

Abstract
The innate and adaptive immune systems in the intestine cooperate to maintain the integrity of the intestinal barrier and to regulate the composition of the resident microbiota. However, little is known about the crosstalk between the innate and adaptive immune systems that contribute to this homeostasis. We find that CD4+ T cells regulate the number and function of barrier-protective innate lymphoid cells (ILCs), as well as, production of antimicrobial peptides (AMPs), Reg3γ and Reg3β. RAG1−/− mice lacking T and B cells had elevated ILC numbers, IL-22 production, and AMP expression which were corrected by replacement of CD4+ T cells. MHCII−/− mice lacking CD4+ T cells also had increased ILCs, IL-22, and AMPs, suggesting that negative regulation by CD4+ T cells occurs at steady state. We utilized transfers and genetically modified mice to show that reduction of IL-22 is mediated by conventional CD4+ T cells and is TCR-dependent. The IL-22-AMP axis responds to commensal bacteria; however, neither the bacterial repertoire nor the gross localization of commensal bacteria differed between MHCII+/− and MHCII−/− littermates. These data define a novel ability of CD4+ T cells to regulate intestinal IL-22-producing ILCs and AMPs.
Introduction
The innate and adaptive immune systems collaborate at mucosal borders such as the lung, skin, and intestine to maintain barrier integrity and homeostasis with commensal microorganisms. The intestine uniquely balances requirements for nutrient breakdown and absorption with protective containment of microorganisms. Innate intestinal epithelial cells and Paneth cells, macrophages, dendritic cells, innate lymphoid cells (ILCs), and secreted mucus and antimicrobial peptides (AMPs), respond to intestinal microbes1,2. T and B lymphocytes, the cells that define the adaptive immune system, contribute to intestinal homeostasis via microbial antigen-specific responses, with secretion of cytokines and bacteria-neutralizing IgA2.
Studies have begun to explore the cooperative interplay between innate and adaptive immunity in the intestine. For example, in the setting of defective innate functions, CD4+ T cells induce protective IgA3, and systemic B cells produce bacteria-specific IgG in response to poorly contained commensals4. Similarly, ILCs prevent systemic invasion of microbes in RAG1−/− mice lacking adaptive immunity5. Thus, adaptive and innate immunity compensate for each other, but whether they directly regulate each other is not well understood.
Mucosal ILCs maintain barrier homeostasis and protect against pathogens through secretion of signature cytokines. Three subclasses of ILCs in the intestine parallel the effector functions of CD4+ helper T cell subsets: ILC1s (classical NK cells) secrete interferon gamma (IFNγ), ILC2s express GATA-3 and secrete IL-5 and IL-13, and ILC3s express RORγt and secrete IL-22 and IL-176. ILC3-derived IL-22 acts on IL-22 receptor-positive intestinal epithelial cells and Paneth cells to increase production of a subset of AMPs, including the Reg3 (regenerating islet-derived 3) family. Reg3γ and Reg3β neutralize gram-positive and -negative bacteria, respectively1, and Reg3γ maintains physical separation between luminal bacteria and the epithelium7. The IL-22-AMP axis contributes to intestinal homeostasis during a variety of challenges to the intestinal barrier8.
Despite the overlap of T cell and ILC function, adaptive immune regulation of ILCs has not been established. We used adoptive transfer and genetic approaches to demonstrate that CD4+ T cells regulate ILC numbers, IL-22 production, and the expression of the downstream AMPs, Reg3γ and Reg3β. This regulation was independent of T cell-dependent IgA, but dependent on antigen-specific TCR signals. The regulation by CD4+ T cells was not mediated by changes in the intestinal microbiota as the presence or absence of CD4+ T cells had no effect on the microbiota composition. Therefore, we have defined a novel ability of CD4+ T cells to regulate this critical innate immune component.
Results
IL-22-dependent innate responses are regulated by the adaptive immune system
To determine if the IL-22/Reg pathway is regulated by the adaptive immune system, we utilized quantitative real-time PCR to compare mRNA expression of the IL-22 responsive AMPs, Reg3g and Reg3b, in total RNA from the distal small intestine, cecum, and proximal large intestine of age-matched RAG1−/− and RAG1-heterozygous (+/−) het mice. AMP mRNA levels were increased in RAG1−/− mice on average six- or four-fold (Reg3g and Reg3b, respectively) in the small intestine, and approximately 100- or 200-fold in the cecum and large intestine (Figure 1a). Reg protein production is regulated by both epithelial and Paneth cell-intrinsic mechanisms and in response to cell-extrinsic production of the cytokine, IL-22. Elevated AMP levels in RAG1−/− mice were uniformly associated with approximately 25-fold increased expression of IL-22 mRNA in the small intestine and 30–40-fold in the large intestine and cecum. Therefore, in the absence of adaptive immunity, the IL-22-dependent bacterial sensing pathway was enhanced.
Figure 1. IL-22-dependent innate responses are enhanced in RAG1−/− mice and reduced after restoration of adaptive immunity.

RAG1−/− mice were reconstituted with 50 x106 cells from WT spleen and MLNs; recipients were analyzed eight weeks later (RAG−/− +Total). Un-manipulated RAG1−/− and RAG1+/− mice were controls. A) Total mRNA from tissue sections from terminal ileum, cecum, or proximal colon was reverse transcribed and analyzed by real-time PCR. Data were analyzed using the CT method normalized to a RAG1+/− for each experiment and GAPDH. Error bars show SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data pooled from three experiments. B) Representative plots showing T cells (TCRβ+) and B cells (CD19+) in Si-LP and spleen. C) Bar graph of representative IgA ELISA small intestine luminal contents. Data in B and C are representative of the three experiments.
To ask if the altered expression of IL-22 and AMPs in RAG1−/− mice could be regulated, the adaptive immune systems of RAG1−/− mice were reconstituted with ~50×106 cells from the spleen and mesenteric LNs of WT mice. T and B cells were reconstituted both peripherally and in the Si-LP and restored small intestine luminal IgA to WT levels by eight weeks after transfer (Figure 1b-c). Transfer of lymphocytes was associated with decreases in SI AMP and IL-22 mRNA levels of approximately two- and ten-fold, respectively (Figure 1a). Similar results were obtained at three and six weeks after transfer (not depicted) and comparable decreases were observed in the cecum and large intestine. Therefore, IL-22-dependent innate responses are regulated by the adaptive immune system.
