

Abstract
There is a growing library of functionalized non-natural substrates for the enzyme protein farnesyltransferase (PFTase). PFTase covalently attaches these functionalized non-natural substrates to proteins ending in the sequence CAAX, where C is a cysteine that becomes alkylated, A represents an aliphatic amino acid, and X is Ser, Met, Ala, or Gln. Reported substrates include a variety of functionalities that allow modified proteins to undergo subsequent bioconjugation reactions. To date the most common strategy used in this approach has been copper catalyzed azide-alkyne cycloaddition (CuAAC). While being fast and bioorthogonal CuAAC has limited use in live cell experiments due to copper’s toxicity.1 Here we report the synthesis of trans-cyclooctene geranyl diphosphate. This substrate can be synthesized from geraniol in six steps and be enzymatically transferred to peptides and proteins that end in a CAAX sequence. Proteins and peptides site-specially modified with trans-cyclooctene geranyl diphosphate were subsequently targeted for further modification via tetrazine ligation. Since tetrazine ligation is bioorthogonal, fast, and is contingent on ring strain rather than the addition of a copper catalyst, this labeling strategy should prove useful for labeling proteins where the presence of copper may hinder solubility or biological reactivity.
Keywords: trans-cyclooctene, tetrazine ligation, protein prenylation, protein farnesyltransferase, bioorthogonal, inverse-electron-demand Diels-Alder
Introduction
Site specific-labeling of proteins is an important area of study in modern biotechnology because it enables the preparation of protein conjugates that can be used to study protein structure, protein-protein interactions, cellular trafficking and the attachment of proteins to other molecules and surfaces. While the benefits of site-specific modification technologies are clear, developing these strategies is difficult due to the many different reactive groups present within the side chains of polypeptides. In order to circumvent the problem of having multiple side chains with comparable reactivity, enzymes that recognize specific peptide sequences can be used to recognize and modify a single amino acid within that sequence for modification.
The enzyme used in this study, protein farnesyltransferase (PFTase), normally transfers farnesyl isoprenoids to site-specific cysteines in proteins ending in the four amino acid sequence CAAX, where C is a cysteine, A is an aliphatic amino acid, and X can be Ser, Met, Ala, or Gln.2 Since the size of the PFTase binding pocket is large and lined with hydrophobic residues the enzyme has been shown to transfer a variety of non-natural substrates with similar size and hydrophobicity to its physiological substrate, farnesyl diphosphate (1).3 Modifications to the isoprenoid scaffold have ranged from making singular atom substitutions (H for F) on the isoprenoid backbone4 to swapping out whole isoprenoid units for reactive groups including alkynes, azides, aldehydes, and benzophenones.5–8 Installation of these functionalities imparts handles that can be used for additional modification via, copper catalyzed azide alkyne cycloaddition (CuAAC), oxime ligation, or Staudinger ligation. These handles have been useful for a number of applications including protein immobilization on solid surfaces, the creation of protein-DNA conjugates, and synthesis of protein-fluorophore adducts.9–12
While these strategies have been successful for site specific protein labeling in vitro, their application to biological systems has limitations. Oxime ligation requires the use of aldehyde or ketone functional groups which are abundant in numerous cellular metabolites. This overlap marks them for side reactivity in cellular applications. Since CuAAC requires the use of copper, its use in studying complex biological systems is limited to experiments that study the outer cell membrane or require treated cells to be lysed before analysis.1,13 Newer copper-free strain promoted azide-alkyne cycloaddition (SPAAC) reactions have allowed reactions within living systems including zebrafish embryos and mammalian cells; however these reactions are much slower than CuAAC (k2 = 0.1 M−1s−1 vs. k = 104 M−1s−1/M copper).14–16 Pioneering work by several research groups has demonstrated that tetrazine cycloaddition is a promising alternative to both CuAAC and SPAAC.17–20 Tetrazine ligation, has a similar rate constant to CuAAC (k2 = 2,000–6,000 M−1s−1)18,21 but is suitable for experiments performed inside live cells because it does not require additional catalysts. Examples of tetrazine ligation include intracellular taxol labeling experiments described by Weissleder and coworkers and fluorescent probe incorporation mediated by enzyme (PRIME) experiments carried out by Ting and coworkers.19,22
Here we report on the synthesis and use of trans-cyclooctene geranyl diphosphate (2). This new alternative PFTase substrate expands the repertoire of non-natural PFTase substrates to include a strained- ring containing substrate that can be site-specifically attached to proteins and peptides ending in the amino acid sequence CVIA. The trans-cyclooctene functionality of the installed substrate allows for subsequent protein or peptide modification via a strain-promoted reaction with functionalized tetrazines 3 and 4 without the use of a potentially toxic copper catalyst. Since both tetrazines and hydrophobic diphosphates have been shown to exhibit cell-penetrating properties, this technology should have applications that allow for the site specific targeting of CAAX box contain proteins inside living cells.19
Materials and Methods
General
Thin layer chromatography was performed on pre-coated silica gel 60 aluminum F254 plates from EMD chemical and visualized by staining with KMnO4. Flash chromatography silica gel was obtained from Mallinckrodt Inc. PPh3 (polymer-supported beads) were purchased from Aldrich. All NMR spectra were acquired on Varian instruments at 25 °C. Preparative HPLC separations were performed using a Beckman mode 127/166 instrument equipped with a Phenomenex C18 column (Luna, 10µm, 10 × 250 mm). HR-MS spectra were obtained using a Bruker BioTOF II instrument. MS analysis on protein samples was performed on an ESI-microTOF-Q from Bruker Daltronics. LC-MS analysis on tetrazine ligated peptides was performed on a Thermo LCQ-Deca ion trap. NAP 5 size exclusion columns were purchased from GE Life Sciences.
