challenge day, animals were placed into individual holding tubes, passed into a Class III biosafety cabinet and aerosol challenged with a presented dose of 5×105 CFU (approximately 100x the mean lethal dose) of Y. pestis CO92. Serial dilutions of starting culture as well as impinger samples were plated on Yersinia Selective Agar (YSA) for enumeration of actual presented dose (Sarovich, et al., 2010). Following aerosol challenge, rats were replaced in housing and monitored for clinical signs of illness, as well as body temperature, heart rate, rhythm, and activity and survival for 7 days post-challenge. Clinical signs of disease included behavior, response to stimuli, ocular discharge, respiratory distress and seizure.
The following criteria were pre-established for euthanasia: presence of seizure activity, severe respiratory distress, forced abdominal respirations, or unresponsive to touch or stimuli. Animals showing one or more of these conditions or those that survived to the end of the 7 day observation period were euthanized by progressive filling of the home cage with CO2, death was assured by bilateral pneumothorax. Both methods are approved by the American Veterinary Medical Association Guidelines on Euthanasia.
Post-mortem analysis for timecourse study
Tissues were collected and fixed in 10% formalin and stained by hematoxylin and eosin (H&E). The slides blinded and read by a veterinarian with expertise in rodent pathology. Quantification of bacterial loads was performed in tissues removed aseptically immediately after euthanasia. Sterile PBS was used for dilution or homogenization of blood or tissues, respectively. Serial dilutions were plated in triplicate onto HIA plates for quantification of bacterial load (CFU per tissue or ml of blood).
Blood was collected and processed within 60 min for complete blood count (white blood cells, neutrophils, lymphocytes, monocytes, eosinophils, basophils, red blood cells, hemoglobin, hematocrit, mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), red blood cell distribution width (RDW), platelets, and mean platelet volume (MPV)) and serum chemistry (alkaline phosphatase (ALP), aspartate amino transferase (AST), Na+, K+, Cl−). In addition, serum and lung homogenate were measured by ELISA for tumor necrosis factor α (TNF-α) production using anti-TNF-α (BioLegend, San Diego, CA).
Statistical Methods
Differences in body temperature, heart rate, and activity were analyzed by one-way ANOVA using a linear statistical model of Infection Stage, Sex and interaction of Infection Stage × Sex using the GLM Procedure of SAS (SAS Institute Inc., Cary, NC) with significance set at P<0.05. Individual treatment means were compared using Fishers Least Significant Difference (LSD) test (P < 0.05).
Results
Fever correlates with lethal outcome in the Brown Norway rat pneumonic plague model
Twenty-eight animals were challenged by inhalation exposure with 100x LD50 Y. pestis CO92, 27 of which had surgical implants to monitor biorhythms and body temperature. Animals were monitored for baseline readings in high containment housing for 22 hours prior to challenge. No changes in heart rate, temperature or activity were apparent immediately following the challenge procedure. Twenty-seven of 28 animals succumbed to the infection within 4 days post-exposure, with one survivor at the end of the 7 day observation period (Figure 1A). The majority of the animals (93%), including the rat that did not have an implant, succumbed to disease before 72 hours post-infection (HPI), with a mean time to death of 58 HPI. Animals that developed lethal disease also developed an increase in body temperature greater than 1°C and this occurred in 85% of the animals before 48 HPI (Figure 1B, table 1). The mean temperature during the fever phase was 39°C ± 0.7°C which was significantly increased compared to pre-exposure mean body temperature (37.5°C ± 0.04°C). The mean time to fever was 40 HPI, with a range of 26 – 53 HPI. Strikingly, the fever period was followed by hypothermia that preceded death (mean temperature 33.1°C ± 0.8°C). Statistical analysis was performed to determine the least significant differences for all study parameters per hour. Based on this analysis the stages of infection were defined as follows: 1) pre-exposure baseline (pre-challenge), 2) asymptomatic (0–36 HPI), 3) fever, defined as temperature greater than 38.5°C (36–46 HPI), 4) hypothermic, defined as temperature lower than the baseline average of 37.5°C (46–82 HPI).
