

Abstract
OBJECTIVE
Angiotensin (Ang) II-induced hypertension is associated with accelerated thrombus development in arterioles. This study assessed the contributions of different components of the coagulation cascade and fibrinolysis to AngII mediated microvascular thrombosis.
METHODS
Light/dye induced thrombus formation (the time of onset and flow cessation) was quantified in cremaster muscle arterioles of AngII infused (2 wk) wild type (WT/AngII) mice, endothelial protein C receptor transgenic mice (EPCR), and mice deficient in plasminogen activator inhibitor-1 (PAI-1). WT/AngII mice were also treated with either tissue factor antibody, antithrombin III, heparin, hirudin, or murine activated protein C (APC).
RESULTS
TF immunoblockade or hirudin treatment did not prevent the AngII-induced acceleration of thrombosis. While antithrombin III treatment prevented the acceleration in both thrombus onset and flow cessation, heparin only improved the time for blood flow cessation. Neither WT mice treated with murine APC nor EPCR transgenic mice were protected against AngII-induced thrombus development. A similar lack of protection was noted in PAI-1deficient mice.
CONCLUSION
These findings implicate a role for thrombin generation pathway in the accelerated thrombosis induced by AngII and suggest that an impaired protein C pathway and increased PAI-1 do not make a significant contribution to this model of microvascular thrombosis.
Keywords: Angiotensin II, coagulation, fibrinolysis, thrombosis, anticoagulants
Introduction
Thrombosis is a major complication of hypertension (HTN) and other risk factors for cardiovascular disease [1,2]. Angiotensin II has been implicated as a mediator of the thrombosis that accompanies HTN based on clinical studies that demonstrate a lower incidence of thrombotic events in patients treated with either angiotensin converting enzyme (ACE) inhibitors or angiotensin II receptor blockers (ARBs) [3]. The increased risk for thrombus development in HTN patients is also associated with abnormalities in platelet function (e.g., increased platelet reactivity to agonist stimulation) [4], coagulation (increased TF expression), and fibrinolysis (increased PAI-1 level) [2,5]. These abnormalities have also been linked to AngII, with ACE inhibitors and/or ARBs showing efficacy in reducing TF activity and PAI-1 levels [6,7] and in inhibiting platelet activation and aggregation in HTN patients [8].
A role for AngII as a mediator of thrombogenesis is also supported by animal studies [9,10]. Models of elevated AngII levels, elicited either genetically or via chronic AngII infusion, have demonstrated increased TF expression and increased plasma PAI-1 levels [11,12]. Furthermore, chronic AngII infusion has been shown to induce platelet-endothelial cell adhesion [13] and to accelerate thrombus formation in both large arteries [9,14] and in arterioles [12]. A variety of cell receptors, including angiotensin II type-1, -2 and -4 receptors, bradykinin-1 and -2 receptors, and the endothelin-1A receptor, have been implicated in the accelerated thrombosis and coagulation abnormalities elicited by AngII [12, 15]. For example, activation of angiotensin-1 type-4 receptors, bradykinin type-2 and endothelin-1 A receptors have all been linked to the increased expression and elevated plasma levels of PAI-1 elicited by AngII, and the same receptors have been implicated in AngII-mediated arteriolar thrombosis [12, 16], suggesting a cause-effect relationship between the elevated PAI-1 level and accelerated arteriolar thrombosis. While PAI-1 and other components of the coagulation and fibrinolytic pathways have also been implicated in AngII-mediated thrombus formation, there is relatively little direct evidence to support their involvement. Hence, the overall objective of this study was to directly assess the contributions of tissue factor, thrombin, the protein C pathway, and PAI-1 to the accelerated thrombus formation observed in arterioles of mice with chronically elevated AngII levels. Both pharmacologic and genetic approaches were used to dissect the roles of the coagulation and fibrinolytic pathways in AngII mediated thrombosis. Our findings support a role for thrombin in the accelerated thrombus development induced by AngII and suggest that neither tissue factor, an impaired protein C pathway or increased PAI-1 do not make a significant contribution to this model of microvascular thrombosis.
Materials and methods
Mice
Male C57BL/6 (wild-type [WT]), PAI-1−/− (B6.129S2-Serpine1tm1Mlg) mice, obtained from Jackson Laboratories (Bar Harbor, ME) and transgenic mice overexpressing the endothelial protein C receptor (EPCR-TgN) (Oklahoma Medical Research Foundation, OK) were used. The EPCR-TgN transgenic mice, backcrossed into C57BL/6 mice, exhibit normal size, weight, viability, fertility, blood cell count and chemistries [17]. EPCR protein levels in all organs of the transgenic mice are at least eight-fold higher than in their WT counterparts. All experimental procedures involving animals were performed according to the criteria outlined by the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of LSU Health Sciences Center-Shreveport.