Recombinant PFTase was overexpressed in E. coli and purified using previously published procedures.23,24 The activity of the PFTase enzyme was measured to be 1.6 umol/min/mg using a previously reported continuous fluorescence assay.25 eGFP-CVIA (12) was overexpressed in E. coli and purified using previously published procedures.26 Concentrations of purified proteins and enzymes were determined using a Bradford assay27 and purity was confirmed using SDS-PAGE analysis. N-Oregon Green-RTRCVIA (9),9 dipyridyl-tetrazine (3),18 benzylamino-tetrazine (4),20 trans-cyclooctene derivative 5,28 and brominated ether 6,29 were synthesized using previously published procedures. Concentrations of stock solutions of trans-cyclooctene geranyl diphosphate (2) were determined using 31P NMR.30
2-(((2E,6E)-8-((R,E)-cyclooct-4-en-1-yloxy)-3,7-dimethylocta-2,6-dien-1-yl)oxy)tetrahydro-2H-pyran (7)
Rel-(1R, 4E, pR)-Cyclooct-4-enol18 (0.060 g, 0.48 mmol), 5, was dissolved in dry THF (2 mL), placed into a 38 mL sealed pressure tube with a threaded type A plug, and cooled to 0 °C. Then NaH (60 % dispersion in mineral oil, 0.141 g, 3.52 mmol) was added and the solution and was stirred for 5 min in the sealed tube. After bubbling ceased, brominated ether 629 (0.171 g, 0.44 mmol) was added, and the tube was resealed and heated for 12 h at 75 °C. At this time the reaction was quenched with H2O (15 mL), extracted with EtOAc (2×20 mL), washed with H2O (2×10 mL), dried over Na2SO4, and concentrated. The oil was then purified by flash chromatography (3:1 hexanes/EtOAc, v/v) to yield 0.088 g of 7 (92.0 % yield). 1HNMR (300 MHz, CDCl3): 1.61 (s, 3H), 1.68 (s, 3H), 1.43-1.76 (m, 5H), 1.76-2.01 (m, 6H), 2.01-2.10 (m, 2H), 2.10-2.25 (m, 5H), 2.29-2.40 (m, 2H), 2.94-3.00 (m, 1H), 3.47-3.54 (m, 1H), 3.68 (d, J = 11.6 Hz, 1H), 3.75 (d, J = 11.6 Hz, 1H), 3.85-3.92 (m, 1H), 4.01 (dd, J = 6.15 Hz, 11.67 Hz, 1H), 4.24 (dd, J = 6.15 Hz, 11.67 Hz, 1H), 4.62 (t, J = 3.5 Hz, 1H), 5.31-5.40 (m, 3H), 5.57 (t, J = 5.57 Hz, 1H); 13CNMR 14.07 (300 MHz, CDCl3): 16.44, 19.64, 25.51, 26.01, 30.72, 31.66, 33.08, 34.56, 37.87, 39.28, 40.84, 62.29, 63.67, 74.07, 84.02, 97.87, 120.78, 127.11, 132.00, 132.76, 135.44, 139.91; HR-ESI-MS calcd for [M+Na]+ C23H38O3Na+, exact mass: 385.2713, found: 385.2712.
