

Summary
Contact-dependent growth inhibition (CDI) is a widespread form of inter-bacterial competition that requires direct cell-to-cell contact. CDI+ inhibitor cells express CdiA effector proteins on their surface. CdiA binds to specific receptors on susceptible target bacteria and delivers a toxin derived from its C-terminal region (CdiA-CT). Here, we show that purified CdiA-CT536 toxin from uropathogenic Escherichia coli 536 translocates into bacteria, thereby by-passing the requirement for cell-to-cell contact during toxin delivery. Genetic analyses demonstrate that the N-terminal domain of CdiA-CT536 is necessary and sufficient for toxin import. The CdiA receptor plays no role in this import pathway; nor do the Tol and Ton systems, which are exploited to internalize colicin toxins. Instead, CdiA-CT536 import requires conjugative F pili. We provide evidence that the N-terminal domain of CdiA-CT536 interacts with F pilin, and that pilus retraction is critical for toxin import. This pathway is reminiscent of the strategy used by small RNA leviviruses to infect F+ cells. We propose that CdiA-CT536 mimics the pilin-binding maturation proteins of leviviruses, allowing the toxin to bind F pili and become internalized during pilus retraction.
Introduction
Contact-dependent growth inhibition (CDI) systems are distributed throughout α–, β– and γ–proteobacteria, where they function in competition between closely related bacteria (Aoki et al., 2010, Aoki et al., 2011, Ruhe et al., 2013a, Ruhe et al., 2013b). CDI is mediated by the CdiB/CdiA family of two-partner secretion proteins. CdiB is an outer-membrane β-barrel protein required for secretion of the CdiA effector. CdiA proteins are filamentous and very large, ranging from ~180 kDa in Moraxella to over 600 kDa in some Pseudomonas species (Aoki et al., 2010, Aoki et al., 2011). CdiA extends from the surface of CDI+ inhibitor cells and interacts with receptors on susceptible target bacteria. CDI has been characterized most extensively in Escherichia coli EC93, and its CdiAEC93 effector binds the highly conserved outer-membrane protein BamA as a receptor (Aoki et al., 2008, Ruhe et al., 2013b). Upon binding the receptor, CdiAEC93 appears to be cleaved to release a C-terminal toxin region (CdiA-CTEC93), which is subsequently translocated into the target cell through a poorly characterized pathway (Aoki et al., 2010, Aoki et al., 2008, Webb et al., 2013). E. coli EC93 inhibitor cells also deliver toxins to one another, but are protected from inhibition by a small CdiIEC93 immunity protein encoded immediately downstream of cdiAEC93 (Aoki et al., 2005). Thus, E. coli EC93 cells deploy CdiA-CTEC93 toxins to inhibit other non-immune strains of E. coli. Because CDI confers a significant growth advantage to inhibitor cells, these systems are thought to mediate inter-strain competition for environmental niches.
CDI loci encode a diverse group of CdiA-CT/CdiI toxin/immunity sequences. There are at least 18 CdiA-CT/CdiI sequence types distributed throughout E. coli strains, and Burkholderia pseudomallei strains carry 10 distinct sequence types (Nikolakakis et al., 2012, Ruhe et al., 2013a). Thus, CDI toxin/immunity pairs are highly variable, even between strains of the same species. In accord with this sequence diversity, CDI toxins exhibit a number of distinct activities. For example, CdiA-CTEC93 dissipates the proton motive force and induces the phage-shock response (Aoki et al., 2009), suggesting that the toxin forms pores in the inner membrane of target cells. Many other CDI toxins have nuclease activities. CDI toxins from Dickeya dadantii 3937 and E. coli EC869 are DNases that degrade target-cell genomic DNA (Aoki et al., 2010, Morse et al., 2012, Webb et al., 2013). CDI+ bacteria also deploy RNases that preferentially cleave tRNA molecules. CdiA-CT536 from uropathogenic E. coli 536 (UPEC 536) is a tRNA anticodon nuclease (Aoki et al., 2010, Diner et al., 2012), and toxins from B. pseudomallei strains cleave tRNA in the anticodon loop, T-loop and aminoacyl-acceptor stem (Nikolakakis et al., 2012). Each CdiA-CT toxin is specifically neutralized by its cognate CdiI protein, but not the immunity proteins from other CDI systems (Aoki et al., 2010, Morse et al., 2012, Nikolakakis et al., 2012). Therefore, CDI toxin/immunity complexity provides the basis for self/non-self discrimination during inter-strain competition.