Angiotensin II infusion
AngII (1 μg/kg/min)-loaded micro-osmotic pumps (Alzet, Cupertino, CA, model 1002) were implanted up to 14 days subcutaneously (intrascapular region) in isofluorane anesthetized mice using sterile procedures. The topical antibiotic (Neosporin) was applied to prevent post-operative infection at the site of implantation. Blood pressure determinations were obtained on day 15 with a computerized tail cuff system (Hatteras Inst, Inc.). We previously demonstrated [12] that WT mice implanted with a saline-loaded pump yield a thrombosis response similar to light/dye injury in the cremaster muscle microvasculature that does not differ from the responses noted in WT mice without an implanted pump. Therefore, WT mice without implanted pumps were used as controls in this study.
Cremaster muscle preparation
The cremaster muscle was exposed for intravital microscopic observation as previously described [12, 18]. Briefly, AngII- or control mice were anesthetized with pentobarbital, and the right carotid artery and jugular vein were cannulated for measurement of arterial pressure and for drug administration (eg, FITC-dextran, supplemental doses of anesthetic), respectively. Arterioles with diameters between 30–40 μm and wall shear rates (WSR) >500 sec−1 were selected for study. An optical Doppler velocimeter (Microcirculation Research Institute, Texas A & M University, College Station, TX) was used to measure red blood cell velocity (VRBC) in the microvessels and WSR was calculated using WSR = 8 (Vmean/DV), where Vmean is mean red blood cell velocity and Dv is vessel diameter.
Light/dye-induced thrombosis was induced as previously described (Senchenkova, 2010, 2011). Briefly, 20 min following cremaster muscle isolation (stabilization period), 10 ml/kg of 5 % FITC-dextran (150,000 MW, Sigma, MO) was administrated i.v. and allowed to circulate for 10 min. Epi-illumination using a 175-W xenon lamp (Lambda LS, Sutter, CA) and a fluorescein filter cube (HQ-FITC, Chroma, VT) was continuously applied to the selected arteriole, and thrombus formation was quantified by determining: 1) the time of onset of platelet deposition/aggregation within the microvessel (onset time), and 2) the time required for complete flow cessation for ≥ 30 sec (cessation time) within the microvessels. The excitation power density was measured daily (ILT 1700 Radiometer, SED033 detector, International Light, MA) and maintained within 1 % of a standardized W/cm2. Epi-illumination was discontinued once blood flow ceased in the vessel under study. Typically, 1 – 3 thrombi were induced in each mouse and the results of the vessels were averaged.
Experimental protocol
To determine whether tissue factor, thrombin, activated protein C (APC) and/or plasminogen activator inhibitor-1 (PAI-1) contribute to the accelerated arteriolar thrombosis induced by chronic AngII infusion, we evaluated light/dye-induced thrombus development in the following experimental groups: 1) wild type (WT) - control mice; 2) WT-AngII-infused mice; 3) WT-AngII-mice receiving 20 mg/kg of rat antimouse tissue factor monoclonal antibody Ab 1H1 (TF Ab) [19] 20 min before photo-exposure (WT-AngII-TF Ab); 4) WT-AngII mice injected with heparin 5 U/mouse (heparin sodium; Sagent Pharmaceuticals, Inc) 10 min before thrombus induction (WT-AngII-heparin) [20]; 5) WT-AngII mice receiving 1 mg/kg of hirudin (Calbiochem, Darmstadt, Germany) [20] 5 min before of epi-illumination of a selected vessel; 6) WT-AngII mice receiving antithrombin III (50 U/kg; Calbiochem) [20] 5 min before photoactivation (WT-AngII-AT III); 7) WT-AngII mice receiving 10 μg/mouse of murine activated protein C (Oklahoma Medical Research Foundation) 10 minutes before photoactivation (WT-AngII-APC) [12, 21]; 8) EPCR-TgN mice implanted with AngII-loaded pump (EPCR-TgN-AngII); and 9) plasminogen activator inhibitor-1 (PAI-1) deficient mice implanted with AngII-loaded pump for 2 weeks (PAI-1−/− -AngII).
Statistical analysis
Statistically significant differences p<0.05 were determined using a one way ANOVA with Newman-Keuls multiple comparison test. The WT-control (n = 12) and WT-AngII (n = 12) data shown in Figures 1–4 represent the same groups. However, the statistical outcomes of the specific interventions described in the Results section were based on a simultaneous comparison of all control and experimental groups. All values are expressed as means ± standard error.
Figure 1.

