

Abstract
Asymmetric dimethylarginine (ADMA) is an endogenously produced nitric oxide synthase (NOS) inhibitor. L-arginine can be metabolized by NOS and arginase, and arginase is the first step in polyamine production necessary for cellular proliferation. We tested the hypothesis that ADMA would inhibit NOS but not arginase activity, and that this pattern of inhibition would result in greater L-arginine bioavailability to arginase and thereby increase viable cell number. Bovine arginase was used in in vitro activity assays with various concentrations of substrate (L-arginine, ADMA, NG-monomethyl-L-arginine (L-NMMA) and NG-nitro-L-arginine methyl ester (L-NAME)). Only L-arginine resulted in measurable urea production (Km = 6.9 ± 0.8 mM; Vmax = 6.6 ± 0.3 μmol/mg protein/min). We then incubated bovine arginase with increasing concentrations of ADMA, L-NMMA, and L-NAME in the presence of 1 mM L-arginine, and found no effect of any of the tested compounds on arginase activity. Using bovine pulmonary arterial endothelial cells (bPAEC) we determined the effects of ADMA on NO and urea production, and found a significantly lower NO production and greater urea production (p<0.003) with ADMA, without changes in arginase protein levels. Additionally, ADMA treatment resulted in ∼30% greater number of viable cells after 48 hours than in control bPAEC. These results demonstrate that ADMA is neither a substrate nor an inhibitor of arginase activity, and that in bPAEC ADMA inhibits NO production and enhances urea production leading to more viable cells. These results may have pathophysiological implications in disorders associated with higher ADMA levels such as pulmonary hypertension.
Keywords: nitric oxide, nitric oxide synthase, pulmonary circulation, pulmonary hypertension
Introduction
Pulmonary hypertension (PH) is a life-threatening disease with no cure that ultimately leads to right heart failure and death. The hallmarks of PAH are elevated pulmonary vascular resistance and vascular remodeling (1, 2). Dysfunction of the endothelial L-arginine-nitric oxide pathway is an important contributor to the pathophysiology seen in PH. L-arginine is the substrate for both nitric oxide synthase (NOS) and arginase. Metabolism of L-arginine by NOS produces L-citrulline and nitric oxide (NO). NO is one of the major endothelium-derived vasoactive mediators that plays a crucial role in the maintenance of low pulmonary vascular tone. The metabolism of L-arginine by arginase produces L-ornithine and urea, and is the first step in polyamine and proline synthesis. Polyamines and proline are critical for cell proliferation, differentiation, tissue repair and growth (3, 4).
Asymmetric dimethylarginine (ADMA) is an endogenous competitive inhibitor of NOS enzymes that competes with L-arginine for binding to the active site of NOS. ADMA is released following the post-translational methylation of arginine residues within proteins by protein arginine methyl-transferases (PRMTs) followed by proteolysis of these arginine-methylated proteins (5). There are three forms of methylated arginine that have been identified in eukaryotes: ADMA, NG-monomethyl-L-arginine (L-NMMA), and symmetric dimethylarginine (SDMA) (5). Only L-NMMA and ADMA are inhibitors of the NOS family of enzymes (5). Additionally, it has been shown that exogenously administered L-NMMA and ADMA, but not SDMA are able to inhibit NOS activity and NO signaling in vitro (6) and in vivo (7). Once released from degraded proteins, the elimination of ADMA from the body is via renal clearance or enzymatic inactivation by dimethylarginine dimethylaminohydrolase (DDAH) (8). ADMA and its effect on L-arginine metabolism have been implicated in the pathogenesis of endothelial dysfunction in pulmonary hypertension (9).
The endothelial impairment associated with pulmonary hypertension has been linked to the dysfunction of the NO pathway and endogenous NOS inhibition may represent a mechanism for the associated pulmonary vascular remodeling (10). Although it has been shown in a rat model of pulmonary hypertension that there is an accumulation of endogenous NOS inhibitors, along with increased arginase activity in the pulmonary artery endothelial cells (11), the direct effects of these endogenous NOS inhibitors on arginase have yet to been reported.
It is not known whether methylated arginine can act as a substrate for arginase or if it can act as a competitive inhibitor of arginase. We hypothesized that ADMA would not inhibit arginase activity. Given that the bioavailability of L-arginine to arginase may be increased secondary to the competitive inhibition of NOS by ADMA, we also hypothesized that ADMA would result in greater viable cell numbers as a result of increased L-arginine metabolism by arginase. To test these hypotheses we utilized an in vitro arginase assay system with bovine arginase, as well as bovine pulmonary arterial endothelial cells.