(2E,6E)-8-((R,E)-cyclooct-4-en-1-yloxy)-3,7-dimethylocta-2,6-dien-1-ol (8)
Protected alcohol 7 (0.106 g, 0.29 mmol) and PPTs (0.007 g, 0.03 mmol) were added to EtOH (5 mL) and refluxed for 14 h at which time the reaction mixture was concentrated and purified by flash chromatography (3:1 hexanes/EtOAc, v/v) to yield 0.059 g of alcohol 8 (73.2 % yield). 1HNMR (300 MHz, CDCl3): 1.41-1.54 (m, 3H), 1.61 (s, 3H), 1.67 (s, 3H), 1.76-1.87 (m, 2H), 1.87-1.99 (m, 2H), 2.01-2.24 (m, 5H), 2.30-2.40 (m, 2H), 2.94-3.00 (m, 1H), 3.68 (d, J = 11.6 Hz, 1H), 3.75 (d, J = 11.6 Hz, 1H), 4.14 (d, 7.1 Hz, 2H), 5.30-5.43 (m, 3H), 5.56 (t, J = 12.6 Hz, 1H); 13CNMR 14.07 (300 MHz, CDCl3): 14.13, 16.27, 25.95, 31.69, 33.08, 34.55, 37.86, 39.13, 40.83, 59.33, 74.15, 84.24, 123.74, 127.03, 132.21, 132.93, 135.45, 139.17; HR-ESI-MS calcd for [M+Na]+ C18H30O2Na+, exact mass: 301.2144, found: 301.2143.
(2E,6E)-8-((R,E)-cyclooct-4-en-1-yloxy)-3,7-dimethylocta-2,6-dien-1-yl dihydrogen diphosphate (2)
PPh3 (polymer supported beads, 0.028 g, 0.09 mmol) was allowed to swell in 3 mL of CH2Cl2 for 20 min at which time alcohol 8 (0.0154 g, 0.06 mmol) and CBr4 (0.020 g, 0.06 mmol) were added and the mixture was gently stirred for 4 h. The mixture was then filtered and concentrated to yield a light brown oil (0.011 g) that was used without further purification. The oil was dissolved in 0.8 mL of CH3CN and (n-Bu4N)3HP2O7 (0.116 g, 0.132 mmol) was added. The reaction was stirred under N2 for 3h and concentrated before the product was converted to its ammonium form using Dowex® AG 50W-X8 exchange resin.31 The resin (10 g) was packed into a column (30 cm×1.5 cm) and washed with three bed volumes of H2O, three bed volumes of H2O/conc. NH4OH (3:1, v/v), and equilibrated with four bed volumes of 25 mM NH4HCO3/isopropanol (49:1, v/v) (solvent A). The crude oil was then dissolved in 20 mL of solvent A and added to the column. This solution was ran through the column and collected. An additional 20 mL of solvent A was then passed through the column and added to the first collected fraction. The combined fractions were then lyophilized and purified by semi preparative RP-HPLC using the following conditions: UV detection 220 nm; flow rate 5.0 ml/mL; gradient 0-40% solvent B in 80 min; solvent A: 25 mM NH4HCO3; solvent B: CH3CN. Product containing fractions (Rt = 50 min) were lyophilized to yield 8.2 mg of trans-cyclooctene containing diphosphate 2 in a 33.5 % two step yield. 1HNMR (300 MHz, D2O): 1.24-1.40 (m, 3H), 1.45 (s, 3H), 1.57 (s, 3H), 1.51-2.24 (m, 11 H), 2.92-3.04 (m, 1H), 3.64 (d, 1H), 3.67 (d, 1H), 4.31 (t, J = 6.6 Hz, 2H), 5.25-5.36 (m, 3H), 5.41-5.60 (m, 1H). 31PNMR (300 MHz, D2O): -9.72(d, 1P), -6.44 (d, 1P). HR-ESI-MS calcd for [M-H]− C18H31O8P2, expected mass: 437.1500, found: 437.1498.
Enzymatic Prenylation of N-Oregon Green-Arg-Tyr-Arg-Cys-Val-Ile-Ala with 2 for HPLC and MS Analysis
A 40.0 mL enzymatic reaction containing 50 mM Tris·HCl, pH 7.0, 10 mM MgCl2, 10 µM ZnCl2, 15 mM DTT, 2.4 µM N-Oregon Green-RTRCVIA (9), 5 µg PFTase, and 20 µM 2 was completed at 30 °C in a water bath. Prior to the reaction all reagents except 2 and enzyme were incubated for 1 h at room temperature to ensure all Cys-Cys disulfide bonds were reduced. After this incubation period 2 and enzyme were added and the reaction was allowed to proceed for 4 h. The reaction mixture was then purified using semi preparative RP-HPLC using the following conditions: fluorescence detection 490 nm; flow rate 5.0 ml/mL; gradient 0-95% solvent B in 65 min; solvent A: 0.1 % TFA in H2O; solvent B: 0.1 % TFA in CH3CN. The enzymatically modified product (10) eluted at Rt = 57 min. HR-ESI-MS calcd for [M+2H]+2 C72H101F2N13O16S+, expected mass: 736.85, found: 736.74.