Figure 1. Rats develop fever prior to 48 hours post-infection and enter hypothermia in the end stage of disease.
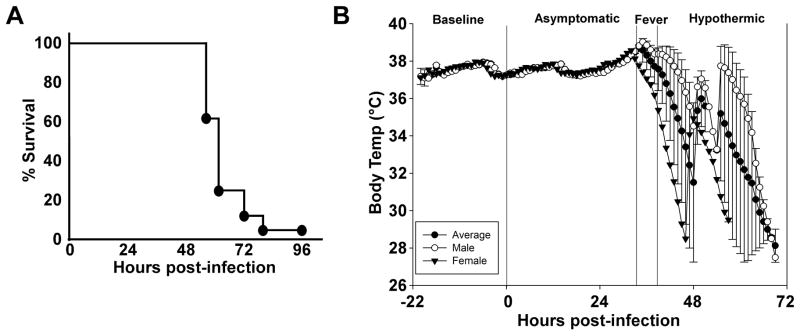
A) Inhalation exposure challenge with 100x mean lethal dose (MLD) of Y. pestis CO92 (6×105 CFU presented dose) in male and female Brown Norway rats with abdominal telemeter implants. Rats were monitored for 7 days; lethality occurred in less than 4 days. B) Period analysis by body temperature (Tb). Mean temperature recordings by the hour as monitored by telemetry implant. Monitoring began 22 hours prior to infection (baseline) and the first 72 hours post-infection are shown (n=9 males, 18 females). All animals were analyzed for statistical significance by period set at P<0.0001. Dashed line between asymptomatic and fever stages as well as fever and hypothermic stages indicates statistical significance between groups. Error bars represent the standard error of the mean.
Table 1.
Mean temperature and heart rate for each animal following inhalation exposure challenge with Y. pestis CO92.
| ID | Weight (g) | Sex | Dose (xMLD) | Tf (°C)a | b-HRb | f-HRc | TTFd | TTDe |
|---|---|---|---|---|---|---|---|---|
| 1 | 265 | M | 161 | 38.9 | 350.1 | 370.6 | 44 | 64.75 |
| 2 | 179 | F | 120 | 38.9 | 373.6 | 404.5 | 38.3 | 56.25 |
| 3 | 178 | F | 120 | 39.1 | 359.5 | 426.2 | 46.75 | 58.5 |
| 4 | 191 | F | 126 | 39 | 366.6 | 375.7 | 41.75 | 52.75 |
| 5 | 274 | M | 165 | 39.1 | 343.4 | 350 | 43.5 | 58.5 |
| 6 | 174 | F | 118 | 38.9 | 365 | 373.6 | 40 | 56.25 |
| 7 | 193 | F | 127 | 39.1 | 372 | 384 | 47.8 | 58.5 |
| 8 | 192 | F | 127 | 39.2 | 352 | 374.8 | 53.3 | 58.5 |
| 9 | 198 | F | 130 | 39 | 360 | 381 | 48.4 | 58.5 |
| 10 | 277 | M | 167 | 38.8 | 347 | 354.3 | 49.7 | 73.5 |
| 11 | 294 | M | 174 | 39 | 335 | 372 | 44.8 | 57.5 |
| 12 | 295 | M | 175 | 39.1 | 331.5 | 366 | 42.8 | 57.75 |
| 13 | 172 | F | 117 | 39 | 384 | 392 | 40.3 | 58.25 |
| 14 | 177 | F | 119 | 39.1 | 353 | 353 | 44 | 58.75 |
| 15 | 274 | M | 165 | 38.9 | 351.7 | 383.5 | 43.5 | 58.75 |
| 16 | 309 | M | 181 | 38.9 | 331 | 359 | 41.7 | 58 |
| 17 | 271 | M | 164 | 39 | 347.3 | 385 | 42 | 58.75 |
| 18 | 171 | F | 116 | 39 | 384 | 394 | 41.7 | 52.25 |
| 19 | 172 | F | 117 | 38.9 | 362 | 375.4 | 41.6 | 71.6 |
| 20 | 177 | F | 119 | No Fever | 350.6 | No Fever | N/A | N/A |
| 21 | 177 | M | 119 | 39 | 347.3 | 367 | 41 | 58.5 |
| 22 | 259 | F | 159 | 39.2 | 355 | 394.3 | 42.3 | 58.25 |
| 23 | 182 | M | 122 | 38.9 | 344 | 366 | 51.6 | 81.5 |
| 24 | 263 | F | 160 | 39.2 | 367.3 | 413.3 | 43.5 | 56 |
| 25 | 186 | F | 124 | 39 | 375.8 | 394 | 40 | 57.25 |
| 26 | 167 | F | 114 | 39 | 345.4 | 371 | 48 | 66 |
| 27 | 178 | F | 120 | 38.9 | 341.5 | 365.2 | 26 | 52 |
Peak fever temperature (Tf)
baseline heart rate (b-HR), beats per minute
fever stage heart rate (f-HR), beats per minute
time to fever (TTF), hours post-infection
time to death (TTD), hours post-infection