Results
Only L-arginine acts as a substrate for arginase
To determine whether the asymmetric methylarginines act as a substrate for arginase, bovine arginase was used in in vitro activity assays with increasing concentrations (0.1 – 30 mM) of tested substrate (L-arginine, D-arginine, ADMA, L-NMMA, and L-NAME). Urea production was measured and Michaelis-Menten kinetics for arginase were determined. Figure 1 demonstrates that L-arginine acted as a substrate for arginase with a Vmax of 6.6 ± 0.3 μmol/mg protein/min and a Km of 6.9 ± 0.8 mM, while D-arginine resulted in no measurable urea production (Figure 1A). When L-arginine, ADMA, L-NMMA and L-NAME were tested as potential substrates for arginase, only L-arginine resulted in measurable urea production (Figure 1B).
Figure 1. Michaelis-Menten kinetics for arginase with different substrates reveals that only L-arginine acts as a substrate for arginase.
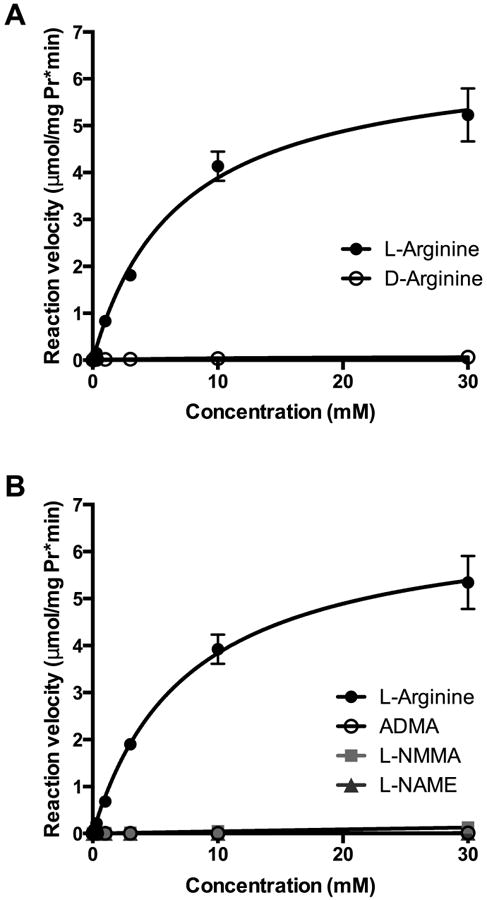
Bovine arginase was used in in vitro activity assays with increasing concentrations (0.1 – 30 mM) of tested substrate (L-arginine, D-arginine, ADMA, L-NMMA, or L-NAME) and urea production was measured. The reaction velocity was calculated as μmol/(mg protein·minute). The Michaelis-Menten equation was applied to determine Vmax and Km values. A. L-arginine acted as a substrate for arginase with a Vmax of 6.6 μmol/mg protein/min and a Km of 6.9 mM, while D-arginine resulted in no measurable urea production (n=3). B. ADMA, L-NMMA and L-NAME were tested as potential substrates for arginase with no measurable urea production (n = 1).
ADMA, L-NMMA or L-NAME do not inhibit arginase activity
To determine the effects of ADMA, L-NMMA and L-NAME on arginase activity, in vitro activity assays were performed. Bovine arginase activity was measured in the presence of 1 mM L-arginine and increasing concentrations of ADMA, L-NMMA, or L-NAME (0.1 – 30 mM). There was no measurable effect on arginase activity for any of the three compounds tested at any concentration tested (Figure 2).
Figure 2. Kinetics of arginase activity with increasing concentrations of ADMA, L-NMMA, or L-NAME demonstrate little inhibitory effect of the methylated arginines on arginase activity.
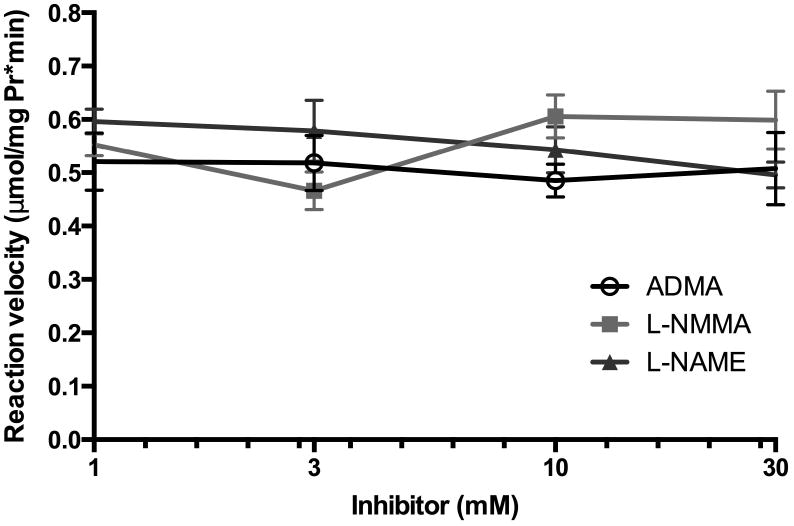
Bovine arginase was used in in vitro activity assays in the presence of 1 mM L-arginine and increasing concentrations of ADMA, L-NMMA, or L-NAME (0.1 – 30 mM) and urea production was measured. The reaction velocity was calculated as μmol/(mg protein·minute) (n = 5).