ILCs are a dominant IL-22-producer in the intestine and a key upstream regulator of Reg protein production by intestinal epithelial and Paneth cells2,8. We compared the number and function of ILCs in the small intestines of RAG1−/− mice with those of age-matched RAG1+/− mice. The total numbers of small intestine lamina propria (Si-LP) and intra-epithelial (Si-IEC) ILCs were increased four- to six-fold in RAG1−/− mice compared to RAG1+/− mice (Figure 2c). The frequency of RORγ t-positive ILC3 among total ILCs was slightly increased in RAG1−/− mice, from approximately 75% to 85% (Figure 2d).
Figure 2. CD4+ T cells are sufficient to reduce IL-22-dependent innate responses.

RAG1−/− mice received 10 x106 sorted CD4+ T cells; recipients were analyzed six to eight weeks later (RAG−/− + CD4). A) Representative flow cytometric analysis of transferred cells, ILCs, and Ki-67 staining on ILC3 cells in the Si-LP of RAG1+/−, RAG1−/− , and RAG1−/− +CD4 mice. B) Representative IgA in the small intestinal lumen eight weeks after transfer of CD4+ T cells. C) Si-LP and Si-IEC ILC numbers. D) The percentage of RORγt+, ILC3 cells among total ILCs. E) Ki-67+ percentage among ILC3 cells. F) Representative flow cytometric analysis of cytokine production by Si-LP ILCs; plots are gated on lineage-cd90.2+ cells. G) Cytokine production by Si-LP ILCs. H) Reg3g, Reg3b, and IL-22 mRNA expression in total terminal ileum (small intestine) tissue, analyzed by quantitative real-time PCR as in figure 1. I) Representative staining of MHCII on Si-LP ILC3 cells in RAG1−/− mice (solid light gray line), RAG1+/− mice (dotted dark gray line), and RAG1−/− +CD4 mice (solid black line). MHCII-negative T cell (light gray filled) and MHCII-positive dendritic cell (thin gray line) controls from RAG1+/− mice are also depicted. J) Percentage of ILC3 cells that are MHCII+. Data in C, D, E, G, and H (IL-22 and Reg3g) were pooled from four experiments. Data in H (Reg3b) and J were each pooled from two of the four experiments. Error bars show SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
We next examined the proliferative capacity of ILC3s. Approximately two to five percent of RORγt+ ILC3 cells were proliferating in RAG1+/− mice, as assessed by Ki-67 positivity; in RAG1−/− mice, the frequency of proliferating ILC3’s was approximately doubled (Figure 2a, e). In addition to the increased ILC numbers and proliferation, the percentage of ILCs producing IL-22 was also elevated; 39% of Si-LP ILCs produced IL-22 in RAG1−/− mice compared with 11% in RAG1+/− mice (P<0.0001), and approximately 5% co-produced IL-22 and IL-17, compared with 1% in RAG1+/− mice (P<0.05) (Figure 2d-e). Therefore, coordinate elevations in AMPs and IL-22 reflect increased ILC numbers, proliferation, and function.
CD4+ T cells are sufficient to regulate IL-22-dependent innate responses in an IFNγ- independent manner
CD4+ T cells are mediators of intestinal homeostasis2 and we hypothesized that they would be sufficient to down-regulate the IL-22/Reg pathway. 10×106 CD4+ T cells FACS-sorted from the spleens and mesenteric LNs of WT mice were transferred to RAG1−/− mice. Cells transferred included naïve, memory, and regulatory T cells (Treg), but excluded NK1.1+ cells. Recipients were analyzed three or six weeks later. CD4+ T cells were found throughout the intestine (comprising 10–40% of cells in the Si-LP), while neither B cells nor luminal IgA were detected in recipient mice (Figure 2b). Therefore, outcomes reflected functions of CD4+ T cells independent of the induction of IgA.
Six weeks after CD4+ T cell transfer, we examined the number and function of Si-LP ILCs. Total ILC numbers in Si-LP and Si-IEC of RAG1−/− recipients were reduced by two-thirds (P<0.0001 and P<0.05) (Figure 2c). In parallel with reduced total ILC numbers, the percentage of ILC3 cells among total ILCs decreased to WT levels (P<0.0001). Both CD4+ T cells and ILCs rely on IL-7 for survival and express IL-7-receptor6,9,10. Expression of IL-7rα on ILC3 cells, however, was not appreciably altered by the addition of CD4+ T cells (Figure 2a and not depicted). Still, the percentage of ILC3 cells that were Ki-67+ was reduced to WT levels (P<0.0001) (Figure 2a, d-e). ILC-derived IL-22 in Si-LP was also reduced two-fold, as was the IL-17/IL-22 double producing population (P<0.001 and P<0.05) (Figure 2f-g). Therefore, CD4+ T cells were sufficient to reduce ILC proliferation, numbers, and IL-22 production.
A recent study demonstrated that a small percentage of ILC3s express MHCII, with functional consequences for CD4+ T cells and inflammation in the Si-LP11. The impact of CD4+ T cells on ILC3 expression of MHCII was therefore assessed. We found that approximately ten percent of ILC3 cells expressed MHCII+ in RAG1+/− mice (Figure 2i-j); MHCII levels on these cells were similar to those of MHCII+ dendritic cells (Figure 2i). In RAG1−/− mice, only 5% of ILC3 cells were MHCII+ (Figure 2j). Six to eight weeks after the transfer of CD4+ T cells, the percentage of ILC3 cells that was MHCII+ in recipient RAG1−/− mice was restored to WT levels (Figure 2j). These data implicate CD4+ T cells in the regulation of MHCII expression on ILC3 cells, in addition to ILC3 numbers and function.
mRNA levels of both AMPs and IL-22 were reduced in parallel with ILC numbers in RAG1−/− recipients of CD4+ T cells, most strikingly in the small intestine, where AMP mRNA levels were lowered by two-fold and IL-22 expression was lowered by three-fold compared to unmanipulated RAG1−/− mice. (Figure 2h). Total intestinal IL-22 mRNA levels were decreased and the percentage of transferred CD4+ T cells producing IL-22 was comparable to endogenous production in RAG1+/− mice (Figure 3). Equivalent percentages of transferred and endogenous CD4+ T cells also generated IL-17. Fewer transferred CD4+ T cells produced IL-2 or IL-10, while significant percentage produced IFNγ (P<0.0001) (Figure 3). Therefore, CD4+ T cells are sufficient to regulate ILCs, their IL-22 production, and levels of downstream AMPs, and this regulation does not require IgA.