Females spent less time with fever compared to males, but there was no significant difference in mean time to death between males and females. Pilot studies examining the fever response in larger male rats following the same challenge protocol yielded similar observations regarding onset of fever and mean time to death (data not shown). Variation in the duration of fever was observed compared to this study, yet in both studies 100% of the animals that later died of plague developed fever followed by hypothermia. Fever and hypothermia are indicators of sepsis, and therefore small variations in the duration of fever and onset of hypothermia would not be expected to affect the outcome of infection (Schorr, et al., 2014).
Period alignment of mean heart rate with temperature showed a small increase in heart rate during the fever stage and a pronounced decrease in heart rate during the hypothermic stage (Figure 2A). Rats were typically active at night and exhibited a circadian rhythm in activity with a period of approximately 24 hours (Figure 2B). Shortly before onset of fever, however, the animals became less active and the peak activity occurred after only 15 hours and was reduced in amplitude. Activity had become very low at the time of fever and was low for the remainder of the infection. These results suggest that a decrease in the animal’s activity may be the earliest outward indication of disease progression.
Figure 2. Period analysis of average temperature, heart rate and activity establishes reduced activity as the most sensitive outward sign of disease progression.

A) Mean heart rate (beats per minute) per hour compared to temperature; B) Average locomotor activity per hour compared to temperature. The bars indicate the Circadian period, with time shown between peak activity.
Sex dependent differences in fever responses in the rat pneumonic plague model
We calculated the mean temperature readings every 60 min of all the animals during the fever stage and found that it was significantly increased compared to the baseline and asymptomatic stages (Figure 3A). Similarly, the mean temperature during the hypothermic stage was significantly lower than the asymptomatic stage. Sex dependent differences were identified in body temperature during the fever and hypothermic stages, with females averaging a small increase in peak body core temperature and more pronounced hypothermia compared to males (Figure 3B). Even though males and females had significantly different fever responses, however, no detectable differences were noted in overall susceptibility to infection as measured by survival and time to death.
Figure 3. Female rats exhibit different clinical signs during late stage pneumonic plague.

Mean values for temperature (A–B), heart rate (C–D) and activity (E–F) were determined from all data points collected from the indicated stage of infection. (A,C,E) Combined data from males and females; (B,D,F) Averages from males (black bars) and females (grey bars). *P<0.05; error bars indicate the standard error of the mean.