ADMA decreased NO production in bPAEC
To determine the effects of asymmetric methylarginine on nitrite production, bPAEC grown to 80-90% confluence were treated with ADMA (100 μM) or vehicle and incubated in 21% O2, 5% CO2, balance N2 for one hour. The cells were then stimulated with the calcium ionophore A23187 for an additional four hours. Nitrite production was measured and normalized to the bPAEC protein concentration. Treatment of bPAEC with ADMA significantly inhibited nitrite production (p < 0.003, Figure 3).
Figure 3. Treatment of bovine PAEC with ADMA results in decreased NO production as evidenced by decreased formation of the stable end-product nitrite.
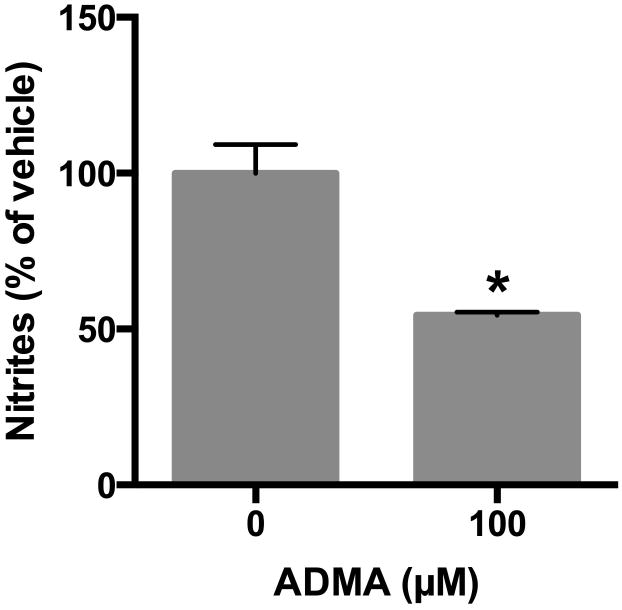
Bovine PAEC were incubated with vehicle or ADMA (100 μM) in PBS and incubated in 21% O2, 5%CO2, balance N2 (normoxia) for one hour, then stimulated with calcium ionophore A23187 for four additional hours, and the PBS was harvested for nitrite assay (n = 4). Nitrite production was measured and normalized to the bPAEC protein concentration. Data are shown as a percentage of vehicle control. * different from vehicle-treated, p < 0.003
ADMA increased urea production in bPAEC
To determine the effects of ADMA on urea production, ADMA (100 μM) or vehicle was added to the media of bPAEC grown to 80-90% confluence and incubated in 21% O2, 5% CO2, balance N2 for 24 hours. Urea production was measured and normalized to protein concentration. Arginase I and II protein was also measured by Western blot analysis. The addition of ADMA resulted in greater urea production than in vehicle-treated controls (p = 0.002, Figure 4A). There was no effect of ADMA on the levels of arginase I or arginase II protein (Figure 4B&C).
Figure 4. Treatment of bPAEC with ADMA results in greater urea production without effecting arginase I or II protein expression.