The CdiA-CT toxin region is typically demarcated by a short, highly conserved peptide sequence. Most CdiA-CT regions are defined by the VENN peptide motif, but CdiA proteins from Burkholderia species contain an analogous (Q/E)LYN sequence (Aoki et al., 2010, Zhang et al., 2011, Nikolakakis et al., 2012, Anderson et al., 2012). The function of this motif has not been explored, but it could play two important roles in CDI. First, the universally conserved Asn residue may catalyze auto-cleavage to release the CdiA-CT for subsequent translocation into target bacteria. Asn side-chains can undergo intra-molecular attack on the peptide backbone, thereby cleaving the peptide chain and producing a cyclic succinimide. Asn residue cyclization is a well characterized reaction that mediates auto-cleavage in several secreted proteins (Dautin et al., 2007, Zarivach et al., 2008). Second, the nucleotide sequence encoding VENN may be important for recombination and subsequent expression of new cdiA-CT/cdiI gene pairs acquired by horizontal transfer. CDI toxins are modular and can be readily exchanged between systems. For example, E. coli CdiA proteins can deliver heterologous toxins from Yersinia pestis CO92 and Dickeya dadantii 3937, provided the CdiA-CTs are fused at the common VENN motif (Aoki et al., 2010, Webb et al., 2013). Thus, bacteria might switch toxin/immunity types by recombining a new cdiA-CT/cdiI sequence onto the 3′-end of the cdiA gene, thereby displacing the original toxin/immunity pair. Moreover, many species carry fragmented cdiA-CT/cdiI gene pairs in tandem arrays downstream of the main cdiBAI gene cluster (Poole et al., 2011). These “orphan” toxin/immunity regions usually contain interspersed sequences that are homologous to transposases and integrases, implying recent acquisition through horizontal gene transfer (Poole et al., 2011, Ruhe et al., 2013a). Taken together, these observations suggest that CdiA proteins have the intrinsic ability to auto-cleave and deliver diverse toxins across the target-cell envelope.
Although CDI toxins are hypothesized to translocate autonomously, CdiAEC93 fragments released by E. coli EC93 cells do not inhibit the growth of other E. coli strains (Aoki et al., 2005). One possible explanation is that only specific CdiA-CT fragments – presumably those released in response to receptor-binding – are able to enter target bacteria. This model suggests that purified toxin may be active against bacteria if it mimics the naturally cleaved CdiA-CT fragment. Therefore, we tested whether purified CdiA-CT toxins inhibit the growth of bacterial cultures. We find that purified CdiA-CT536 (from UPEC 536) inhibits E. coli cell growth, whereas CdiA-CT3937 (from D. dadantii 3937) has no effect. Structure-function analyses show that the N-terminal domain of CdiA-CT536 is necessary and sufficient for cell import, but surprisingly toxin translocation does not require BamA. Instead, conjugative F pili are required for the import of purified CdiA-CT536 toxin. We provide evidence that CdiA-CT536 binds to F pilin and hypothesize that this interaction allows the toxin to be internalized during pilus retraction. F-dependent RNA phages exploit a similar pathway to transport their genomes into cells, suggesting that CdiA-CT536 mimics RNA phage to enter F+ cells.
Results
Purified CdiA-CT536 inhibits E. coli growth
To test whether CdiA-CT toxins enter bacteria in the absence of cell-to-cell contact, we treated E. coli cultures with purified toxins from Dickeya dadantii 3937 (CdiA-CT3937) and uropathogenic E. coli 536 (CdiA-CT536). Each toxin was first purified as a complex with its cognate His6-tagged CdiI protein and then separated from the immunity protein under denaturing conditions. CdiA-CT536 and CdiA-CT3937 refold efficiently and regain nuclease activity when denaturant is removed by dialysis (Aoki et al., 2010, Diner et al., 2012). We added each toxin (100 nM final concentration) to E. coli X90 cultures and monitored cell growth. Purified CdiA-CT3937 had no discernable effect on growth, but cells treated with CdiA-CT536 were inhibited compared to the buffer-treated control (Fig. 1A). We also tested the CdiA-CT/CdiI536 complex, and somewhat surprisingly found that cells treated with the toxin/immunity complex were inhibited to the same extent as cells treated with CdiA-CT536 toxin alone (Fig. 1A). This result suggests that only the toxin enters cells and immunity protein remains in the media. Alternatively, the CdiA-CT/CdiI536 complex might inhibit growth by a novel mechanism that is independent of the toxin’s tRNase activity. To differentiate between these possibilities, we added purified CdiA-CT536 to cells that express plasmid-borne immunity genes. Cells expressing cdiI536 were not inhibited by added CdiA-CT536 toxin (Fig. 1B), indicating that immunity protein is protective when present in the cytoplasm. This protection is specific, because cells expressing non-cognate cdiI3937 were inhibited to the same extent as cells that carry no immunity gene (Fig. 1B). To confirm that purified CdiA-CT536 inhibits growth through its anticodon nuclease activity, we isolated RNA from the toxin-treated cultures and examined transfer RNA by northern blot. This analysis revealed cleaved tRNAICGArg in all CdiA-CT536-treated cells except those expressing the cognate cdiI536 immunity gene (Fig. 1C). Together, these results show that exogenous CdiA-CT536 translocates into E. coli cells, where it cleaves tRNA to inhibit growth.