The mean heart rate was indistinguishable from baseline during the asymptomatic stage but increased during the fever stage (Figure 3C). Similarly, the mean heart rate was significantly decreased during the hypothermic stage compared to baseline. Sex dependent differences in heart rate were apparent throughout the infection (Figure 3D). Whereas females averaged a lower heart rate than males during the baseline, asymptomatic and fever stages, females averaged a higher heart rate during the hypothermic stage. A similar analysis was conducted on activity. While no sex dependent differences in activity were detectable at baseline nor during the hypothermic stage, males were significantly more active than females during the asymptomatic and fever stages (Figure 3E–F). In summary, females averaged a higher temperature, lower heart rate and lower activity during the fever stage. During the hypothermic stage, females averaged a lower temperature and heart rate and activity was minimal in both sexes. As males and females were aged-matched and males were approximately 50% larger than females, it is conceivable that these differences are related to animal size rather than sex or age. Importantly, no sex dependent differences were detectable in the mean time to death suggesting these different clinical responses do not affect the outcome of infection. Overall, the data appear similar to human pneumonic plague where no sex dependent differences in disease susceptibility are apparent.
Electrocardiography indicates heart disease in most of the rats during hypothermic stage
We examined the end stage electrocardiograms for 18 rats to identify any cardiac abnormalities that could have contributed to death. Arrhythmias were observed in 16 of these animals (Table 2). The most common abnormality observed during the end stage was bradycardia, which was sometimes accompanied by the absence of the p-wave, known as the Osborne wave (Figure 4A–C) (Osborn, 1953). The severity of bradycardia varied, and in some cases was absent even though all animals became hypothermic during the end stage. These conditions are often associated with hypothermia. More severe arrhythmias such as premature ventricular complex and atrial standstill, indicative of heart disease, were also present in some of the animals (Figure 4D). These data are consistent with heart failure as likely contributing to the death of these animals.
Table 2.
End stage cardiac failure in Brown Norway rats following inhalation exposure to Y. pestis CO92.
| Animal IDa | Time to deathb | Atrial standstill w/junctional escape | AV block w/complete aventricular dissoc. | Osborne waves | Sinus Rhythm | Sinus bradycardia | VPCd | Sympathetic surge |
|---|---|---|---|---|---|---|---|---|
| 2F | 56.25 | + | ||||||
| 3F | 58.5 | + | ||||||
| 4F | 52.75 | + | ||||||
| 6F | 56.25 | + | + | |||||
| 10M | 73.5 | + | + | |||||
| 11M | 57.5 | + | ||||||
| 12M | 57.75 | + | + | |||||
| 13F | 58.25 | +/− | +/− | |||||
| 16M | 58 | + | ||||||
| 18F | 52.25 | + | + | |||||
| 19F | 71.6 | + | ||||||
| 22F | 58.25 | + | + | |||||
| 23M | 81.5 | + | ||||||
| 24F | 56 | + | ||||||
| 25F | 57.25 | + | ||||||
| 26F | 66 | + | ||||||
| 27F | 52 | + |
M: Male; F: female
hours post-infection
BPM: beats per minute
Ventricular premature complex (VPC)
Figure 4. Sample electrocardiograms (ECG) from the end stage disease showing heart disease as likely contributing factors in death from pneumonic plague.

Male and female Brown Norway rats on day 3 post-infection. (A) Normal heart rate (HR) of surviving animal (HR=360 beats/min (bpm)); end stage ECGs: (B) sinus bradycardia (HR=300 bpm), (C) Osborne wave (HR=60 bpm), and (D) ventricular premature complex (HR=180 bpm). A 10 second interval is shown in all 4 panels.
Bacterial load, tissue pathology and sepsis correlate with clinical time course of disease
In order to correlate disease progression with the appearance of fever, we challenged animals that did not have transmitter implants and monitored for bacterial load, histopathology and blood chemistry between 1 and 48 hours post-infection. At 100x mean lethal dose, 96% of the animals succumbed to disease within 4 days, the majority of which occurred before 72 HPI (Figure 5A). This lethality and disease kinetics appear indistinguishable from the natural history study described above, which was conducted at the same challenge dose. At 100-fold lower challenge dose, however, only 65% of the animals succumbed to infection, most of which died in less than 72 hours, similar to the higher challenge dose.
Figure 5. Rapid bacterial growth and fulminant disease are not always present at 48 HPI.