Creation of 11 by tetrazine ligation between trans-cyclooctene modified peptide 10 and dipyridyl-tetrazine (3)
A small scale tetrazine ligation reaction was prepared containing 20 µM trans-cyclooctene modified peptide 10 (3 µL of a 196 µM solution) and 50 µM dipyridyl-tetrazine 3 (1.5 µL of a 1 mM solution) in 20 mM NaOAc (27 µL). The reaction was allowed to sit at room temperature for 10 min after which time it was analyzed by LC-MS using the following conditions: (column-Microsorb-MV, 5µM, 4.6×250 mm; flow rate 1.0 mL/min; gradient 0-95% solvent B in 65 min; solvent A: 0.1 % TFA in H2O; solvent B: 0.1 % TFA in CH3CN). Tetrazine ligated peptide 11 was observed at Rt = 46.5 min and was confirmed by MS analysis. MS calcd for [M+2H]+2 C84H108F2N18O16S, expected mass: 848.4, found 848.5.
Kinetic analysis of enzymatic modification of N-Dansyl-GCVIA with trans-cyclooctene geranyl diphosphate (2)
To determine the KM,app for trans-cyclooctene analogue 2 a previously published fluorescent assay was used.32 Briefly 100 μL of a solution consisting of 125 mM Tris HCl, pH=7.5, 25 mM MgCl2, 25 μM ZnCl2, 0.1% n-dodecyl-β-D-maltoside, 12.5 mM DTT, 5 μM of N-Dansyl- GCVIA and various concentrations of 2 was added to individual wells of a black 96-well plate. An additional 130 μL of water was added to each of the wells. The PFTase enzyme was added to a buffer of 50 mM Tris HCl, pH=7.5, 50 μM ZnCl2, 5 mM MgCl2, 20 mM KCl, 1 mM DTT and 1.0 mg/mL BSA to create a solution with a 62.5 nM enzyme concentration. The reaction was monitored for fluorescence intensity on a Beckman Coulter DTX 880 Multimode Detector using the A 340/10 filter (ex.=340 nM) for the excitation and the F 353/25 filter (em.=505 nM) for emission. An initial fluorescence reading was taken before the addition of PFTase, and then the reaction was initiated with the addition of 20 μL of the PFTase containing solution described above. The reaction was monitored until the fluorescence intensity reached a maximum, which took approximately 1 h. The data was exported from the Multimode Analysis Software to Microsoft Excel where initial rate data analysis was performed. The final rate constants were determined using the non-linear regression feature of KaleidaGraph 3.6.
Enzymatic Modification of eGFP-CVIA (12) to create trans-cyclooctene modified protein 13
A 11.0 mL enzymatic reaction containing 50 mM Tris, 10 mM MgCl2, 10 µM ZnCl2, 15 mM DTT, 2.4 µM eGFP-CVIA (12), 24 µM 2, and 25 µg of PFTase was performed in a 30 °C water bath. Prior to the reaction all reagents except 2 and enzyme were incubated for 1 h at room temperature to ensure all Cys-Cys disulfide bonds were reduced. After this incubation period 2 and enzyme were added and the reaction was allowed to proceed for 4 h. At this time the reaction was concentrated to 500 µL and excess 2 was removed via a NAP 5 column. MS analysis was then performed on an ESI-micro-TOF-Q. Protein samples were injected at a concentration of 20 µM-40 µM into a flow of H2O/CH3CN/HCO2H (64.9:35.0:0.1) at a flow rate of 0.1 mL/min. Ion series from m/z peaks were processed and deconvoluted using Bruker DataAnalysis software. Calculated [M+H] Mr = 27595.2, observed m = 27596.0; relative error 0.003%.
Creation of 14 by tetrazine ligation of trans-cyclooctene modified eGFP-CVIA (13) and benzylamino-tetrazine (4)
A small scale tetrazine ligation reaction was prepared containing 33 µM trans-cyclooctene modified eGFP-CVIA (13, 200 µL of a 35 µM solution dissolved in 20 mM Tris pH 7.0) and 630 µM benzylamino-tetrazine (4, 10 µL of a 630 µM solution dissolved in DMSO). The reaction was allowed to stand at room temperature for 4 h and was placed at 4 °C overnight at which time it was subjected to MS analysis using an ESI-micro-TOF-Q. Protein samples were injected at a concentration of 20 µM-40 µM into a flow of H2O/CH3CN/HCO2H (64.9:35.0:0.1) at a flow rate of 0.1 mL/min. Ion series from m/z peaks were processed and deconvoluted using Bruker DataAnalysis software. Calculated [M+H] Mr = 27754.3, observed m = 27753.0; relative error 0.005%.