Effects of tissue factor (TF) immunoblockade on light-dye-induced thrombus formation in cremaster muscle arterioles of wild type mice infused with angiotensin II (AngII) for 2 wks. WT = control wild type (WT) mice (n=12); WT-AngII = WT mice implanted with AngII-loaded pump for 2 weeks (n=12); WT-AngII-TF Ab = WT mice with AngII-loaded pump treated with a TF neutralizing antibody 20 min before photoactivation (n=7). ** Indicates p<0.01 vs WT; *** indicates p<0.001 vs WT.
Figure 4.
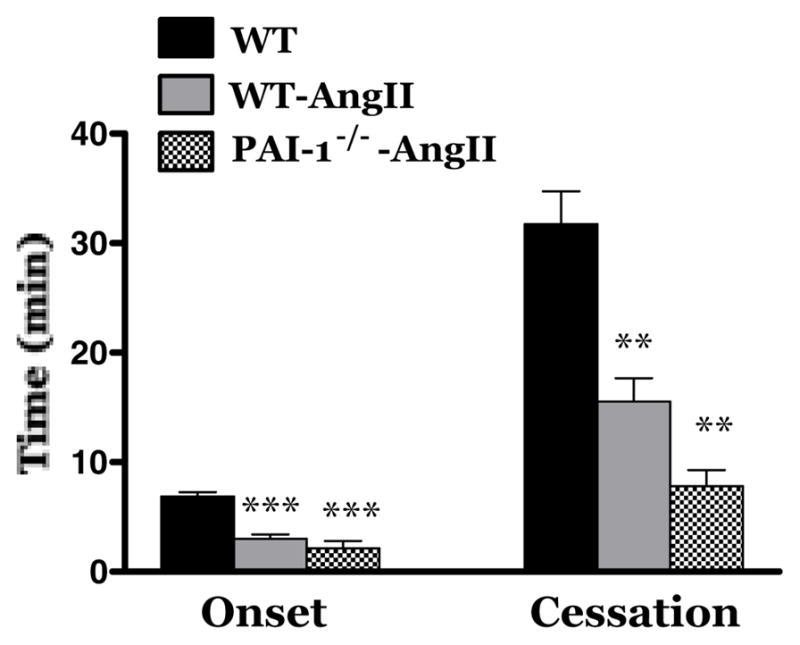
Light/dye-induced thrombosis in plasminogen activator inhibitor-1 (PAI-1) deficient mice implanted with AngII-loaded pump for 2 weeks. WT mice (n=12); WT-AngII mice (n=12); PAI-1−/−-AngII (n=4). ** Indicates p<0.01 vs WT; *** indicates p<0.001 vs WT.
Results
Implantation of an AngII loaded pump elicited a significant increase in blood pressure (162.4 ± 6 mmHg), compared to control (104.5 ± 3.3 mmHg) mice. As shown in Figure 1, and previously reported by this laboratory [12, 22], 2 wk infusion of AngII is associated with an acceleration of light/dye-induced thrombus formation in cremaster muscle arterioles. The accelerated thrombosis is manifested as significant reductions in both the time of onset of the thrombus and time to flow cessation within the injured arterioles. Tissue factor is known to contribute to the initiation of thrombus development [23]. Muller et al have reported [24] that Ang II type 1 receptor (AT1r) antagonism prevents tissue factor expression. We have previously shown that neither AT1r immunoblockade nor genetic deficiency of AT1r protects arterioles against enhanced microvascular thrombosis elicited by AngII infusion [12] Consistent with these observations in our finding (Figure 1) that immunoblockade of tissue factor has no significant effect on the accelerated thrombus development elicited by chronic AngII infusion.
The contribution of thrombin to AngII enhanced, light/dye-induced thrombus formation was evaluated using distinct anti-thrombin agents, i.e., heparin, hirudin and antithrombin III (Figure 2). As reported previously in our laboratory neither heparin, hirudin nor ATIII alters the onset and/or flow cessation time of thrombus formation following light/dye-induced injury in cremaster muscle arterioles of control mice [20]. In AngII-infused hypertensive mice, heparin pretreatment (panel A) protected against the initiation of thrombus formation (onset time) but did not alter the time to flow cessation. Hirudin pretreatment had no effect on either parameter (panel B). Antithrombin III pretreatment (Panel C) was highly effective in preventing the acceleration of both onset time and time to flow cessation normally elicited by AngII. The role of the protein C pathway in AngII dependent thrombus formation was evaluated using AngII-infused WT mice treated with murine APC and using AngII-infused EPCR-TgN mice (Figure 3). We have previously reported that both EPCR-TgN mice and WT mice receiving murine APC exhibit a normal thrombogenic response in cremaster arterioles after light/dye injury [43]. In this study, we observed that treatment with murine APC had no effect on the time of onset of the thrombus and the time of blood flow cessation (Panel A). A comparison of the AngII mediated responses between WT and EPCR-TgN mice also revealed no differences in either onset time or time to flow cessation.