Groups of 35 male Brown Norway rats were challenged by inhalation exposure with a presented dose of 5×105 CFU (A–H) or 5×103 CFU (A only) Y. pestis CO92. A) Survival was monitored over 7 days. B–J) Groups of 5–6 animals were euthanized at the indicated time points, blood was collected and lungs and liver were harvested for determination of bacterial load (B), TNF-α (C), complete blood count: neutrophils (D), platelets (E), red blood cells (F), hematocrit (G); blood chemistry: Na+ (H); or histopathology (I–K). (I) Lungs from 5 animals were scored for severity of lesions (0=no lesions, 3= severe pathology); 3 sections per tissue were examined in single blind fashion. (J–L) Representative images from diseased animals at 48 HPI are shown; (J) lungs, scale bar indicates 100μm; (K) liver, arrows point to focal inflammation and apoptotic cells, scale bar indicates 50μm; (L) Diff-Quik stain of blood showing the characteristic safety pin appearance of Y. pestis bacteria. Data were collected in 2 independent trials (n=11). Bars indicate the median (B–I). Data in D–H were evaluated for statistical significance using one-way ANOVA; *P<0.05, ns: not significant.
At the high challenge dose, bacteria were below the limit of detection in the lungs of over 70% of the animals at 1 and 24 HPI, and only 1 animal had bacteria that could be detected in the liver, but not the blood nor spleen (Figure 5B, data not shown). At 48 HPI, bacteria were recovered from lungs of 70% of the animals, most of which also had bacteria in the liver and blood. Interestingly, even though the low challenge dose resulted in 50% lethality within 72 HPI, only 2 of 12 animals had detectable bacteria in the lungs at 48 HPI, one of which had also disseminated to the liver (data not shown).
Serum TNF-α is often found in animals with Gram-negative sepsis. Although this cytokine was detected in some of these animals, more than 50% of the animals had no detectable TNF-α in the lung homogenate or serum at 48 HPI (Figure 5C). Of note, this time point is after the end of the fever stage observed in the biotelemetry study, suggesting that the majority of the animals were experiencing severe disease and were in the hypothermic stage. Neutrophils were enumerated following Diff-Quik staining and a complete blood count (CBC). At 24 HPI, significantly fewer neutrophils were found in the blood compared to 1 HPI, consistent with the early anti-inflammatory response that is a hallmark of pneumonic plague (Figure 5D). At 48 HPI, animals with high bacterial load also had increased neutrophils circulating in the blood. Platelet counts typically decrease upon the onset of severe sepsis (Schorr, et al., 2014). As might be predicted based on the uniform development of fever by 48 HPI, a significant decrease in platelet count was observed compared to 1 HPI (Figure 5E). Unexpectedly, however, platelet counts were also significantly decreased at 24 HPI compared to 1 HPI. Red blood cells were also significantly decreased, with a median value of 6.5×106 RBCs compared to 7.5×106 RBCs at 1 HPI (normal range for rats is 7.4–10.6×106 RBCs ml−1) (Stammers, 1928). Hemoglobin and hematocrit levels were also significantly decreased at 24 and 48 HPI, with normal MCV and RDW at both time points (Figure 5F–G, data not shown). Thus, from the CBC, we observed a decrease in neutrophils, erythrocytes and thrombocytes at 24 HPI, suggesting the rats experience mild to moderate pancytopenia shortly after infection. Sodium and chloride levels were significantly decreased at 48 HPI, but were normal at 24 HPI (Figure 5H, data not shown). Potassium, AST and ALP levels were normal at all time points (data not shown). Together the data suggest the development of sepsis at 48 HPI.