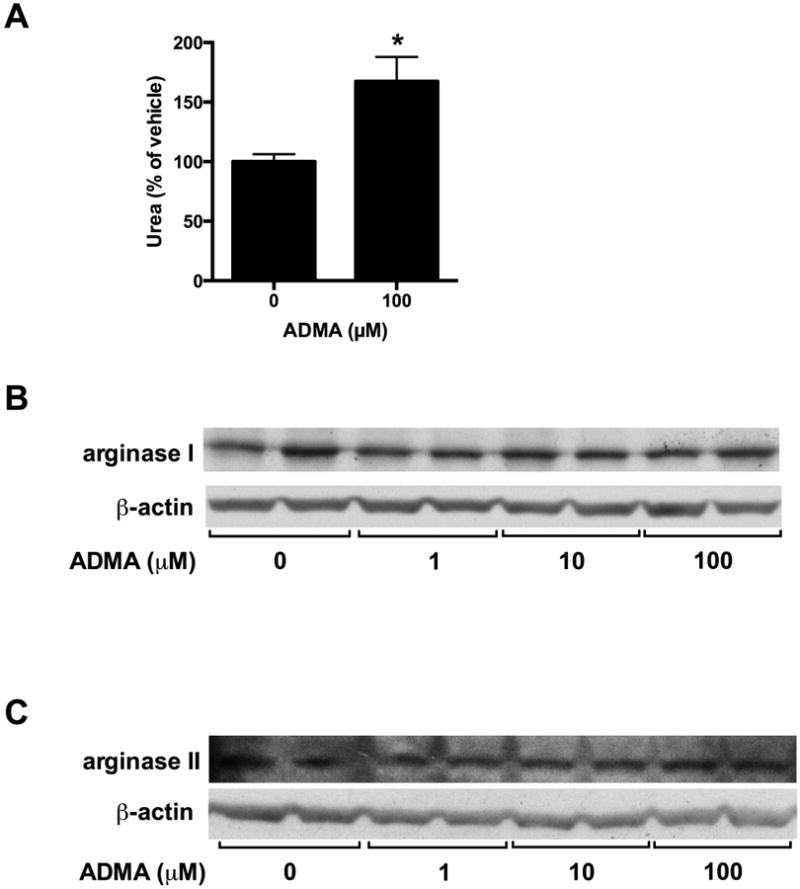
Bovine PAEC were incubated with vehicle or varying concentrations of ADMA, then incubated in 21% O2, 5%CO2, balance N2 (normoxia) for 24 hours and the media was harvested for urea assay (n=5). Urea production was measured and normalized to the bPAEC protein concentration. Data are shown as a percentage of vehicle control (A). Arginase I and II protein expression was analyzed by Western blot and normalized to β-actin. Representative Western blots are shown for arginase I (B) and arginase II (C).
* different from vehicle-treated, p = 0.002
ADMA increases viable cell number
To determine the impact of ADMA on viable cell numbers, bPAEC were seeded onto six-well plates at a density of 50,000 cells per well and ADMA was added to the medium. The cells were incubated for 48 hours in 21% O2, 5% CO2, balance N2 and viable cell numbers were determined using trypan blue exclusion. In control bPAEC after 48 hours the number of viable cells in each well was 651,500 ± 67,561, demonstrating a 13-fold increase in viable cell number in 48 hours. ADMA treatment resulted in ∼30% more viable cells after 48 hours (p < 0.0007) compared to vehicle-treated control cells (Figure 5).
Figure 5. Treatment of bPAEC with ADMA results in greater numbers of viable cells after 48 hours in culture.
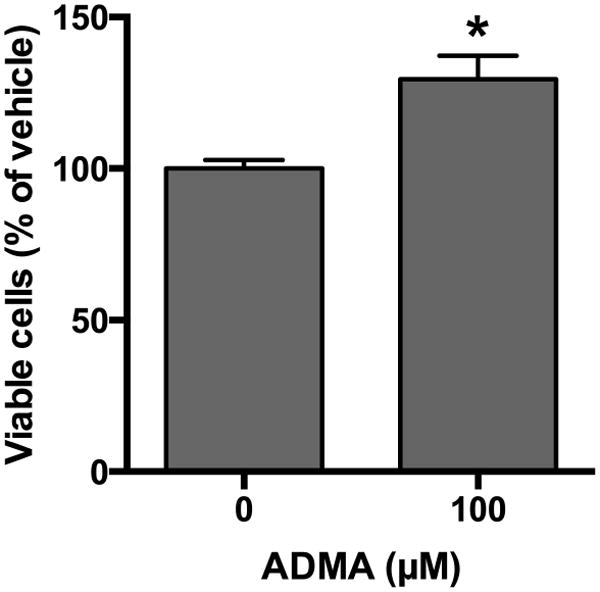
Bovine PAEC were seeded (5 × 104 cells/well) and incubated with vehicle or ADMA (100 μM) in 21% O2, 5%CO2, balance N2 (normoxia) for 48 hours (n=5-10). Viable cell numbers were determined using trypan blue exclusion and cells were counted using a hemacytometer. Data are shown as viable cells as a percentage of vehicle control. * different from vehicle-treated, p = 0.0007
Discussion
The main objective of the present study was to determine the effects of ADMA, an endogenously produced inhibitor of NOS activity, on arginase activity and resultant viable cell number. The major findings of this study were that 1) asymmetric methylarginines were not a substrate for arginase metabolism to urea, 2) asymmetric methylarginines did not inhibit arginase activity, 3) ADMA significantly increased urea production in bPAEC, likely secondary to its inhibitor effect on NOS, and 4) ADMA significantly increased the number of viable cells. Taken together, these findings support the notion that although the asymmetric methylarginines were not substrates for arginase and had no direct impact on arginase activity, ADMA increased the bioavailability of L-arginine to arginase resulting in increased arginase activity as measured by urea production. The increase in arginase activity was associated with an increase in viable cell number. These findings are consistent with our hypothesis that ADMA would result in greater viable cell numbers as a result of increased L-arginine bioavailability and metabolism by arginase.