Figure 1. Purified CdiA-CT536 inhibits E. coli cell growth.

A) Purified CdiA-CT (CT) or CdiA-CT/CdiI (CT/CdiI) complex was added to E. coli X90 cultures at 30 min (indicated by the arrow) and cell growth monitored by measuring the optical density at 600 nm (OD600). B) E. coli X90 cells carrying plasmid-borne cdiI536 or cdiI3937 immunity genes were treated with purified CdiA-CT536 where indicated the arrow. C) Northern blot analysis of CdiA-CT536 treated cells. Total RNA was isolated from the cells in panel B and tRNAICGArg analyzed by northern blot. The migration positions of full-length and cleaved tRNAICGArg are indicated.
The N-terminal domain of CdiA-CT536 is required for cell import
Like many CdiA-CTs, CdiA-CT536 is composed of two domains (Diner et al., 2012, Morse et al., 2012). The tRNase activity of CdiA-CT536 is contained within the C-terminal 145 residues (Diner et al., 2012), which corresponds to Gly3098 – Ile3242 of full-length CdiA536 (Fig. 2A). However, E. coli cells were not inhibited when treated with the purified tRNase domain (Fig. 2B). This result suggests that the N-terminal domain of CdiA-CT536 is required for cell import; or alternatively, the C-terminal tRNase domain may be insufficient to inhibit cell growth. To test inhibition activity, we constructed a plasmid that produces the tRNase domain under control of the arabinose-inducible PBAD promoter and asked whether this construct could be introduced into E. coli cells in the presence of L-arabinose. As a control, we also transformed the plasmid into E. coli ΔcysK cells, because CysK is required to activate the CdiA-CT536 nuclease (Diner et al., 2012). The construct expressing the tRNase domain yielded no stable transformants in the cysK+ background, but was readily introduced into ΔcysK cells (Fig. 3). Furthermore, cysK+ cells were transformed at high-efficiency with either the empty vector plasmid (pCH450) or a construct encoding CdiA-CT536 with the His178Ala mutation that abolishes tRNase activity (Diner et al., 2012) (Fig. 3). Together, these results demonstrate that the C-terminal tRNase domain is toxic when expressed inside cells and suggest that the N-terminal domain plays a role in cell import.
Figure 2. The N-terminal domain of CdiA-CT536 is required for import.

A) CdiA-CT536 constructs. Predicted domain organization for the C-terminal region of CdiA536 is depicted with the corresponding residue numbers. Residues Val3016 – Asn3019 comprise the VENN peptide motif. DUF638 is a domain of unknown function corresponding to Pfam PF04829. B) E. coli X90 cells were treated with the indicated CdiA-CT536 fragments and cell growth monitored by OD600 measurements.
Figure 3. CdiA-CT536 toxicity.

Arabinose-inducible cdiA-CT536 expression plasmids were introduced into E. coli X90 and transformants selected on LB-agar supplemented with tetracycline and L-arabinose. CdiA-CT536 requires activation by CysK, therefore E. coli ΔcysK cells were used to control for transformation efficiency. Construct nomenclature corresponds to that introduced in Fig. 2A. The His178Ala mutation ablates tRNase activity and plasmid pCH450 is the empty vector.
The VENN peptide motif is predicted to be part of a larger domain of unknown function, termed DUF638, which is found in many CdiA proteins (Aoki et al., 2010). Given the conservation of the VENN motif, we asked whether this sequence is required for CdiA-CT536 import. Cells treated with CdiA-CT536 lacking this sequence (VENN-less, residues Leu3023 – Ile3242) were inhibited to the same extent as cells treated with the full CdiA-CT536 containing residues Val3016 – Ile3242 (Fig. 2A & 2B). Surprisingly, a larger fragment (containing residues Gly2969 – Ile3242) that includes the entire DUF638 region had no effect on cell growth (Figs. 2A & 2B), even though this protein is toxic when produced inside cysK+ cells (Fig. 3). Although CdiA-CT sequences are diverse, their N-terminal domains often contain paired cysteine residues that presumably form disulfide linkages. We reasoned that this predicted disulfide bond could be critical for toxin import, and therefore mutated Cys3028 and Cys3034 to serine residues to generate a “Cys-less” toxin (Fig. 2A). Cells treated with the Cys-less CdiA-CT536 were not inhibited (Fig. 2B), yet this toxin variant still inhibited growth when produced inside E. coli cells (Fig. 3). The preceding experiments results suggest a role for N-terminal domain of CdiA-CT536 in cell import. However, it also possible that some of the toxin variants are unstable in the growth medium and do not inhibit cell growth because they are rapidly degraded. To address this possibility, we monitored CdiA-CT536 antigen using immunoblot analysis, which revealed that each toxin was stable in shaking-broth cultures for up to five hours at 37 °C (Fig. S1). Together, these data indicate that the N-terminal domain of CdiA-CT536 is required for import. However, the DUF368 region appears to block CdiA-CT536 translocation, perhaps by interacting with the N-terminal region and masking cell-import epitopes.