We analyzed formalin fixed tissues of these animals by histochemistry. Not surprisingly, the most common pathology occurred in the lungs. Lungs for five of the animals were scored for severity of disease according to inflammation, alveolar destruction and bacterial colonization. The most severe pathology occurred in animals with the highest bacterial titer, but overall only mild to moderate pathology was observed in the lungs of all animals at 48 HPI (Figure 5I–J). Mild to moderate pathology including inflammation, necrosis of inflammatory cells and apoptosis of hepatocytes was found in the liver of animals with high bacterial load in this tissue but extracellular bacteria were not observed in this tissue (Figure 5K). Extracellular bacteria could be readily observed in the blood of animals with high titer bacteremia (Figure 5L). Although bacteria were sometimes recovered from the heart homogenate, we did not observe bacterial colonization of the heart in the histological analysis (data not shown). Overall, the pathological analysis showed mild to moderate disease severity with evidence of inflammation in multiple tissues at 48 HPI. Given that the majority of these animals were likely within 12 hours from death, these data indicate that the onset of disease is sudden and rapidly overtakes the host. Together with the data collected in the natural history study, it appears that the development of fever in Brown Norway rats precedes the onset of overt disease.
Discussion
The US Food and Drug Administration has established the Animal Rule to be applied to development of products against diseases where large scale human clinical trials for efficacy cannot be performed due to a small amount of naturally occurring human disease. Pneumonic plague is a disease that has historically caused devastating human outbreaks, but in modern times is rare (Butler, 2013). New vaccines and therapeutics to prevent pneumonic plague will therefore be subjected to the Animal Rule (FDA, 2004). For an experimental model to meet Animal Rule requirements, it must be shown to be highly similar to the human disease, at clinical, pathological and microbiological levels. The use of rodent models for large scale efficacy testing such as may be required for pre-clinical and clinical studies under the Animal Rule can greatly reduce development costs but in order to do so, systems for biotelemetric monitoring of clinical disease in the high-containment laboratory are required.
Pneumonic plague and other diseases caused through the respiratory route that can be rapidly lethal present many challenges for researchers. High-containment, high-capacity rodent housing necessitates keeping the animals near one another which interferes with radio signals sent by transmitter units and presents challenges for clinical observation as well as data collection. Monitoring for non-lethal endpoints typically involves visual inspection and in this work, we showed that telemetry monitoring from outside containment provided a similar, if not better, assessment of animal health. Moreover, this method minimizes the need to stress the animals by handling their cages to assess their health. With this technology, we performed a high-powered study in which we simultaneously monitored 3 parameters at controlled frequencies. Novel observations were made that improve our understanding of plague pathogenesis and these may ultimately influence the development of post-exposure treatments. Of the 18 animals analyzed for end stage cardiac disease, only 2 had normal heart rhythm at the time of death. While further studies are required to determine the underlying causes, our results suggest anemia, thrombocytopenia and sepsis could be contributing factors.
Previous work in the non-human primate model showed no correlation between inhaled dose, the timing of bacteremia and the timing of fever, similar to our observations in the rat model (Layton, et al., 2011). The fever in the primate model took longer to develop, reached a higher temperature and lasted for longer whereas rats appeared more sensitive to the rapid progression of disease. Hypothermia also developed in non-human primates and followed the fever stage before death. The consistency of end stage hypothermia in rats and non-human primates supports further evaluation of this condition as a non-lethal experimental endpoint. Overall, rats and non-human primates respond similarly to aerosol challenge by Y. pestis and it appears the rat is a suitable small animal model of pneumonic plague that would reasonably predict efficacy of candidate vaccines and therapeutics in humans.
Aerosol challenge models of biohazards such as Y. pestis must be performed in a high containment setting to enable precise dose delivery while preserving the safety of laboratory workers and the surrounding environment. The NIH/NIAID Regional Biocontainment Laboratories were constructed to support development of medical products against morbid respiratory infectious diseases that would likely fall under the Animal Rule. In this work, we demonstrated some of the capabilities of these laboratories to support research and development, and the knowledge that can be gained from these powerful experimental rodent models.
Acknowledgments
We are grateful to Dr. Sarah Hansen, Sherri Neff and the veterinary staff of the University of Missouri Office of Animal Resources for performing implant surgeries. We are also indebted to Dr. David Bland, Miqdad Dhariwala and Joshua Willix for assistance with the BSL3 protocols, and to the University of Missouri Laboratory for Infectious Disease Research Aerobiology Core for assistance with the inhalation exposure challenge. Blood chemistry and CBC were performed by Comparative Clinical Pathology Services, LLC; tissue processing and staining were performed by IDEXX-RADIL. In addition, we are grateful to Dr. Joe Campbell, Dr. Craig Franklin, Dr. Dan Hassett, and Dr. Ami Patel for helpful discussions as well as to Jenny Rohrer and the technical team at Data Science International for assistance with data collection and analysis. This work was supported by NIH/NIAID HHSN272201000035I (DMA).