Figure 3. Transferred CD4+ T cells preferentially produce IFNγ.

Cytokine production by endogenous or transferred sorted CD4+ T cells (RAG+/- or RAG−/− +CD4, respectively) from mice analyzed six to eight weeks after cell transfer. A) Representative flow cytometic analysis from Si-LP; plots are gated on CD4+TCRβ+ cells. B) Cytokine production by Si-LP CD4+ T cells. Data are pooled from either two (IL-2, IL-10) or all four (IFNγ, IL-17, IL-22) of the experiments shown in Figure 2. Error bars show SEM, *P<0.05, **P<0.01, ****P<0.0001
The increased IFNγ production by CD4+ T cells transferred to RAG1−/− mice raised the possibility that the cytokine was regulating ILC3s; this possibility was assessed by transferring 10×106 IFNγ −/− CD4+ T cells to RAG1−/− mice. Fix or six weeks later, the transferred cells could be found in the Si-LP and, as expected, were not producing IFNγ (Figure 4a). ILC numbers still fell by approximately two thirds, comparable to the transfer of WT CD4+ T cells (Figure 4b). The percentage of ILC3 cells that were Ki-67+ also decreased (P<0.0001) (Figure 4c). The percentages of IL-22 and IL22/17 double-producing ILCs were restored to WT levels (P<0.0001) (Figure 4d). Thus, the ability of CD4+ T cells to down-regulate ILC numbers, proliferation, and function was not dependent on IFNγ. Surprisingly, RAG1−/− mice that received IFNγ −/− CD4+ T cells developed wasting and grossly visible, pan-intestinal colitis (not depicted)12. Therefore, CD4+ T cells are sufficient to regulate ILCs, their IL-22 production, and levels of downstream AMPs. This regulation does not require IgA or IFNγ production by CD4+ T cells, and, importantly, occurs even in the presence of gross intestinal inflammation.
Figure 4. CD4+ T cell regulation of ILCs is independent of IFNγ.

RAG1−/− mice received 10 x106 sorted IFNγ −/− CD4+ T cells; recipients were analyzed five or six weeks later (RAG−/− +IFNγ −/− CD4). (A) Percent of transferred or endogenous CD4+ T cells in the Si-LP producing IFNγ. C) Numbers of Si-LP and ILCs in Si-LP of indicated mice. D) Percentage of ILC3 cells that are Ki-67+. E) Cytokine production by Si-LP ILCs in the indicated mice. Data in A-E are pooled from two experiments.
CD4+ T cells are important in steady state regulation of ILCs/AMPs
To ask if CD4+ T cells regulate ILC numbers, IL-22 production, and additionally the Reg proteins at steady state, we made use of mice with more selective adaptive immune deficiencies; major histocompatibility class II (MHCII) −/− mice lack MHCII expression and MHCII-restricted CD4+ T cells (Figure 5a). However, MHCII−/− and MHCII+/− mice do contain quantitatively similar amounts of IgA in the small intestine (not depicted) in agreement with previous work showing that IgA production can be T-independent13. ILC numbers were increased approximately two-fold in MHCII−/− mice compared to MHCII+/− controls, as was the percent of ILCs producing IL-22 (Figure 5b-c). The percent of ILCs producing IL-22 in MHCII−/− mice was variable (7–31%). Consistent with these observations, both IL-22 and Reg3g mRNA in ileum and large intestine tissue, although also variable, were also increased (Figure 5d). These data are in contrast to RAG1−/− mice, where ILC numbers and ILC-derived IL-22 and AMPs were consistently elevated.
Figure 5. IL-22-dependent innate responses are enhanced in the absence of TCR-stimulated CD4+ T cells.

A) Representative plots of CD4+ and CD8+ T cells in the Si-LP of MHCII+/-, MHCII−/− , K14 (β2m WT or +/−), and β2m−/− (all MHCII+/−) mice. B) ILC numbers and C) ILC-derived IL-22 after PMA/ionomycin stimulation in the indicated mice. D) Reg3g and IL-22 mRNA expression in total intestinal tissue, analyzed by quantitative real-time PCR as in figure 1. Error bars show SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are pooled from six or seven experiments.
MHCII−/− mice contain CD8+ T cells, as well as, NKT cells and other atypical MHCI-restricted CD4+ T cells. We used beta-2 microglobulin (β2m)−/− mice that lack normal expression of MHCI and these T cell populations to assess their contribution to ILC regulation. ILC numbers in β2m−/− mice were intermediate between MHCII+/−/β2m+/− (WT) and MHCII−/− mice though not statistically different from those in MHCII/β2m+/− mice, suggesting that β2m-restricted cells have a minimal effect on ILC numbers (Figure 5b). The percent of ILCs producing IL-22 in β2m−/− mice was equivalent to those in MHCII+/− animals (12% ± 0.8%), confirming that β2m-restricted cells are dispensable for regulation of IL-22 production by ILCs (Figure 5c). These data suggest that MHCII-restricted CD4+ T cells (but not MHCI-restricted CD8+ T cells) are important to control ILC numbers, their IL-22 production, and levels of Reg3γ. However, the variability in ILC function in MHCII−/− mice suggests that this pathway is not absolutely required.