Figure 2.

Effects of thrombin inhibition on AngII-enhanced thrombosis. Panel A: A comparison of untreated and heparin treated (WT-AngII-heparin) WT mice implanted with AngII loaded pumps. WT mice (n=12); WT-AngII mice (n=12); WT-AngII-heparin (n =5) receiving heparin 10 min prior to photoexposure. Panel B: A comparison of untreated WT mice (n=12) and WT-AngII (n=5) receiving hirudin 5 min prior epi-illumination hypertensive mice. Panel C: A comparison of untreated and antithrombin III (ATIII, administered 5 min prior to photoactivation) treated WT mice. WT controls (n=12); WT-AngII (n=12); WT-AngII-ATIII (n = 5). ** Indicates p<0.01 vs WT; *** indicates p<0.001 vs WT; # indicates p<0.05 vs WT-AngII; ## indicates p<0.01 vs WT-AngII.
Figure 3.
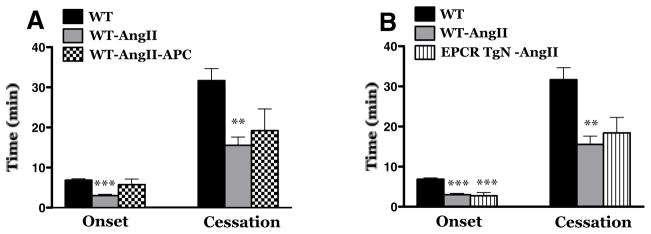
Effects of treatment with murine activated protein C (APC) (Panel A) or genetic overexpression of the endothelial protein C receptor (EPCR-TgN) (Panel B) on AngII-accelerated light/dye-induced thrombus formation in cremaster muscle arterioles. WT mice (n=12); WT-AngII mice (n=12); WT-AngII-APC (n=6); EPCR-TgN-AngII (n=6). ** Indicates p<0.01 vs WT; *** indicates p<0.001 vs WT.
We and others have previously reported that AngII elevation is associated with an increased plasma PAI-1 concentration and an enhanced expression in endothelial cells [12, 25–27]. To directly assess the contribution of PAI-1 to AngII dependent thrombus development in arterioles, we compared thrombus formation between AngII-infused WT and PAI-1−/− mice (Figure 4). This comparison revealed no differences in thrombus formation between arterioles of WT and PAI-1−/− mice exposed to chronically elevated AngII levels. These findings suggest that the increased PAI-1 detected in this AngII-dependent model of experimental hypertension does not make a significant contribution to the corresponding enhancement of thrombus development in the microvasculature.
Discussion
Hypertension is associated with a procoagulant, prothrombogenic state [8, 28] that is ameliorated by drugs that target the renin-angiotensin system [2]. The changes in hemostatic biomarkers in hypertensive patients are generally consistent with subclinical activation of the coagulation system and impaired fibrinolysis [6]. The enhanced thrombus development that accompanies HTN is evidenced in both large and microscopic arteries [9, 12]. While it generally assumed that the hemostatic abnormalities that are associated with HTN underlie the accelerated thrombus formation in this condition, there are relatively few studies that directly address the causal relationship between the coagulation/fibrinolytic systems and HTN-enhanced thrombogenesis, particularly in the microcirculation. In this study, we examined the contributions of tissue factor, thrombin, protein C and PAI-1 to AngII-enhanced thrombosis in arterioles. Overall, our findings implicate thrombin in AngII-enhanced microvascular thrombosis and suggest that impairment of the protein C pathway and fibrinolysis are unlikely contributors to this response.
The initiation of coagulation begins with exposure of active tissue factor to plasma, formation of the TF/VIIa factor complex, and the activation of factors IX and X [29]. There are several lines of circumstantial evidence that links TF to AngII dependent coagulation/thrombosis: 1) exposure of endothelial cells, monocytes and other cell types to AngII results in nuclear factor kappa-B (NFkB) dependent TF synthesis [7, 30], 2) circulating TF levels (both soluble and cell/microparticle-associated) are elevated in hypertensive patients [31], and 3) drugs that target the renin-angiotensin system reduce TF levels in animal models of, and patients with, HTN [7, 24]. The results of the present study do not provide evidence for implicating TF in the accelerated onset time, which reflects the initiation phases of thrombogenesis, elicited by AngII infusion. The identity of the factor that initiates AngII accelerated microvascular thrombosis remains unclear.