Footnotes
The authors declare no conflict of interest for this work.
References
- Agar S, Sha J, Foltz S, et al. Characterization of the rat pneumonic plague model: Infection kinetics following aerosolization of Yersinia pestis CO92. Micobes Infect. 2009;11:205–214. doi: 10.1016/j.micinf.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Agar S, Sha J, Foltz S, et al. Characterization of a mouse model of plague after aerosolization of Yersinia pestis CO92. Microbiology. 2008;154:1939–1948. doi: 10.1099/mic.0.2008/017335-0. [DOI] [PubMed] [Google Scholar]
- Anderson D, Ciletti N, Lee-Lewis H, et al. Pneumonic plague pathogenesis and immunity in Brown Norway rats. Am J Path. 2009;174:910–921. doi: 10.2353/ajpath.2009.071168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G, Leary S, Williamson E, Titball R, Welkos S, Worsham P, Friedlander A. Recombinant V antigen protects mice against pneumonic and bubonic plague caused by F1-capsule positive and negative strains of Yersinia pestis. Infect Immun. 1996;64:4580–4585. doi: 10.1128/iai.64.11.4580-4585.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos R, Watkins L, Rudy J, Maier S. Characterization of the sickness response in young and aging rats following E. coli infection. Brain Behav Immun. 2009;23:450–454. doi: 10.1016/j.bbi.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler T. Plague gives surprises in the first decade of the 21st century in the United States and worldwide. Am J Trop Med Hyg. 2013;89:788–793. doi: 10.4269/ajtmh.13-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne W, Welkos S, Pitt M, et al. Antibiotic treatment of experimental pneumonic plague in mice. Antimic Agents & Chem. 1998;42:675–681. doi: 10.1128/aac.42.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius C, Quenee L, Overheim K, et al. Immunization with recombinant V10 protects cynomolgus macaques from lethal pneumonic plague. Infect Immun. 2008;76:5588–5597. doi: 10.1128/IAI.00699-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Yu C, Yan Y, et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci. 2013;110:577–582. doi: 10.1073/pnas.1205750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis K, Fritz D, Pitt M, Welkos S, Worsham P, Friedlander A. Pathology of experimental pneumonic plague produced by fraction 1-positive and fraction 1-negative Yersinia pestis in African green monkeys (Cercopithecus aethiops) Arch Pathol Lab Med. 1996;120:156–163. [PubMed] [Google Scholar]
- DeBord K, Anderson D, Marketon M, et al. Immunogenicity and protective immunity against bubonic plague and pneumonic plague by immunization of mice with the recombinant V10 antigen, a variant of LcrV. Infect Immun. 2006;74:4910–4914. doi: 10.1128/IAI.01860-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA. FDA Center for Biologics and Evaluation public workshop on animal models and correlates of protection for plague vaccines. Gaithersburg, MD: 2004. [Google Scholar]
- Gage K, Kosoy M. Natural history of plague: perspectives from more than a centrury of research. Ann Rev Entomol. 2005;50:505–528. doi: 10.1146/annurev.ento.50.071803.130337. [DOI] [PubMed] [Google Scholar]
- Gater S, Peters K, Kocsis A, Dhariwala M, Anderson D, Anderson P. Host stress and immune responses during aerosol challenge of Brown Norway rats with Yersinia pestis. Front Cell Infect Microbiol. 2012;2:147. doi: 10.3389/fcimb.2012.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine H, Chuvala L, Riggins R, et al. Natural history of Yersinia pestis pneumonia in aerosol-challenged BALB/c mice. Antimicrob Agents Chemother. 2013;57:2010–2015. doi: 10.1128/AAC.02504-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch B, Rosso M, Schwan T, Carniel E. High-frequency conjugative transfer of antibiotic resistance genes to Yersinia pestis in the flea midgut. Mol Microbiol. 2002;2:349–354. doi: 10.1046/j.1365-2958.2002.03159.x. [DOI] [PubMed] [Google Scholar]