Would depletion of CD4+ T cells from adult WT mice have the same effect as the developmental defect in MHCII−/− mice? WT mice were treated with CD4-depleting antibody (GK1.5), leading to the eradication of CD4+ T cells from the Si-LP (Figure 6a). Six weeks after depletion, both ILC numbers and ILC3 proliferation trended upwards, but differences failed to reach statistical significance (Figure 6b-c), while the percentage of ILC3 cells expressing MHCII fell by one third (P=0.014) (Figure 6d). Similarly, the percentages of ILCs secreting IL-22 and IL-22/IL-17 trended upwards; although, they were not statistically significantly increased (Figure 6e). However, the upward shifts in both ILC numbers and IL-22 production were associated with a 2.5 fold increase in tissue mRNA expression of Reg3g in CD4-depleted mice (Figure 6f). Overall, these data are consistent with the variably increased ILC numbers, IL-22 secretion, and tissue AMP and IL-22 mRNA expression observed in MHCII−/− mice, but again suggest that CD4+ T cells are not absolutely required in WT mice to down-regulate ILC function at steady state.
Figure 6. Depletion of CD4+ T cells in WT mice.
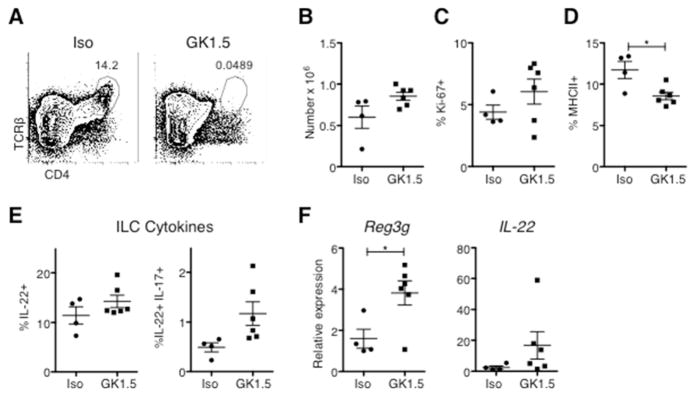
Mice were treated with CD4-depleting antibody (GK1.5) or an isotype control for six weeks prior to Si-LP analysis. A) Flow cytometry of CD4+ T cells in the Si-LP of treated mice. (B-F) Graphs show B) ILC numbers, C) the percentage of ILC3 cells that are Ki-67+, D) the percentage of ILC3 cells that are MHCII+, E) cytokine production by ILCs after stimulation with PMA and ionomycin. F) Reg3g, Reg3b, and IL-22 mRNA expression in total terminal ileum (small intestine) tissue, analyzed by quantitative real-time PCR. Data are pooled from two independent experiments, *P<0.05.
CD4+ T cells do not alter the large intestine microbiota or bacterial localization
In our specific pathogen free mouse facility, both IL-22 production and secondary AMP production in RAG1−/− mice are antibiotic-sensitive (Figure 7a), suggesting a dependence on local intestinal bacteria. In antibiotic-treated RAG1−/− mice, ILC numbers additionally fell by approximately one third, and IL-22 production decreased by two fold (Figure 7b).
Figure 7. Impact of CD4+ T cells and adaptive immunity on the large intestine microbiota.

A) Reg3g, Reg3b, and IL-22 mRNA expression in total terminal ileum (small intestine) tissue of antibiotic-treated or control mice, analyzed by quantitative real-time PCR as in figure 1, pooled from two experiments. Error bars show SEM, **P<0.01, ***P<0.001. B) ILC numbers and IL-22 production in antibiotic-treated or control RAG1−/− mice, pooled from two experiments. C) Quantitative PCR of major bacterial groups in MHCII+/− and MHCII−/− littermates (MHCII+/− n=9–12, MHCII−/− n=13–14), analyzed using genomic reference DNA standards. D) Principle coordinate plot of a representative MHCII litter, with +/− genotype shown in red and −/− in blue. E) Principle coordinate analyses of all MHCII+/− or MHC−/− mice sequenced, colored by genotype (+/− in red and −/− in blue), mother (purple and green), and litter (blue and brown tones). F-H) The same analyses performed for MHCII mice, applied to RAG1+/− or RAG1−/− littermates (for E, RAG1+/− n=47, RAG1−/− n=35).
To address two different explanations for the enhanced response to the commensal bacterial microbiota, we asked if CD4+ T cells altered either the repertoire of commensal bacteria and/or host sensing of the commensal bacteria. To address differences in bacterial repertoire, we defined the composition of the commensal intestinal bacteria in MHCII−/− mice and their MHCII+/− littermates. Because measures in the small intestine revealed relatively few bacteria (not depicted), we focused on the large intestine. To minimize environmental and husbandry differences that can drive microbial diversity14, pregnant mothers were separated into their own cages prior to giving birth, and littermates were weaned into individual cages at three to four weeks of age and euthanized at six weeks, at which point total colonic DNA was prepared from each individual animal. Quantitative PCR analyses showed that the expression of MHCII and the presence of CD4+ T cells had no statistically significant effect on the total numbers of either Eubacteria or major bacterial families, with the exception of a less than two-fold effect on lactobacilli (Figure 7c). Additionally, deep sequencing analysis of the16S rDNA genes of colonic commensals found that qualitative differences in bacterial repertoire between MHCII+/− and MHCII−/− genotypes did not achieve significance, as measured by principle coordinate analyses of litters (Figure 7d). In contrast, there were statistically significant differences in the numbers and bacterial repertoires of RAG1−/− mice and their littermate controls. (Figure 7f-g). In the setting of these small differences, we could not detect quantitative changes in RAG1−/− mice with reconstitution of the adaptive immune system (not depicted). In all the strains examined, litter and maternal effects on bacterial composition dominated over genotype (Figure 7e,h). These data suggest that the effects of CD4+ T cells on ILC biology are not secondary to altered microbial composition.