Both HTN patients and animal models of HTN are associated with enhanced thrombin generation, as evidenced by reported increases plasma thrombin-antithrombin (TaT) complexes [32, 33]. Furthermore, the elevated TaT complexes in HTN patients are reduced by treatment with an ACE inhibitor [32], suggesting that AngII mediates the enhanced thrombin production in HTN. This possibility is supported by a report describing elevated plasma TaT levels in human subjects subjected to an intravenous infusion (sufficient to increase mean arterial pressure by 23 ± 2 mmHg) of AngII [34]. While it remains unclear how AngII promotes thrombin generation, this response may simply arise from the known actions of the peptide on TF synthesis. In the present study, we provide evidence, using three mechanistically distinct anti-thrombin agents, that thrombin contributes to AngII-enhanced thrombosis in arterioles. We noted that treatment with heparin protects against accelerated onset of the thrombus, but not for blood flow cessation. While hirudin administration had no effect on the AngII accelerated thrombosis response, antithrombin III treatment effectively prevented the AngII-mediated acceleration of both onset time and time to flow cessation. The much greater effectiveness of ATIII compared to heparin and hirudin in our study is consistent with a previous report that described a significantly greater antithrombotic efficacy of ATIII than heparin and hirudin in arterioles of the ear in hairless mice after light/dye injury [35]. The greater efficacy of ATIII may relate to the ability of this anticoagulant to also inhibit other factors (e.g., factors IXa, Xa, XII) in the coagulation cascade [36, 37] or its ability to promote prostaglandin release [38] and inhibit NFkB activation in endothelial cells [39].
The protein C pathway is an important endogenous anticoagulant system that serves to limit thrombin production through activated protein C-mediated inhibition of coagulation factors (Va, VIIIa). The binding of thrombin to thrombomodulin (TM) on endothelial cells initiates the protein C pathway, with thrombin-TM complexes activating protein C via a proteolytic process that is accelerated over 10-fold by engagement of the endothelial protein C receptor [40]. When APC dissociates from EPCR it can interact with protein S to inactivate factors Va and VIIIa. Since the expression of EPCR and TM are significantly down-regulated by cytokines (e.g., TNF α, IL-1 β) that exhibit increased expression and plasma levels during AngII infusion [41, 42], it is possible that inactivation of the protein C pathway could account for the accelerated thrombogenesis that is associated with chronically elevated AngII levels. In our experiments involving administration of exogenous activated protein C to AngII infused mice, we did not note any protection against the AngII- accelerated thrombosis response. Similarly, AngII-infused EPCR-TgN mice did not exhibit protective phenotype for AngII accelerated thrombus formation. Collectively, these findings suggest that down-regulation of the protein C pathway is not a major cause of the accelerated thrombosis elicited by AngII. This finding contrasts with a previously published report from our laboratory that describes significant protection by APC and EPCR-TgN against the accelerated arteriolar thrombosis elicited in a murine model of experimental colitis [43]. The lesser role of the protein C pathway in the AngII model may reflect a less intense systemic inflammatory response (and less cytokine release) compared to colonic inflammation.
Impaired fibrinolysis has been implicated in the thrombosis associated with HTN [2, 27]. PAI-1 levels are elevated in hypertensive subjects [27] and in AngII-dependent animal models of HTN [12]. AngII has been shown to induce a dose-dependent increase in PAI-1 secretion by endothelial and vascular smooth muscle cells, a response that has been linked to the angiotensin II type-4 receptor [16]. Since we have recently reported that PAI-1 levels are significantly elevated in our AngII model and that AT4 receptor (AT4r) blockade effectively inhibits the flow cessation component of AngII accelerated arteriolar thrombosis [12], we hypothesized that PAI-1 (resulting from AT4r activation) contributes to the AngII dependent arteriolar thrombogenesis. However, our findings in PAI-1−/− mice do not support this hypothesis and suggest that the elevated plasma PAI-1 does not make a significant contribution to AngII dependent arteriolar thrombosis. Our findings contrast the results obtained in models of atherothrombosis wherein large artery thrombosis is prolonged in both AT4r deficient mice and in mice treated with a PAI-1 antagonist [16]. The absence of protection by PAI-1 deficiency in arterioles of AngII infused mice is not likely related to the lack of expression of this fibrinolytic factor in microvessels since PAI-1 is known to be expressed in arterioles and it has been shown to make a significant contribution to thrombolysis in rat mesenteric arterioles [44]. The absence of change in the AngII mediated response in PAI-1−/− mice may reflect the rapid nature of the light/dye model for studying thrombus development. An acute thrombosis model such as the light/dye method may generate clots within a time-frame that does not allow for the involvement of PAI-1 in thrombus regression. However, others have reported a prolongation of the time to complete occlusion induced by ferric chloride injury in 100 um testicular arterioles treated with the PAI-1 inhibitor TM5001 [45].