Discussion
Synthesis of trans-cyclooctene geranyl diphosphate
Synthesis of trans-cyclooctene geranyl diphosphate (2) was completed in six steps in its longest linear sequence. Following previously published procedures, bromide 6 was obtained in three steps from commercially available geraniol.29 This allylic halide reacted with deprotonated (E)-cyclooct-4-enol (5) to produce strain-ring containing ether 7. Deprotection of 7 gave alcohol 8 which was transformed to a bromide via an Appel reaction before being converted to trans-cyclooctene geranyl diphosphate (2) using [(nBu)4)N]3HP2O7. Trans-cyclooctene geranyl diphosphate (2) was purified by ion-exchange chromatography and RP-HPLC. NMR based phosphorous analysis was used to obtain accurate concentrations of 2 using a phosphoric acid standard. 30
Evaluation of Compound 2 as a Substrate for PFTase
First, compound 2 was investigated as an alternative substrate for PFTase using an HPLC-based assay. An Oregon Green-containing CAAX box peptide (9), was synthesized as previously reported9 and used as a peptide substrate for PFTase enzyme. This fluorescent peptide substrate was chosen due to its high fluorescence intensity and water solubility. Fluorescent peptide 9 (2.4 µM) was incubated with non-natural substrate 2 (20 µM), and PFTase (5 µg) for 4 h hours at room temperature. At this time the reaction mixture was purified using semi-preparative HPLC by isolating a newly appearing peak that had a retention time much later than unmodified peptide 9. This was expected due to the increase in hydrophobicity in the enzymatically alkylated product. ESI- MS analysis of product containing fractions confirmed that the modification had occurred on peptide 9 to create trans-cyclooctene containing peptide 10.
Utilization of Tetrazine Ligation on trans-cyclooctene containing peptides
After trans-cyclooctene modification, peptide 10 was tested for its ability to react with a tetrazine via an inverse electron demand Diels-Alder reaction. Trans-cyclooctene containing peptide 10 (20 µM) was incubated with dipyridyl-tetrazine (3) (50 µM) for 15 minutes and subjected to analysis by ESI-MS. The ESI-MS analysis showed a mass of 848.55 which corresponded to the [M+2H]2+ peak of tetrazine ligated peptide 11 (see supplementary materials). The mass of unligated peptide 10 was not found in a full mass scan of the sample. The absence of observable starting material suggests that the reaction proceeded to completion within 15 minutes. That rapid reaction time is constant with the high second order rate constant associated with tetrazine ligation (k2 = 2000–6000 M s−1).18,19 A control reaction containing trans-cyclooctene modified peptide 10 that was not exposed to tetrazine was also analyzed by ESI-MS. This analysis showed a mass of 735.61 which corresponded to the [M+2H]2+ peak of the peptide. This result indicates that if starting material remained, it would have likely been observed in the ESI-MS measurements.
Enzymatic rate of trans-cyclooctene geranyl diphosphate incorporation
The rate of peptide modification using trans-cyclooctene geranyl diphosphate (2) was compared to that of the natural PFTase substrate, farnesyl diphosphate (1), using a continuous florescence-based assay. For the assay a fixed concentration of N-Dansyl-GCVIA (5 µM) was incubated with a range of concentrations of alternative substrate 2 (0–25 µM) and PFTase (5 nM). As N-Dansyl-GCVIA is enzymatically modified with the trans-cyclooctene containing isoprenoid (2), the change in hydrophobicity around the environmentally sensitive fluorophore causes a change in fluorescence intensity. KM,app can be calculated from this series of measurements since the rate of this change is dependent on the concentration of enzymatic substrate. The experimentally determined KM,app for compound 2 was found to be 0.77 µM (Figure 2) while the reported KM,app value for PFTase’s physiological substrate, farnesyl diphosphate (1), is reported to be 0.42 µM.33 This corresponds to roughly a two-fold difference between the enzyme’s natural substrate (1) and trans-cyclooctene geranyl diphosphate (2) with the former manifesting the greater affinity. Kcat/KM,app for trans-cyclooctene geranyl diphosphate was calculated to be 0.13 µM−1s−1. This is within two orders of magnitude of the reported 4.5 µM−1s−1 Kcat/KM,app value for PFTase’s natural substrate, FPP.34
Figure 2.