To explore the possibility of altered bacterial sensing in the absence of CD4+ T cells, we utilized fluorescence in situ hybridization (FISH) to eubacterial 16S to assess bacterial localization in the large intestine in littermate or extensively co-housed mice; differences in bacterial localization could alter bacterial sensing in the absence of compositional differences. Blinded images were scored based on the proximity of bacteria relative to epithelium (Figure 8a). Overall bacterial localization was not different in MHCII−/− mice compared to MHCII+/− mice, nor in RAG1−/− mice compared to RAG1+/− mice or CD4+ T cell-reconstituted RAG1−/− mice (Figure 8b-c). These data suggest that effects of CD4+ T cells on altered ILC biology is not driven by large-scale differences in bacterial localization.
Figure 8. Large intestine bacterial localization is not altered by the presence of CD4+ T cells.
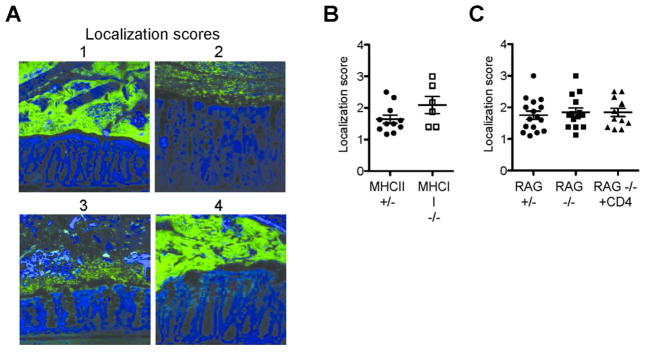
A) Representative images of FISH staining of large intestine demonstrating localization scores: 1-no bacterial contact with epithelium and preserved mucus layer; 2-bacterial penetration into mucus layer without epithelial contact; 3-epithelia contact; 4-extensive epithelial contact. Images were acquired and analyzed in a blinded manner, with three to six images acquired per large intestine. Image scores were averaged to acquire a single score per mouse. B) Localization scores from MHCII+/− and MHCII−/− mice. C) Localization scores from RAG1+/− mice, RAG1−/− mice, and six-week CD4+ T cell-reconstituted RAG1−/− mice. Error bars show SEM.
CD4+ T cell regulation of ILCs requires MHCII-TCR interactions
The effect of CD4+ T cells on IL-22-producing ILCs in the small intestine may require antigen-dependent T cell activation, or, alternatively, be mediated by antigen-independent competition with ILCs for a niche and survival factors. To distinguish between these possibilities, we asked if CD4+ T cells require antigen-specific TCR signals to regulate the IL-22/Reg pathway. We made use of K14-Aβb mice (K14), in which MHCII is restricted to thymic cortical epithelium; CD4+ T cells are positively selected but are not exposed to any peripheral TCR signals15. Positive selection leads to increased frequencies of CD4+ T cells in the Si-LP of K14 mice as compared to the residual CD4+ T cell population in MHCII−/− mice (Figure 5a). ILC numbers and the percentage of ILCs producing IL-22 in K14 mice were equivalent to those in MHCII−/− mice; both strains had increased numbers of ILCs compared to MHCII+/− mice (Figure 5b-c). Similarly, levels of IL-22 and Reg3g mRNA were increased in K14 and MHCII−/− mice (Figure 5d). The comparable elevations in MHCII−/− and K14 mice in ILC numbers, function, and AMP levels strongly suggest that CD4+ T cells require TCR signals to regulate ILCs, IL-22, and AMPs.
Tregs are not sufficient to regulate ILC-derived IL-22 in the small intestine
Foxp3+ Tregs dampen immune responses and prevent intestinal inflammation in multiple settings16. To determine if the CD4+ effect on ILC regulation was mediated by Tregs, GFP+ Tregs sorted from Foxp3-GFP reporter mice were transferred into RAG1−/− mice in numbers comparable to the number of Tregs in the initial inoculum of 10×106 total CD4+ T cells (~0.5–1×106). Transferred cells comprised one to five percent of cells in the Si-LP three to six weeks after transfer (Figure 9a). The purity of the Treg population was reduced by 40–80% after transfer, consistent with past descriptions of reconstitution of lymphopenic mice17 (Figure 9a). ILC numbers were inconsistent both three and six weeks after transfer (Figure 9b). However, the percentage of ILCs producing IL-22, as well as levels of IL-22 and AMP mRNA remained elevated in the small intestine (Figure 9c-d). Thus, although Tregs may influence the numbers of Si-LP ILCs, Tregs do not mediate the negative regulation exerted on ILC function by CD4+ T cells in the small intestine.
Figure 9. Tregs are not sufficient to reduce IL-22 dependent innate responses.
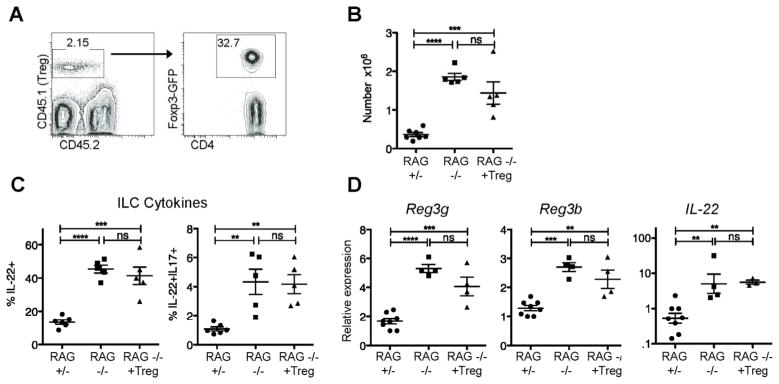
RAG1−/− mice that received 0.5–1×106 Tregs were analyzed six weeks after transfer. A) Representative plots from Si-LP showing Treg purity. B) Si-LP ILC numbers and C) percentage of ILCs that are RORγt+ pooled from two experiments. D) ILC-derived cytokines after stimulation with PMA/ionomycin from the same experiments shown in B-C. E) Reg3g, Reg3b, and IL-22 mRNA expression in total terminal ileum (small intestine) tissue, analyzed by quantitative real-time PCR pooled from two experiments, analyzed as in figure 1. Error bars show SEM, **P<0.01, ***P<0.001, ****P<0.0001
Discussion
Past studies have elucidated mechanisms by which the microbiota and the innate immune system regulate CD4+ T cell responses2. Our studies establish that conventional CD4+ T cells negatively regulate innate responses. We found that conventional CD4+ T cells reduce ILC numbers, proliferation, and IL-22 production in the intestine, as well as expression of the IL-22-responsive AMPs, Reg3γ and Reg3β. This regulation required neither IgA nor changes in the intestinal commensal microbiota, but required antigen-specific TCR signals. CD4+ T cells and ILCs have functional overlap; however, CD4+ T cells and ILCs do not simply have redundant functions, as transferred CD4+ T cells did not produce significant amounts of IL-22 and the total level of IL-22 mRNA in CD4+ T cell-reconstituted RAG1−/− mice was significantly decreased.