L-arginine is the substrate for both NOS and arginase. In terms of NOS, the reported Km value for arginine is ∼10 μM, and therefore the concentrations of L-arginine in vivo far exceed the Km values. However, it has been shown repeatedly that adding excess L-arginine increases NO production (12-14), a phenomenon that has been termed the “L-arginine paradox” (8, 15). The presence of an endogenous competitive NOS inhibitor, specifically ADMA, may underlie the “L-arginine paradox” since increasing the L-arginine concentration would compete away the ADMA inhibitor effect on NOS (16). For arginase, the Km values for arginine are in the mM range; therefore, in the ranges of L-arginine tested in these studies, one would expect an increase in arginase activity with increasing L-arginine concentrations. Our results are also consistent with previous studies demonstrating that arginase is the major intracellular metabolic pathway for L-arginine (4, 17-19), since the levels of urea produced were measured in μmoles while the levels of NO produced were measured in nmoles.
Consistent with previous in vitro studies showing inhibition of NOS activity (20), as expected we found that ADMA inhibited NO production in bPAEC (Figure 3). On the other hand, we found no direct effects of ADMA on arginase activity in vitro (Figures 1 and 2). Indeed, we found that urea production was elevated and viable cell numbers were greater at the highest concentration of ADMA utilized. Given that ADMA did not act as a substrate, had no inhibitor effect on arginase, and did not induce arginase protein expression, the increase in urea production is most likely due to an increase in L-arginine availability to arginase due to inhibition of NOS by ADMA. We have previously found in bPAEC that NOS and arginase compete for L-arginine and that when one L-arginine metabolic pathway is inhibited the activity of the other is increased (17).
The biological significance of enhanced arginase activity due to increased substrate availability, in the face of elevated ADMA levels remains unclear. The ADMA-induced increase in arginase activity may favor tissue repair after an insult by stimulating ornithine production. For example, in murine macrophages and mice it has been found that Th2 cytokines are potent inducers of arginase, whereas Th1 cytokines are potent inducers of iNOS (21, 22). Given the key role of arginase in polyamine and proline synthesis, the increase in arginase activity and inhibition of NOS activity caused by ADMA may allow for cellular proliferation. Supporting this concept, Ignarro and colleagues (23) reported that treatment of vascular smooth muscle cells with NO donors decreased cellular proliferation, whereas overexpression of arginase I in these cells enhanced cellular proliferation. Furthermore, it has been shown recently in human pulmonary microvascular endothelial cells that inhibiting arginase decreased cellular proliferation (19). Thus, the ADMA-induced inhibition of NOS and increase in arginase activity may be a mechanism to prevent tissue damage associated with excessive NO production and augment tissue repair in lung diseases associated with NO over-production.
In contrast, an increase in levels of ADMA could prove to be detrimental. Our finding of increased arginase activity due to substrate bioavailability in ADMA-treated bPAEC is consistent with findings in a monocrotaline model of pulmonary hypertension in rats wherein ADMA content and arginase activity were found to be greater in pulmonary artery endothelial cells isolated from treated animals than in those isolated from controls (11). The ADMA-induced increase in arginase activity would be expected to have negative consequences in the pulmonary circulation and potentially lead to pulmonary hypertension. The ADMA-induced increase in arginase activity may increase vascular cell proliferation by producing L-ornithine, the first step in polyamine and proline synthesis. In this regard, if ADMA levels are high enough to inhibit NO production and increase arginase activity, then this would contribute to both vascular constriction and vascular remodeling. A recent study of endothelial cells from humans with primary pulmonary hypertension found higher levels of arginase protein, greater arginase activity, and lower levels of NO production than in endothelial cells from normal controls, despite similar levels of eNOS protein between the two groups (24). Thus, elevated levels of ADMA may be involved in the pathogenesis of pulmonary hypertension through the combined effects of decreased NO production and increased arginase activity leading to vasoconstriction and vascular cell proliferation as shown in Figure 6.
Figure 6. Proposed model for the role of ADMA in the pathogenesis of pulmonary hypertension.

Elevated levels of ADMA may result in decreased NO production and increased arginase activity leading to vasoconstriction and vascular cell proliferation.
There is sufficient evidence to suggest that endogenous NOS inhibitors are involved in the pathogenesis of endothelial dysfunction seen in pulmonary hypertensive diseases. We have elucidated the effects of the asymmetric methylarginines, specifically ADMA, on arginase activity. We conclude that ADMA has no direct effects on arginase protein expression or arginase activity, and in the endothelial cell, ADMA results in increased urea production and viable cell numbers. Our findings support the notion that ADMA is a potential target for preventing the endothelial dysfunction seen in pulmonary vascular diseases.