In conclusion, the findings of this study indicate that the accelerated microvascular thrombosis mediated by chronic AngII infusion involves components of the common (thrombin) coagulation pathway, with tissue factor, the protein C anticoagulation pathway and fibrinolysis playing no role in this response. While the molecular mechanisms that link AngII to activation of the coagulation cascade remain undefined, our observation that ATIII effectively prevents the accelerated thrombosis mediated by AngII suggest that this anti-thrombin agent may prove effective for the treatment HTN patients who are at high risk for thrombus development.
Perspectives.
The accelerated thrombus development that accompanies AngII-induced hypertension involves activation of different components of the coagulation cascade. Our findings suggest that ATIII has greater potential as a therapeutic strategy than other anticoagulant agents for prevention of enhanced thrombus development associated with AngII-dependent hypertension.
Acknowledgments
Supported by a grant (DNG) from the National Heart Lung & Blood Institute (HL26441-32), CTE is an Investigator of the Howard Hughes Medical Institute
The present study is supported by a grant (DNG) from the National Heart Lung & Blood Institute (HL26441-32). EYS is supported by a Malcolm Feist Cardiovascular Research Postdoctoral Fellowship. CTE is an Investigator of the Howard Hughes Medical Institute.
References
- 1.Brown NJ, Vaughan DE. Prothrombotic effects of angiotensin. Adv Intern Med. 2000;45:419–29. [PubMed] [Google Scholar]
- 2.Chan MY, Andreotti F, Becker RC. Hypercoagulable states in cardiovascular disease. Circulation. 2008;118(22):2286–97. doi: 10.1161/CIRCULATIONAHA.108.778837. [DOI] [PubMed] [Google Scholar]
- 3.Nadar S, Lip GY. The prothrombotic state in hypertension and the effects of antihypertensive treatment. Curr Pharm. 2003;9(21):1715–32. doi: 10.2174/1381612033454559. [DOI] [PubMed] [Google Scholar]
- 4.Monton M, Jimenez A, Nunez A, Lopez-Blaya A, Farre J, Gomez J, et al. Comparative effects of angiotensin II AT-1-type receptor antagonists in vitro on human platelet activation. J Cardiovasc Pharmacol. 2000;35(6):906–13. doi: 10.1097/00005344-200006000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Nishimura H, Tsuji H, Masuda H, Nakagawa K, Nakahara Y, Kitamura H, et al. Angiotensin II increases plasminogen activator inhibitor-1 and tissue factor mRNA expression without changing that of tissue type plasminogen activator or tissue factor pathway inhibitor in cultured rat aortic endothelial cells. Thromb Haemost. 1997;77(6):1189–95. [PubMed] [Google Scholar]
- 6.Matsumoto T, Horie M. Angiotensin-converting enzyme inhibition and fibrinolytic balance. Hypertens Res. 2011;34(4):448–9. doi: 10.1038/hr.2011.3. [DOI] [PubMed] [Google Scholar]
- 7.Celi A, Cianchetti S, Dell’Omo G, Pedrinelli R. Angiotensin II, tissue factor and the thrombotic paradox of hypertension. Expert Rev Cardiovasc Ther. 2010;8(12):1723–9. doi: 10.1586/erc.10.161. [DOI] [PubMed] [Google Scholar]
- 8.Remkova A, Remko M. The role of renin-angiotensin system in prothrombotic state in essential hypertension. Physiol Res. 2010;59(1):13–23. doi: 10.33549/physiolres.931525. [DOI] [PubMed] [Google Scholar]
- 9.Kaminska M, Mogielnicki A, Stankiewicz A, Kramkowski K, Domaniewski T, Buczko W, et al. Angiotensin II via AT1 receptor accelerates arterial thrombosis in renovascular hypertensive rats. J Physiol Pharmacol. 2005;56(4):571–85. [PubMed] [Google Scholar]
- 10.Ishikawa M, Sekizuka E, Yamaguchi N, Nakadate H, Terao S, Granger DN, et al. Angiotensin II type 1 receptor signaling contributes to platelet-leukocyte-endothelial cell interactions in the cerebral microvasculature. Am J Physiol Heart Circ Physiol. 2007;292(5):H2306–15. doi: 10.1152/ajpheart.00601.2006. [DOI] [PubMed] [Google Scholar]