A Fluorescence-based enzyme assay was used to calculate the rate for enzymatic modification using different concentrations of 2. The plot of rate versus substrate concentration showed that compound 2 was a substrate for PFTase with a KM,app of 0.76 μM. Each measurement was performed in triplicate and the average plotted with the SD indicated by the error bars.
This measured KM,app for 2 is similar to other FPP mimics reported by Spielmann and coworkers that have incorporated aryl rings onto the terminal ends of synthetic isoprenoid diphosphates.35 The transfer of 2 is especially significant due to the bulky trans-cyclooctene moiety onto the terminal end of the analog. Due to its size, a trans-cyclooctene modification does not seem to be an intuitive isoprenoid mimic; however due to trans-cyclooctene’s ability to take up a crown conformation it adopts a relatively compact molecular volume comparable to an isoprenoid unit. This agrees with Spielmann’s conclusions that size and shape are the most important factors in PFTase’s ability to transfer non-natural isoprenoids to proteins and peptides ending in a “CAAX” motif. In addition, trans-cyclooctene’s hydrophobic characteristics appear to be well suited for interaction with the PFTase active site which is lined with nonpolar aromatic residues.
Modification of eGFP-CVIA using trans-cyclooctene diphosphate (2)
After showing CAAX box containing peptides can be modified using non-natural analog 2, we moved to show this chemistry also works using a model protein. Since this chemistry works on modifying proteins, it may be potentially useful for targeting prenylatable proteins in living cells. Both non-natural isoprenoids36 and fluorescently tagged tetrazines22 have been shown to exhibit cell penetrating properties. Thus a tetrazine/ trans-cyclooctene geranyl diphosphate bioorthogonal pair may prove useful for labeling lipid modified proteins in living unfixed cells.
In order to demonstrate that enzymatic modification of a protein with trans-cyclooctene geranyl diphosphate (2) was possible, a model protein, eGFP-CVIA (12, 2.4 µM), was incubated with 2 (20 µM) and PFTase under typical prenylation reaction conditions. After 4 h the reaction mixture was concentrated and purified using a NAP 5 size exclusion column to remove any unreacted substrate. The identity of trans-cyclooctene modified eGFP-CVIA (13) was confirmed by performing ESI-MS measurements on both unreacted eGFP-CVIA and the protein containing fraction isolated from the size exclusion column. ESI-MS of unreacted eGFP-CVIA produced an ion series (Fig. 3A) that was deconvoluted to yield a mass of 27334.0 (Fig. 3B). ESI-MS analysis of eGFP-CVIA that was enzymatically modified with trans-cyclooctene geranyl diphosphate (2) yielded a new mass that matches that of trans-cyclooctene modified protein 13 (calculated [M+H] Mr = 27595.2, observed m = 27596.0; relative error 0.003%) (Fig. 3C). Another mass at 27469 was observed. It is hypothesized that this mass is the result of proteolyzed protein or enzyme but this has not been confirmed.
Figure 3.

(A) ESI-MS of eGFP-CVIA (12). (B) Deconvoluted mass spectrum of eGFP-CVIA. (C) Deconvoluted mass spectra of eGFP-CVIA that was subjected to enzymatic modification using trans-cyclooctene geranyl diphosphate (2) and PFTase. (D) Deconvoluted mass spectra of trans-cyclooctene labeled eGFP-CVIA that underwent tetrazine ligation with benzylamino-tetrazine (4).
Next trans-cyclooctene-containing eGFP-CVIA (13) was incubated with benzylamino-tetrazine (4). This unsymmetrical tetrazine (4) was used due its superior aqueous solubility compared with dipyridyl-tetrazine (3) and recent reports on its superior use for in vivo studies.19 After purification using a NAP 5 size exclusion column, protein containing fractions were analyzed using ESI-MS. Analysis of benzylamino-tetrazine treated samples produced a deconvoluted mass that matched that of tetrazine ligated protein 14 (calculated [M+H] Mr = 27754.3, observed m = 27753.0; relative error 0.005%) (Fig 3D). This confirmed that eGFP-CVIA can be enzymatically modified with trans-cyclooctene isoprenoid 2 and undergo subsequent bioconjugation via tetrazine ligation.