Materials and methods
Arginase activity
For the determination of arginase activity, test tube experiments were prepared and assays performed as previously described (25). Arginase, from bovine liver (Sigma-Aldrich, St. Louis, MO) was added to 10% (vol/vol) MnCl2 with 0.2 M Tris Buffer (pH 9.0) for a total volume of 150 μL. The mixtures were vortexed and incubated at 55°C for five minutes. Increasing concentrations of substrate [0.1 – 30 mM; L-arginine, D-arginine, ADMA, L-NMMA, and NG-nitro-L-arginine methyl ester (L-NAME) (Sigma-Aldrich)] were added to the heat-activated bovine arginase in each test tube, vortexed, and incubated at 37°C for one hour. The samples were colorimetrically assayed in duplicate for urea as previously described (18). Each sample (300 μL) was added to 1.5 ml chromogenic reagent [5 mg thiosemicarbazide, 250 mg diacetyl monoxime, and 25 mg FeCl3 in 150 ml 16.7% (vol/vol) H2SO4 and 13.3% (vol/vol) H3PO4]. The mixtures were vortexed, boiled at 100°C for five minutes, then cooled to room temperature. Each sample with the added chromogenic reagent (200 μL) was pipetted into a microplate in duplicate and the difference in absorbance at 530 nm was compared with a urea standard curve. The reaction velocity was plotted against the substrate concentration to calculate the Vmax and Km values using the Michaelis-Menten equation (Sigmaplot 11.0, Jandel Scientific, Carlsbad, CA).
To determine whether ADMA, L-NAME, or L-NMMA were inhibitors of arginase, reaction rates were measured in the presence of L-arginine (1 mM) with concentrations of the various candidate inhibitors (0 – 30 mM).
Cell culture
Bovine pulmonary arterial endothelial cells (bPAEC; Lonza, Allendale, NJ) were grown in 21% O2, 5% CO2, balance N2 at 37°C in endothelial growth media (EGM, Lonza) as previously described (17). Bovine PAEC between passages 3 and 8 were used in experiments. The cells were grown to 80-90% confluence in T-75 flasks, washed, and fresh media was placed on the cells. ADMA (100 μM) or vehicle was added to the cells, and then incubated in 21% O2, 5% CO2, balance N2 at 37°C for 24 hours. For the nitrite assays, the cells were washed twice with PBS, and fresh PBS containing 0.7 mM Ca2+, 1.1 mM Mg2+, 2.7 mM K+ and 6.1 mM glucose were then added to the cells. The cells were pre-treated with ADMA (100 μM) or vehicle for one hour, followed by stimulation with calcium ionophore A23187 (3 μM) for an additional four hours. At the end of the experimental period, the media or PBS was harvested and stored in 1 ml aliquots, and frozen at -80°C.
Protein isolation
Following treatment, bPAEC were harvested for total protein as previously described (17). Briefly, bPAEC were washed with HEPES balanced salt solution (HBSS, Lonza), and 750 μL of lysis buffer (0.2 M NaOH, 0.2% SDS) were added to each flask. Thirty minutes before use, the following protease inhibitors were added to each milliliter of lysis buffer: 0.2 μl aprotinin [10 mg/ml double distilled (dd) H2O], 0.5 μl of leupeptin (10 mg/ml ddH2O), 0.14 μl pepstatin A (5 mg/ml methanol), and 5 μl of phenylmethylsulfonyl fluoride (34.8 mg/ml methanol). This was sterile filtered in a syringe and added to each T-75 flask of bPAEC. The bPAEC were scraped and placed in sterile centrifuge tubes on ice. The supernatant was stored at -80°C. Total protein concentration was determined by the Bradford method with a commercially available assay (Bio-Rad, Hercules, CA).
Western blot
The lysed bPAEC were assayed for arginase I and arginase II protein by Western blot analysis as previously described (18, 26). Aliquots of cell lysate (15-20 μg of protein) were diluted 1:1 with SDS sample buffer, heated to 95°C for 5 minutes, and electrophoresed on polyacrylamide gel. Separated proteins were electrotransferred to PVDF membranes, incubated with 5% nonfat milk for one hour, and then incubated overnight with primary antibody, arginase I (1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), or arginase II (1:500; Santa Cruz) overnight and then washed three times with PBS-T. The membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:15,000; Bio-Rad). The protein bands were visualized using enhanced chemiluminescence (ECL Plus reagent, Amersham Pharmacia Biotech, Piscataway, NJ) and quantified using densitometry (Sigma Gel, Jandel Scientific, San Rafael, CA). To control for protein loading, the blots were stripped using a stripping buffer containing 62.5 mM Tris HCl (pH 6.8), 2% SDS, and 100 mM 2-β-mercaptoethanol, and the blots were reprobed for β-actin (1:5,000; Abcam, Cambridge, MA) as described above.