We propose that CD4+ T cell regulation of ILCs is TCR-dependent, as demonstrated by elevated ILC numbers in function in K14 mice. The TCR signals could differentiate or expand naïve populations, or activate pre-existing memory populations that subsequently regulate ILCs. While AMP expression correlated with ILC production of IL-22, separate regulatory mechanisms may also operate. A recent study showed that ILC3s express MHCII, and that this expression regulates intestinal homeostasis11 and we observed differential expression of MHCII on ILC3 cells of RAG1+/− and −/− mice. Furthermore, CD4+ T cells were sufficient to restore levels of MHCII on ILC3 cells in RAG1−/− to WT levels. Hepworth et al. suggested that MHCII on ILC3 cells affected CD4+ T cell function. Our data implicate CD4+ T cells in the converse regulation of MHCII on ILC3s; although, they do not address whether that regulation is via cognate CD4+ T cell-ILC3 interactions. Nonetheless, the putative requirement for TCR signals on CD4+ T cells to regulate ILC3 cells makes this direct interaction an attractive potential mechanism.
The ability of CD4+ T cells to regulate ILCs and AMPs may have resided within Tregs which prevent colitis in RAG1−/− mice reconstituted with effector CD4+ T cells16. However, Tregs did not reduce IL-22 or AMPs in RAG1−/− mice, while variably altering ILC numbers. Perhaps this variability stemmed from outgrowth of conventional CD4+ T cells from the original transferred population. Alternatively, Tregs and ILCs both utilize IL-2 and TLR2 signals16,18, raising the possibility of signaling crosstalk between these populations. Transfer of a larger number of Tregs may have further altered ILCs. Nonetheless, these data suggest that Tregs do not play a significant role in the regulation of ILC3s.
CD4+ T cells transferred to RAG1−/− mice produced more IFNγ than any other cytokine; yet, WT mice contain relatively few IFNγ -producing Th1 cells in the Si-LP at steady state. Indeed, we found that CD4+ T cell-mediated regulation of ILCs was IFNγ – independent. Consistent with past work12, RAG1−/− recipients of IFNγ −/− CD4+ T cells developed colitis and wasting; these data show that regulation of ILCs by CD4+ T cells still occurs in overt inflammation. A recent study suggested that ILC3 cells may downregulate Th17 cells19. However, the impact of IL-17 on ILCs is uncertain and future studies should directly address its role. Conversely, epithelial cell-derived IL-25 may down-regulate ILC3-derived IL-2220; the potential for CD4+ T cell action in this pathway could also be explored.
Our results do not rule out that the possibility that CD4+ T cells and ILCs additionally compete for an anatomic niche or for a soluble factor. While IL-7rα levels on ILC3 cells were not appreciably altered by the presence of CD4+ T cells, IL-7 might still regulate such a niche; both ILC and CD4+ T cell survival depend on IL-7 and in the presence of exogenous IL-7, ILCs expand6,9,10. In RAG1−/− mice, IL-22 mRNA levels are common-gamma chain dependent21. However, it is equally likely that CD4+ T cells could compete with ILCs for as yet undefined metabolites.
Because ILCs and AMP-producing epithelial cells respond to commensal and pathogenic bacteria and are sensitive to changes in intestinal microorganisms, CD4+ T cells could affect ILCs and AMPs secondarily to regulating either microbial composition or sensing. In contrast to previous studies examining the microbial consequences of alterations in subsets of CD4+ T cells22,23, we found no significant evidence that CD4+ T cells alter the composition of the intestinal microbiota. Eliminating specific CD4+ T cell subsets could induce greater immune malfunction and secondary microbial dysbiosis than loss of all CD4+ T cells. However, in agreement with Pamer and colleagues, maternal and litter effects dominated over genotype differences in our hands14. We similarly did not find altered bacterial localization that could lead to altered bacterial sensing at the epithelium. However, these studies lacked the sensitivity to reliably identify bacteria invading across the epithelium, which could also contribute to altered sensing. Future studies should also examine the small intestine, which contains fewer bacteria but where Reg3γ plays the greatest role in bacterial localization7 and where our observed ILC effects were most robust. These data suggest that CD4+ T cell regulation of ILCs and AMPs occurs independently of gross changes in the composition or localization of the commensal microbiota.
We and others find that the microbiota has a stimulatory effect on IL-22 and AMPs21,24–26. In contrast, a recent report by Eberl and colleagues found an inhibitory role for commensal microbiota on IL-22 producing ILCs except in extreme inflammation20. Consistent with our data, Sawa et al. observed increased IL-22 production by ILCs in RAG1−/− mice, though surprisingly this did not translate into increased levels of AMPs. The regulation of ILC homeostasis and function by commensals remains unclear27.
While confusing, these distinct data support the idea that regulatory pathways that determine IL-22 production by ILCs are both dependent and independent of the commensal microbiota; regulation by CD4+ T cells could be a microbiota-independent pathway. While CD4+ T cells decreased ILC numbers and IL-22 production independent of IgA, our results do not negate a possible complementary role for this or other mucosal actors, such as γδ T cells, that control bacteria at the mucosal surface or prevent intestinal injury13,28. Indeed, in contrast to RAG1−/− mice, MHCII−/− mice had quite variable levels of ILC-derived IL-22, and depletion of CD4+ T cells in WT mice did not statistically significantly alter ILCs, though Reg3g was increased. One explanation could lie in the ability of other cell subsets to influence the signals delivered to ILCs independently of CD4+ T cells. Why this influence is variable is unclear. It is possible that ILCs respond to a specific microbiota species with differing levels across MHCII−/− mice; sequencing analyses may not be sufficiently robust to detect such differences.