- Inglesby T, Dennis D, Henderson D, et al. Plague as a biological weapon: medical and public health management. Working group on civilian biodefense. JAMA. 2000;283:2281–2290. doi: 10.1001/jama.283.17.2281. [DOI] [PubMed] [Google Scholar]
- Koster F, Perlin D, Park S, et al. Milestones in progression of primary pneumonic plague in Cynomolgus macaques. Infect Immun. 2010;78:2946–2955. doi: 10.1128/IAI.01296-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathem W, Crosby S, Miller V, Goldman W. Progression of primary pneumonic plague: A mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci. 2005;102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layton R, Mega W, McDonald J, Brasel T, Barr E, Gigliotti A, Koster F. Levofloxacin cures experimental pneumonic plague in African green monkeys. PloS Negl Trop Dis. 2011;5:e959. doi: 10.1371/journal.pntd.0000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn J. Experimental hypothermia: respiratory and blood pH changes in relation to cardiac function. Am J Physiol. 1953:389–398. doi: 10.1152/ajplegacy.1953.175.3.389. [DOI] [PubMed] [Google Scholar]
- Pollitzer R. Plague. World Health Organization; Geneva, Switzerland: 1954. [Google Scholar]
- Price P, Jin J, Goldman W. Pulmonary infection by Yersinia pestis rapidly establishes a permissive environment for microbial proliferation. Proc Natl Acad Sci USA. 2012;109:3083–3088. doi: 10.1073/pnas.1112729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarovich D, Colman R, Price E, et al. Selective isolation of Yersinia pestis from plague-infected fleas. J Microbiol Methods. 2010;82:95–97. doi: 10.1016/j.mimet.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorr C, Zanotti S, Dellinger R. Severe sepsis and septic shock: Management and performance improvement. Virulence. 2014;5:1–10. doi: 10.4161/viru.27409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebbane F, Gardner D, Long D, Gowen B, Hinnebusch B. Kinetics of disease progression and host response in a rat model of bubonic plague. Am J Path. 2005;166:1427–1439. doi: 10.1016/S0002-9440(10)62360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stammers A. The blood count and body temperature in normal rats. J Physiol. 1928;61:329–336. doi: 10.1113/jphysiol.1926.sp002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surgalla M, Beesley E. Congo red-agar plating medium for detecting pigmentation in Pasteurella pestis. App Microbiol. 1969;18:834–837. doi: 10.1128/am.18.5.834-837.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, Webber D, Sellors W, et al. Characterization and deposition of respirable large- and small- particle bioaerosols. App Env Microbiol. 2008;74:6437–6443. doi: 10.1128/AEM.01194-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Cui Y, Wang Z, et al. A dog-associated primary pneumonic plague in Qinghai Province, China. Clin Infect Dis. 2011;52:185–190. doi: 10.1093/cid/ciq107. [DOI] [PubMed] [Google Scholar]
- Welch T, Fricke W, McDermott P, et al. Multiple antimicrobial resistance in plague: An emerging public health risk. PLoS One. 2007;2:1–6. doi: 10.1371/journal.pone.0000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welkos S, Friedlander A, Davis K. Studies on the role of plasminogen activator in systemic infection by virulent Yersinia pestis strain CO92. Microb Path. 1997;23:211–223. doi: 10.1006/mpat.1997.0154. [DOI] [PubMed] [Google Scholar]
- Williamson E, Eley S, Stagg A, Green M, Russell P, Titball R. A single dose sub-unit vaccine protects against pneumonic plague. Vaccine. 2001;19:566–571. doi: 10.1016/s0264-410x(00)00159-6. [DOI] [PubMed] [Google Scholar]