Nitrite assay
The samples of PBS were assayed in duplicate for nitrite using a chemiluminescence NO analyzer (model 280i, Sievers Instruments, Boulder, CO) as previously described (17, 22). Briefly, 100 μl of sample was placed in a reaction chamber containing a mixture of NaI in glacial acetic acid to reduce nitrite to NO. The NO gas was carried into the NO analyzer using a constant flow of helium gas. The analyzer was calibrated using a NaNO2 standard curve.
Urea assay
The medium samples were assayed in duplicate for urea concentration colorimetrically, as previously described (19, 27). Briefly, 100 μl of sample was added to 3 ml of chromogenic reagent [5 mg of thiosemicarbazide, 250 mg of diacetyl monoxime, 37.5 mg of FeCl3 in 150 ml 25% (vol/vol) H2SO4, 20% (vol/vol) H3PO4]. After 1 h at 37°C the mixtures were vortexed and then boiled at 100°C for five minutes. The mixtures were cooled to room temperature, and the absorbance (530 nm) was determined and compared with a urea standard curve.
Viable cell number
To determine the number of viable cells, bPAEC were seeded in six-well plates (5 × 104 cells per well) in EGM and incubated in 21% O2, 5% CO2, balance N2 at 37°C for 48 hours. Cells were treated with vehicle or ADMA (100 μM) prior to incubation. At the end of the experiment, the adherent cells were trypsinized, and viable cells were counted using trypan blue exclusion as previously described (19). Given the inter-experiment variability in cell proliferation for pulmonary endothelial cells as previously shown (19, 28, 29), data for each experiment (performed at five separate times) were normalized to control. The data are shown as a percentage of vehicle control.
Statistical analysis
Values are given as means ± SEM. Data were analyzed using unpaired t-test (GraphPad Prism, San Diego, CA). Differences were considered significant when p < 0.05.
Acknowledgments
This work was supported by grant 1K08HL105677-01A2 (BC) from the National Heart Lung and Blood Institute of the NIH
References
- 1.Humbert M, Morrell NW, Archer SL, et al. Cellular and Molecular Pathobiology of Pulmonary Arterial Hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 2.Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. 2002;45:173–202. doi: 10.1053/pcad.2002.130041. [DOI] [PubMed] [Google Scholar]
- 3.Janne J, Alhonen L, Leinonen P. Polyamines: from molecular biology to clinical applications. Annals of medicine. 1991;23:241–59. doi: 10.3109/07853899109148056. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Meinginger CJ, Hawker JR, et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Physiol Endocrinol Metab. 2001;280:E75–E80. doi: 10.1152/ajpendo.2001.280.1.E75. [DOI] [PubMed] [Google Scholar]
- 5.Leiper J, Nandi M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nature reviews Drug discovery. 2011;10:277–91. doi: 10.1038/nrd3358. [DOI] [PubMed] [Google Scholar]
- 6.MacAllister RJ, Fickling SA, Whitley GS, Vallance P. Metabolism of methylarginines by human vasculature; implications for the regulation of nitric oxide synthesis. Br J Pharmacol. 1994;112:43–8. doi: 10.1111/j.1476-5381.1994.tb13026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kielstein JT, Impraim B, Simmel S, et al. Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation. 2004;109:172–7. doi: 10.1161/01.CIR.0000105764.22626.B1. [DOI] [PubMed] [Google Scholar]
- 8.Boger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. The Journal of nutrition. 2004;134:2842S–7S. doi: 10.1093/jn/134.10.2842S. discussion 53S. [DOI] [PubMed] [Google Scholar]
- 9.Pullamsetti S, Kiss L, Ghofrani HA, et al. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J. 2005;19:1175–7. doi: 10.1096/fj.04-3223fje. [DOI] [PubMed] [Google Scholar]
- 10.Millatt LJ, Whitley GS, Li D, et al. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia-induced pulmonary hypertension. Circulation. 2003;108:1493–8. doi: 10.1161/01.CIR.0000089087.25930.FF. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki A, Doi S, Mizutani S, Azuma H. Roles of accumulated endogenous nitric oxide synthase inhibitors, enhanced arginase activity, and attenuated nitric oxide synthase activity in endothelial cells for pulmonary hypertension in rats. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1480–7. doi: 10.1152/ajplung.00360.2006. [DOI] [PubMed] [Google Scholar]