We can begin to speculate how CD4+ T cell-mediated regulation of ILCs will fit into the regulatory networks governing innate- and adaptive-immune cooperation to maintain homeostasis at steady state; whereas, most studies to date have examined pathologic settings29–31. This work demonstrates a non-redundant role for conventional CD4+ T cells in the regulation of IL-22-producing ILC3s in the intestine. Perhaps this regulation checks the potential for inappropriate innate-driven inflammation, substituting a more tailored adaptive response. Further study of this pathway, should help explain the maintenance of intestinal homeostasis and could reveal additional therapeutic targets for treatment of its breakdown in disease.
Materials and Methods
Mice
WT CD45.1, RAG1, C57BL/6J were originally purchased from Jackson Laboratories (Bar Harbor, ME). β2m/Aβb −/− mice32 were acquired from Taconic (Hudson, NY). MHCII−/− (Aβb)33, K14-Aβb15, and Foxp3-GFP mice34 were bred in house. All mice were housed under specific pathogen free conditions (norovirus and Helicobacter-colonized) in accordance with the University of Pennsylvania animal care and use guidelines, and fed autoclaved Labdiet Rodent Diet 1050 (Animal Specialties and Provisions). Mice used in ILC and transfer experiments were two to five months old and were littermates or extensively cohoused. Breeding and husbandry of mice used for quantitative bacterial PCR and sequencing analyses are in Results.
Antibodies, flow cytometry, and cell sorting
Antibodies were purchased from eBioscience (San Diego, CA), Biolegend (San Diego, CA), BD (Franklin Lakes, NJ), or Invitrogen (Grand Island, NY). Live/Dead Fixable Aqua (Invitrogen) or DAPI was used for live/dead discrimination. ILCs were defined as CD45.2+, CD90.2+, lineage negative. Lineage markers included TCRβ, CD3, CD11c, and B220 or CD19, Gr-1, CD11b, and CD5. The Foxp3/Transcription Factor Staining Buffer Set (eBioscience) was used for transcription factor stains. For intracellular cytokine stains, samples were fixed with 1.6% PFA and stained in 0.5% saponin after stimulation. Samples were collected on a BD LSRII or FACs Canto and analyzed using Flowjo software (Treestar). For cell sorting, CD4+ T cells were enriched by negative selection as previously described35, and further sorted on DAPI- CD4+ B220-CD8β-IAb-CD11c-CD11b-Gr1-NK1.1- cells. For Treg transfers, CD4-enriched cells from Foxp3-GFP mice were additionally sorted on GFP+ cells. Sorting was performed on a BD Aria.
CD4 depletion
Mice were injected IP with 1mg GK1.5 (BioXcell, West Lebanon, NH) or Rat IgG isotype control (Sigma, St. Louis, MO) every two weeks for six weeks.
Antibiotic Treatment
Drinking water was supplemented with ampicillin (0.5mg/ml), gentamicin (0.5mg/ml), metronidazole (0.5mg/ml), neomycin (0.5mg/ml), and vancomycin (0.25mg/ml) plus sucralose sweetener for two weeks as previously described36.
Preparation of lamina propria and intraepithelial cells
Single cell suspensions of lamina propria were prepared by standard techniques, utilizing epithelial stripping in 1mM DTT and 5mM EDTA followed by digestion with 0.1mg/ml Liberase TL and Dnase (Roche, Indianapolis, IN). IEC were obtained from the stripped epithelial fraction after centrifugation in 30% Percoll (GE Healthcare Life Sciences, Pittsburgh, PA). In experiments with MHCII−/− , β2m−/− , or K14 mice, Peyer’s patches were first removed. Cytokine production was assessed after stimulation of LP cells with PMA 50ng/ml and Ionomycin 500ng/ml in the presence of Brefeldin A 1μg/ml, at 1×106 cells/ml for 3.5 hours at 37°C.
RNA Isolation and reverse transcription
Tissue pieces were cleared of stool, placed in RNAlater (Ambion, Grand Island, NY) for storage, and homogenized using a PowerGen700 Homogenizer (Fisher, Waltham, MA). RNA isolations and reverse transcription were carried out using the RNeasy kit (QIAGEN, Valencia, CA) and the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Grand Island, NY), respectively.
Bacterial DNA Isolation
Large intestines were homogenized in sterile PBS. Bacterial DNA isolation was performed using the QIAmp DNA Stool Kit (QIAGEN) with optional high temperature step37.
Quantitative and real-time PCR
The following primers were utilized37: total Eubacteria (Total), and group specific primers for Bacteroides (Bact), Lactobacilli/enterococcus (Lact), and E. rectale/C. coccoides (Erec). Additional primer sequences were: Reg3g F: TTCCTGTCCTCCATGATCAAAA, Reg3g R: CATCCACCTCTGTTGGGTTCA; Reg3b F, TCCCAGGCTTATGGCTCCTA; Reg3b R, GCAGGCCAGTTCTGCATCA; IL-22 F TCCGAGGAGTCAGTGCTAAA, IL-22 R AGAACGTCTTCCAGGGTGAA; GAPDH F TCATCAACGGGAAGCCCATCAC, GAPDH R AGACTCCACGACATACTCAGCACCG. SYBR-green (Applied Biosystems) was utilized for all reactions.
IgA ELISA
IgA ELISAs were performed similarly to published techniques38. Briefly, plates were coated with 1:500 goat anti-mouse Ig(H+L) (Southern Biotech, Birmingham, AL), blocked with 10% soymilk (8th continent, Santa Ana, CA) plus 0.05% Tween 20, incubated with small intestine lumen supernatants, followed by 1:2000 goat anti-mouse IgA(α) detection antibody conjugated to horseradish peroxidase. Plates were developed with o-Phenylenediamine and read at 405nm.