Conclusions
This work reports the synthesis of the first strained-ring containing PFTase substrate. Trans-cyclooctene geranyl diphosphate (2) binds to PFTase with an affinity similar to that of the natural substrate FPP (1) and can be efficiently transferred to proteins and peptides ending in CAAX motifs. Site-specific modification with a trans-cyclooctene handle allows these proteins and peptides to undergo subsequent bioconjugation with tetrazines. Enzymatic transfer and subsequent tetrazine ligation were confirmed by mass spectrometry, HPLC analysis, and spectrofluorometric assays. Since tetrazine ligation is bioorthogonal, has a high second order rate constant, and does not require the use of metal or copper catalysts, this strategy has the potential to be useful for site-specific labeling of proteins inside living cells. Future work will probe the utility of this bioconjugation strategy for applications in cell culture as well as whole organisms.
Supplementary Material
Figure 1.

Farnesyl diphosphate (1), trans-cyclooctene geranyl diphosphate (2), dipyridyl-tetrazine (3), and benzylamino-tetrazine (4).
Scheme 1.
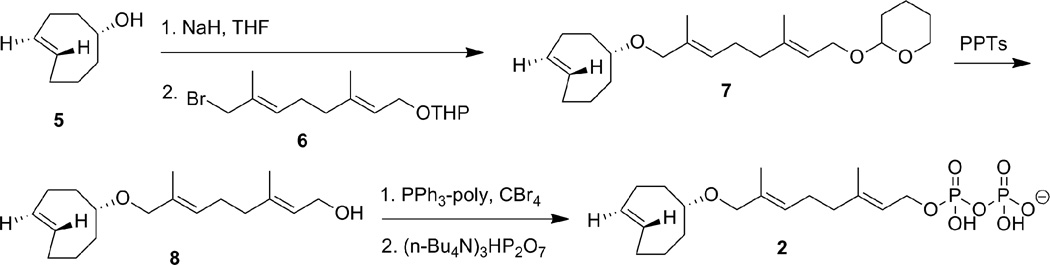
Synthesis of trans-cyclooctene geranyl diphosphate (2).
Scheme 2.

Enzymatic modification of a CAAX-box containing peptide with trans-cyclooctene geranyl diphosphate (2) and subsequent tetrazine ligation with dipyridyl-tetrazine (3).
Scheme 3.

Enzymatic modification of eGFP-CVIA with trans-cyclooctene geranyl diphosphate (2) and subsequent tetrazine ligation with benzylamino-tetrazine (4).
Acknowledgements
The authors thank Dr. Daniel Mullen for guidance on peptide synthesis, Brock Matter, Rebecca Guza, Sean Murray, and Dr. Peter Villalta for assistance in obtaining ESI-MS data, and Letitia Yao for assistance in NMR. Portions of the mass spectrometry done in this work was completed at the University of Minnesota Masonic Cancer Center, a comprehensive cancer center designated by the National Cancer Institute supported in part by P30A77598. This work was supported by the National Institutes of Health Grant Nos. GM058842, GM084152, CA104609, T32M008347 and by an award from Research Corporation for Science Advancement. This research project was funded by a 3M Faculty/Student Collaborative Grant through the Center of Excellence for Women, Science, and Technology of the University of St. Catherine, St. Paul, Minnesota.
Abbreviations
- CuACC
copper catalyzed azide alkyne cycloaddition
- DTT
dithiothreitol; EtOH, ethanol
- HR-ESI-MS
high resolution electrospray ionization mass spectrometry
- LC-MS
liquid chromatography mass spectrometry
- PFTase
protein farnesyltranferase
- PPTs
pyridinium p-toluenesulfonate
- SPAAC
strain promoted azide-alkyne cycloaddition
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
Footnotes
Supplementary Materials: 1H NMR spectra and HPLC traces available in supplementary materials.