- 12.Creager MA, Gallagher SJ, Girerd XJ, Coleman SM, Dzau VJ, Cooke JP. L-arginine improves endothelium-dependent vasodilation in hypercholesterolemic humans. J Clin Invest. 1992;90:1248–53. doi: 10.1172/JCI115987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drexler H, Zeiher AM, Meinzer K, Just H. Correction of endothelial dysfunction in coronary microcirculation of hypercholesterolaemic patients by L-arginine. Lancet. 1991;338:1546–50. doi: 10.1016/0140-6736(91)92372-9. [DOI] [PubMed] [Google Scholar]
- 14.Girerd XJ, Hirsch AT, Cooke JP, Dzau VJ, Creager MA. L-arginine augments endothelium-dependent vasodilation in cholesterol-fed rabbits. Circ Res. 1990;67:1301–8. doi: 10.1161/01.res.67.6.1301. [DOI] [PubMed] [Google Scholar]
- 15.Tsikas D, Boger RH, Sandmann J, Bode-Boger SM, Frolich JC. Endogenous nitric oxide synthase inhibitors are responsible for the L-arginine paradox. FEBS letters. 2000;478:1–3. doi: 10.1016/s0014-5793(00)01686-0. [DOI] [PubMed] [Google Scholar]
- 16.Mugge A, Harrison DG. L-arginine does not restore endothelial dysfunction in atherosclerotic rabbit aorta in vitro. Blood vessels. 1991;28:354–7. doi: 10.1159/000158881. [DOI] [PubMed] [Google Scholar]
- 17.Chicoine LG, Paffett ML, Young TL, Nelin LD. Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L60–8. doi: 10.1152/ajplung.00194.2003. [DOI] [PubMed] [Google Scholar]
- 18.Nelin LD, Nash HE, Chicoine LG. Cytokine treatment increases arginine metabolism and uptake in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1232–L9. doi: 10.1152/ajplung.2001.281.5.L1232. [DOI] [PubMed] [Google Scholar]
- 19.Toby IT, Chicoine LG, Cui H, Chen B, Nelin LD. Hypoxia-induced proliferation of human pulmonary microvascular endothelial cells depends on epidermal growth factor receptor tyrosine kinase activation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L600–6. doi: 10.1152/ajplung.00122.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faraci FM, Brian JE, Jr, Heistad DD. Response of cerebral blood vessels to an endogenous inhibitor of nitric oxide synthase. Am J Physiol. 1995;269:H1522–7. doi: 10.1152/ajpheart.1995.269.5.H1522. [DOI] [PubMed] [Google Scholar]
- 21.Morris SM, Jr, Kepka-Lenhart D, Chen LC. Differential regulation of arginases and inducible nitric oxide synthase in murine macrophage cells. Am J Physiol. 1998;275:E740–7. doi: 10.1152/ajpendo.1998.275.5.E740. [DOI] [PubMed] [Google Scholar]
- 22.Nelin LD, Wang X, Zhao Q, et al. MKP-1 switches arginine metabolism from nitric oxide synthase to arginase following endotoxin challenge. Am J Physiol Cell Physiol. 2007;293:C632–40. doi: 10.1152/ajpcell.00137.2006. [DOI] [PubMed] [Google Scholar]
- 23.Ignarro LF, Buga GM, Wei LH, Bauer PM, Wu G, del Soldato P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci. 2001;98:4202–8. doi: 10.1073/pnas.071054698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu W, Kaneko FT, Zheng S, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004;18:1746–8. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 25.Chen B, Calvert AE, Cui H, Nelin LD. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1151–9. doi: 10.1152/ajplung.00183.2009. [DOI] [PubMed] [Google Scholar]
- 26.Cui H, Chen B, Chicoine LG, Nelin LD. Overexpression of cationic amino acid transporter-1 increases nitric oxide production in hypoxic human pulmonary microvascular endothelial cells. Clinical and experimental pharmacology & physiology. 2011;38:796–803. doi: 10.1111/j.1440-1681.2011.05609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelin LD, Chicoine LG, Reber KM, English BK, Young TL, Liu Y. Cytokine-induced endothelial arginase expression is dependent on epidermal growth factor receptor. Am J Respir Cell Mol Biol. 2005;33:394–401. doi: 10.1165/rcmb.2005-0039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chicoine LG, Stenger MR, Cui H, et al. Nitric oxide suppression of cellular proliferation depends on cationic amino acid transporter activity in cytokine-stimulated pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L596–604. doi: 10.1152/ajplung.00029.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stanley KP, Chicoine LG, Young TL, et al. Gene transfer with inducible nitric oxide synthase decreases production of urea by arginase in pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L298–306. doi: 10.1152/ajplung.00140.2005. [DOI] [PubMed] [Google Scholar]