454 Sequencing
Sequence determination was carried out using the 454/Roche pyrosequencing method39. DNA samples were amplified using bar coded DNA primers that annealed to the 16S rRNA gene within the V1V2 region essentially as described40. Sequence reads were analyzed with QIIME pipeline41 using UniFrac42,43. Sequence reads have been deposited in the NCBI Sequence Read Archive and are available under the SRA Project accession SRP021545.
Fluorescence in situ hybridization (FISH)
16S FISH was performed as previously described44. Briefly, 5μm longitudinal sections were hybridized to 16s rRNA eubacterial probe ([AminoC6+Alexa488]GCTGCCTCCCGTAGGAGT[AmC7-Q+Alexa488], (Eurofins MWG Operon, Huntsville, AL) at 1μM. Images were acquired on a Zeiss LSM 710 confocal microscope and analyzed using Fiji (ImageJ) software45.
Statistics
Significance was determined by one-way ANOVA with a Bonferroni post-test (Figures 1, 2, 4, and 7) or unpaired Student’s T-test (Figures 3 and 5a). Graphs shown on a log scale were log-transformed before analysis. For quantitative bacterial PCRs, a separate analysis of covariance was performed for each dependent measure (log10 bacterial count), with genotype as the fixed independent variable and parent and litter as random effects. This allowed comparison between the +/− and −/− genotype, while controlling for litter and parent effects. Analyses were performed using SAS software.
Acknowledgments
This work was supported by a pilot grant from the Penn-CHOP Joint Center for Digestive, Liver and Pancreatic Medicine P30-DK050306, the Penn Genome Frontiers Institute, and the University of Pennsylvania Center for AIDS Research (CFAR) P30 AI 045008, and R01-AI57757 (NHS). LLK was supported by the Training Program in Rheumatic Disease 5T32AR007442-25.
Footnotes
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. 2012;12:503–516. doi: 10.1038/nri3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 3.Khounlotham M, et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity. 2012;37:563–573. doi: 10.1016/j.immuni.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slack E, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonnenberg GF, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spits H, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 7.Vaishnava S, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 9.Schmutz S, et al. Cutting edge: IL-7 regulates the peripheral pool of adult ROR gamma+ lymphoid tissue inducer cells. J Immunol. 2009;183:2217–2221. doi: 10.4049/jimmunol.0802911. [DOI] [PubMed] [Google Scholar]
- 10.Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol. 2011;12:478–484. doi: 10.1038/ni.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hepworth MR, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498:113–117. doi: 10.1038/nature12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida M, et al. Differential localization of colitogenic Th1 and Th2 cells monospecific to a microflora-associated antigen in mice. Gastroenterology. 2002;123:1949–1961. doi: 10.1053/gast.2002.37049. [DOI] [PubMed] [Google Scholar]
- 13.Cerutti A, Rescigno M. The biology of intestinal immunoglobulin A responses. Immunity. 2008;28:740–750. doi: 10.1016/j.immuni.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ubeda C, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. The Journal of Experimental medicine. 2012;209:1445–1456. doi: 10.1084/jem.20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laufer TM, DeKoning J, Markowitz JS, Lo D, Glimcher LH. Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex. Nature. 1996;383:81–85. doi: 10.1038/383081a0. [DOI] [PubMed] [Google Scholar]
- 16.Nutsch KM, Hsieh CS. T cell tolerance and immunity to commensal bacteria. Curr Opin Immunol. 2012;24:385–391. doi: 10.1016/j.coi.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komatsu N, et al. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA. 2009;106:1903–1908. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crellin NK, et al. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity. 2010;33:752–764. doi: 10.1016/j.immuni.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Qiu J, et al. Group 3 Innate Lymphoid Cells Inhibit T-Cell-Mediated Intestinal Inflammation through Aryl Hydrocarbon Receptor Signaling and Regulation of Microflora. Immunity. 2013;39:386–399. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sawa S, et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 21.Satoh-Takayama N, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Kawamoto S, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336:485–489. doi: 10.1126/science.1217718. [DOI] [PubMed] [Google Scholar]
- 23.Josefowicz SZ, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–399. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keilbaugh SA, et al. Activation of RegIII / and interferon expression in the intestinal tract of SCID mice: an innate response to bacterial colonisation of the gut. Gut. 2005;54:623–629. doi: 10.1136/gut.2004.056028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandl K, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455:804–807. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanos SL, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tait Wojno ED, Artis D. Innate lymphoid cells: balancing immunity, inflammation, and tissue repair in the intestine. Cell Host Microbe. 2012;12:445–457. doi: 10.1016/j.chom.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ismail AS, et al. Gammadelta intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc Natl Acad Sci USA. 2011;108:8743–8748. doi: 10.1073/pnas.1019574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu R, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012;37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zenewicz LA, et al. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buonocore S, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grusby MJ, et al. Mice lacking major histocompatibility complex class I and class II molecules. Proc Natl Acad Sci USA. 1993;90:3913–3917. doi: 10.1073/pnas.90.9.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grusby MJ, Johnson RS, Papaioannou VE, Glimcher LH. Depletion of CD4+ T cells in major histocompatibility complex class II-deficient mice. Science. 1991;253:1417–1420. doi: 10.1126/science.1910207. [DOI] [PubMed] [Google Scholar]
- 34.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 35.Allenspach EJ, Lemos MP, Porrett PM, Turka LA, Laufer TM. Migratory and lymphoid-resident dendritic cells cooperate to efficiently prime naive CD4 T cells. Immunity. 2008;29:795–806. doi: 10.1016/j.immuni.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill DA, et al. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2010;3:148–158. doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salzman NH, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bry L, Brigl M, Brenner MB. CD4+-T-Cell Effector Functions and Costimulatory Requirements Essential for Surviving Mucosal Infection with Citrobacter rodentium. Infect Immun. 2006 doi: 10.1128/IAI.74.1.673-681.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKenna P, et al. The Macaque Gut Microbiome in Health, Lentiviral Infection, and Chronic Enterocolitis. PLoS Pathog. 2008;4:e20. doi: 10.1371/journal.ppat.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and Qualitative β Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Appl Environ Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hand TW, et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science. 2012;337:1553–1556. doi: 10.1126/science.1220961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]