- 11.Doller A, Gauer S, Sobkowiak E, Geiger H, Pfeilschifter J, Eberhardt W. Angiotensin II induces renal plasminogen activator inhibitor-1 and cyclooxygenase-2 expression post-transcriptionally via activation of the mRNA-stabilizing factor human-antigen R. Am J Pathol. 2009;174(4):1252–63. doi: 10.2353/ajpath.2009.080652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senchenkova EY, Russell J, Almeida-Paula LD, Harding JW, Granger DN. Angiotensin II-mediated microvascular thrombosis. Hypertension. 2010;56(6):1089–95. doi: 10.1161/HYPERTENSIONAHA.110.158220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vital SA, Terao S, Nagai M, Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation. 2010;17(8):641–9. doi: 10.1111/j.1549-8719.2010.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mogielnicki A, Chabielska E, Pawlak R, Szemraj J, Buczko W. Angiotensin II enhances thrombosis development in renovascular hypertensive rats. Thromb Haemost. 2005;93(6):1069–76. doi: 10.1160/TH04-10-0701. [DOI] [PubMed] [Google Scholar]
- 15.Li JS, Touyz RM, Schiffrin EL. Effects of AT1 and AT2 angiotensin receptor antagonists in angiotensin II-infused rats. Hypertension. 1998;31(1 Pt 2):487–92. doi: 10.1161/01.hyp.31.1.487. [DOI] [PubMed] [Google Scholar]
- 16.Numaguchi Y, Ishii M, Kubota R, Morita Y, Yamamoto K, Matsushita T, et al. Ablation of angiotensin IV receptor attenuates hypofibrinolysis via PAI-1 downregulation and reduces occlusive arterial thrombosis. Arterioscler Thromb Vasc Biol. 2009;29(12):2102–8. doi: 10.1161/ATVBAHA.109.195057. [DOI] [PubMed] [Google Scholar]
- 17.Li W, Zheng X, Gu J, Hunter J, Ferrell GL, Lupu F, et al. Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost. 2005;3(7):1351–9. doi: 10.1111/j.1538-7836.2005.01385.x. [DOI] [PubMed] [Google Scholar]
- 18.Stokes KY, Clanton EC, Russell JM, Ross CR, Granger DN. NAD(P)H oxidase-derived superoxide mediates hypercholesterolemia-induced leukocyte-endothelial cell adhesion. Circ Res. 2001;16;88(5):499–505. doi: 10.1161/01.res.88.5.499. [DOI] [PubMed] [Google Scholar]
- 19.Kirchhofer D, Moran P, Bullens S, Peale F, Bunting S. A monoclonal antibody that inhibits mouse tissue factor function. J Thromb Haemost. 2005;3(5):1098–9. doi: 10.1111/j.1538-7836.2005.01253.x. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida H, Russell J, Granger DN. Thrombin mediates the extraintestinal thrombosis associated with experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2008;295(5):G904–8. doi: 10.1152/ajpgi.90400.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagai M, Yilmaz CE, Kirchhofer D, Esmon CT, Mackman N, Granger DN. Role of coagulation factors in cerebral venous sinus and cerebral microvascular thrombosis. Neurosurgery. 2010;66(3):560–5. doi: 10.1227/01.NEU.0000365745.49583.FD. discussion 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Senchenkova EY, Russell J, Kurmaeva E, Ostanin D, Granger DN. Role of T lymphocytes in angiotensin II-mediated microvascular thrombosis. Hypertension. 2011;58(5):959–65. doi: 10.1161/HYPERTENSIONAHA.111.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owens AP, 3rd, Mackman N. Tissue factor and thrombosis: The clot starts here. Thromb Haemost. 2010;104(3):432–9. doi: 10.1160/TH09-11-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Müller DN, Mervaala EM, Dechend R, Fiebeler A, Park JK, Schmidt F, Theuer J, Breu V, Mackman N, Luther T, Schneider W, Gulba D, Ganten D, Haller H, Luft FC. Angiotensin II (AT(1)) receptor blockade reduces vascular tissue factor in angiotensin II-induced cardiac vasculopathy. Am J Pathol. 2000;157(1):111–2225. doi: 10.1016/S0002-9440(10)64523-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaughan DE, Lazos SA, Tong K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest. 1995;95(3):995–1001. doi: 10.1172/JCI117809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerins DM, Hao Q, Vaughan DE. Angiotensin induction of PAI-1 expression in endothelial cells is mediated by the hexapeptide angiotensin IV. J Clin Invest. 1995;96(5):2515–20. doi: 10.1172/JCI118312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ridker PM, Gaboury CL, Conlin PR, Seely EW, Williams GH, Vaughan DE. Stimulation of plasminogen activator inhibitor in vivo by infusion of angiotensin II. Evidence of a potential interaction between the renin-angiotensin system and fibrinolytic function. Circulation. 1993;87(6):1969–73. doi: 10.1161/01.cir.87.6.1969. [DOI] [PubMed] [Google Scholar]