References
- 1.Sletten EM, Bertozzi CR. Angew. Chem. Int. Ed. 2009;48:6974. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casey PJ, Seabra MC. J. Biol. Chem. 1996;271:5289. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 3.Kale Tamara A, Hsieh Shih-an J, Rose Matt W, Distefano Mark D. Curr Top Med Chem. 2003;3:1043. doi: 10.2174/1568026033452087. [DOI] [PubMed] [Google Scholar]
- 4.Dolence JM, Poulter CD. Proc Natl Acad Sci U S A. 1995;92:5008. doi: 10.1073/pnas.92.11.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosokawa A, Wollack JW, Zhang Z, Chen L, Barany G, Distefano MD. Int. J. Pept. Res. Ther. 2007;13:345. [Google Scholar]
- 6.Rose MW, Xu J, Kale TA, O'Doherty G, Barany G, Distefano MD. Biopolymers . 2005;80:164. doi: 10.1002/bip.20239. [DOI] [PubMed] [Google Scholar]
- 7.Rashidian M, Song JM, Pricer RE, Distefano MD. J. Am. Chem. Soc. 2012;134:8455. doi: 10.1021/ja211308s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaon I, Turek TC, Weller VA, Edelstein RL, Singh SK, Distefano MD. J. Org. Chem. 1996;61:7738. doi: 10.1021/jo9602736. [DOI] [PubMed] [Google Scholar]
- 9.Wollack JW, Silverman JM, Petzold CJ, Mougous JD, Distefano MD. ChemBioChem. 2010;10:2934. doi: 10.1002/cbic.200900566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duckworth BP, Chen Y, Wollack JW, Sham Y, Mueller JD, Taton TA, Distefano MD. Angew. Chem. Int. Ed. 2007;46:8819. doi: 10.1002/anie.200701942. [DOI] [PubMed] [Google Scholar]
- 11.Grammel M, Hang HC. Nat. Chem. Biol. 2013;9:475. doi: 10.1038/nchembio.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rashidian M, Dozier JK, Distefano MD. Bioconjugate Chem. 2013;24:1277. doi: 10.1021/bc400102w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 14.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc. Natl. Acad. Sci. 2007;104:16793. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Presolski SI, Hong V, Cho S-H, Finn MG. J. Am. Chem. Soc. 2010;132:14570. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dommerholt J, Schmidt S, Temming R, Hendriks LJA, Rutjes FPJT, van HJCM, Lefeber DJ, Friedl P, van DFL. Angew. Chem. Int. Ed. 2010;49:9422. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Versteegen RM, Rossin R, Ten HW, Janssen HM, Robillard MS. Angew Chem nt Ed Engl. 2013 doi: 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- 18.Blackman ML, Royzen M, Fox JM. J. Am. Chem. Soc. 2008;130:13518. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devaraj Neal K, Hilderbrand Scott A, Upadhyay R, Mazitschek R, Weissleder R. Angew Chem. 2010;2010:2869. doi: 10.1002/anie.200906120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karver MR, Weissleder R, Hilderbrand SA. Bioconjugate Chem. 2011;22:2263. doi: 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devaraj NK, Upadhyay R, Haun JB, Hilderbrand SA, Weissleder R. Angew. Chem. Int. Ed. 2009;48:7013. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu DS, Tangpeerachaikul A, Selvaraj R, Taylor MT, Fox JM, Ting AY. J. Am. Chem. Soc. 2012;134:792. doi: 10.1021/ja209325n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeGraw AJ, Hast MA, Xu J, Mullen D, Beese LS, Barany G, Distefano MD. Chem. Biol. Drug Des. 2008;72:171. doi: 10.1111/j.1747-0285.2008.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dozier JK, Distefano MD. Anal. Biochem. 2012;421:158. doi: 10.1016/j.ab.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wollack JW. 2009 [Google Scholar]
- 26.Duckworth BP, Xu J, Taton TA, Guo A, Distefano MD. Bioconj. Chem. 2006;17:967. doi: 10.1021/bc060125e. [DOI] [PubMed] [Google Scholar]
- 27.Bradford MM. Anal Biochem. 1976;72:248. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 28.Royzen M, Yap GPA, Fox JM. J. Am. Chem. Soc. 2008;130:3760. doi: 10.1021/ja8001919. [DOI] [PubMed] [Google Scholar]
- 29.Chen APC, Chen Y-H, Liu H-P, Li Y-C, Chen C-T, Liang P-H. J. Am. Chem. Soc. 2002;124:15217. doi: 10.1021/ja020937v. [DOI] [PubMed] [Google Scholar]
- 30.Lenevich S, Distefano MD. Anal. Biochem. 2011;408:316. doi: 10.1016/j.ab.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, Poulter CD. J. Org. Chem. 1986;51:4768. [Google Scholar]
- 32.Cassidy PB, Dolence JM, Poulter CD. Methods Enzymol. 1995;250:30. doi: 10.1016/0076-6879(95)50060-x. [DOI] [PubMed] [Google Scholar]
- 33.Mathis JR, Poulter CD. Biochemistry. 1997;36:6367. doi: 10.1021/bi9629182. [DOI] [PubMed] [Google Scholar]
- 34.Dolence JM, Cassidy PB, Mathis JR, Poulter CD. Biochemistry. 1995;34:16687. doi: 10.1021/bi00051a017. [DOI] [PubMed] [Google Scholar]
- 35.Subramanian T, Liu S, Troutman JM, Andres DA, Spielmann HP. ChemBioChem. 2008;9:2872. doi: 10.1002/cbic.200800248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeGraw AJ, Palsuledesai C, Ochocki JD, Dozier JK, Lenevich S, Rashidian M, Distefano MD. Chem. Biol. Drug Des. 2010;76:460. doi: 10.1111/j.1747-0285.2010.01037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.