- 28.Lip GY. Hypertension and the prothrombotic state. J Hum Hypertens. 2000;14(10–11):687–90. doi: 10.1038/sj.jhh.1001051. [DOI] [PubMed] [Google Scholar]
- 29.Wolberg AS. Thrombin generation assays: understanding how the method influences the results. Thromb Res. 2007;119(6):663–5. doi: 10.1016/j.thromres.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 30.He M, He X, Xie Q, Chen F, He S. Angiotensin II induces the expression of tissue factor and its mechanism in human monocytes. Thromb Res. 2006;117(5):579–90. doi: 10.1016/j.thromres.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 31.Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113(5):722–31. doi: 10.1161/CIRCULATIONAHA.105.567297. [DOI] [PubMed] [Google Scholar]
- 32.Ekholm M, Wallen NH, Johnsson H, Eliasson K, Kahan T. Long-term angiotensin-converting enzyme inhibition with ramipril reduces thrombin generation in human hypertension. Clin Sci (Lond) 2002;103(2):151–5. doi: 10.1042/cs1030151. [DOI] [PubMed] [Google Scholar]
- 33.Sawada K, Naiki M, Yago H, Matsushita K, Ohtsuki T, Kitagawa K, et al. Hypertension associated with reduced plasma thrombomodulin levels and a hypercoagulable state in rats. Clin Exp Hypertens. 2003;25(2):73–84. doi: 10.1081/ceh-120017928. [DOI] [PubMed] [Google Scholar]
- 34.Larsson PT, Schwieler JH, Wallen NH. Platelet activation during angiotensin II infusion in healthy volunteers. Blood Coagul Fibrinolysis. 2000;11(1):61–9. [PubMed] [Google Scholar]
- 35.Sorg H, Hoffmann JN, Menger MD, Lindenblatt N, Goehring P, Vollmar B. Antithrombin is as effective as heparin and hirudin to prevent formation of microvascular thrombosis in a murine model. Thromb Haemost. 2006;96(3):371–7. [PubMed] [Google Scholar]
- 36.High KA. Antithrombin III, protein C, and protein S. Naturally occurring anticoagulant proteins. Arch Pathol Lab Med. 1988;112(1):28–36. [PubMed] [Google Scholar]
- 37.Rosenberg RD. Actions and interactions of antithrombin and heparin. N Engl J Med. 1975;292(3):146–51. doi: 10.1056/NEJM197501162920307. [DOI] [PubMed] [Google Scholar]
- 38.Hoffmann JN, Vollmar B, Inthorn D, Schildberg FW, Menger MD. Antithrombin reduces leukocyte adhesion during chronic endotoxemia by modulation of the cyclooxygenase pathway. Am J Physiol Cell Physiol. 2000;279(1):C98–C107. doi: 10.1152/ajpcell.2000.279.1.C98. [DOI] [PubMed] [Google Scholar]
- 39.Oelschlager C, Romisch J, Staubitz A, Stauss H, Leithauser B, Tillmanns H, et al. Antithrombin III inhibits nuclear factor kappaB activation in human monocytes and vascular endothelial cells. Blood. 2002;99(11):4015–20. doi: 10.1182/blood.v99.11.4015. [DOI] [PubMed] [Google Scholar]
- 40.Esmon CT. Protein C anticoagulant pathway and its role in controlling microvascular thrombosis and inflammation. Crit Care Med. 2001;29(7 Suppl):S48–51. doi: 10.1097/00003246-200107001-00018. discussion -2. [DOI] [PubMed] [Google Scholar]
- 41.Nawroth PP, Stern DM. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. 1986;163(3):740–5. doi: 10.1084/jem.163.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faust SN, Levin M, Harrison OB, Goldin RD, Lockhart MS, Kondaveeti S, et al. Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N Engl J Med. 2001;345(6):408–16. doi: 10.1056/NEJM200108093450603. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida H, Russell J, Stokes KY, Yilmaz CE, Esmon CT, Granger DN. Role of the protein C pathway in the extraintestinal thrombosis associated with murine colitis. Gastroenterology. 2008;135(3):882–8. doi: 10.1053/j.gastro.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rupin A, Martin F, Vallez MO, Bonhomme E, Verbeuren TJ. Inactivation of plasminogen activator inhibitor-1 accelerates thrombolysis of a platelet-rich thrombus in rat mesenteric arterioles. Thromb Haemost. 2001;86(6):1528–31. [PubMed] [Google Scholar]
- 45.Izuhara Y, Takahashi S, Nangaku M, Takizawa S, Ishida H, Kurokawa K, van Ypersele de Strihou C, Hirayama N, Miyata T. Inhibition of plasminogen activator inhibitor-1: its mechanism and effectiveness on coagulation and fibrosis. Arterioscler Thromb Vasc Biol. 2008;28(4):672–7. doi: 10.1161/ATVBAHA.107.157479. [DOI] [PubMed] [Google Scholar]