The N-terminal domain of CdiA-CT536 shares significant sequence identity with the corresponding region of CdiAECL from Enterobacter cloacae ATCC 13047 (ECL), but the C-terminal nuclease domains are not related in sequence (Fig. 4A). Moreover, the C-terminal domain of CdiA-CTECL has a distinct RNase activity that cleaves 16S ribosomal RNA (Beck et al., 2014). We reasoned that if the N-terminus of CdiA-CT536 mediates cell import, then purified CdiA-CTECL (corresponding to residues Ala3087 – Asp3321) should also inhibit E. coli growth. We found that the purified CdiA-CT/CdiIECL complex had no effect on E. coli X90 cells (data not shown), but the isolated CdiA-CTECL toxin inhibited cell growth (Fig. 4B). This result may indicate that CdiIECL does not dissociate from CdiA-CTECL during translocation, or alternatively the bound immunity protein may prevent toxin import altogether. Regardless, the purified CdiA-CTECL toxin entered cells because the cdiIECL immunity gene prevented growth inhibition, whereas non-cognate cdiI536 provided no protection (Fig. 4B). Thus, the homologous N-terminal domains of CdiA-CT536 and CdiA-CTECL appear to function similarly in cell import.
Figure 4. CdiA-CTECL from Enterobacter cloacae inhibits E. coli cell growth.
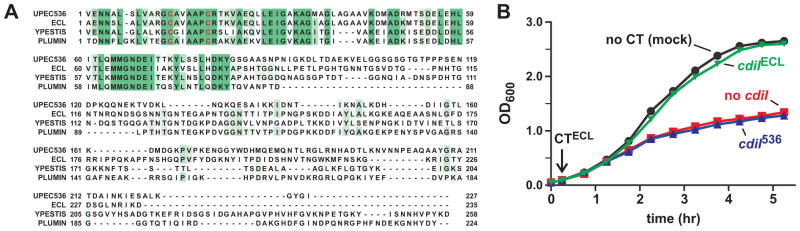
A) Alignment of the CdiA-CT regions from UPEC 536 (Uniprot: Q0T963), Enterobacter cloacae ATCC 13047 (D5CBA0), Yersinia pestis 91001 (Q74T84) and Photorhabdus luminescens TT01 (Q7MB60). Proteins were aligned using Clustal-W and rendered with Jalview 2.8 (Waterhouse et al., 2009) at 30% sequence identity. The conserved cysteine residues within the N-terminal domain are depicted in red. B) Purified CdiA-CTECL was added to E. coli X90 cultures when indicated (downward arrow) and cell growth monitored by OD600 measurements. Where indicated, cells carried plasmid-borne cdiI536 (cdiI536) or cdiIECL immunity genes.
The N-terminal domain of CdiA-CT536 is sufficient for cell import
The shared N-terminal domain of CdiA-CT536 and CdiA-CTECL is required for toxin import, suggesting that it carries tethered nuclease domains into the cell. We tested this hypothesis by asking whether the N-terminal domain still supports translocation when fused to a heterologous passenger domain. We fused residues Val1 – Tyr82 of CdiA-CT536 to the C-terminal nuclease domain of colicin E5 (ColE5-CT, residues Ala451 – Gln556) (Fig. 5A). We chose the ColE5-CT domain as a passenger because it is similar in size and activity to the CdiA-CT536 tRNase domain (Fig. 5A) (Ogawa et al., 1999, Lin et al., 2005). Although full-length colicin E5 enters and kills E. coli cells (Ogawa et al., 1999), the isolated ColE5-CT domain has no inhibition activity because it lacks receptor-binding and translocation domains (Figs. 5A & 5B). We fused the N-terminal domain of CdiA-CT536 to the ColE5-CT nuclease domain and tested the purified fusion protein on E. coli cultures. The fusion had weak activity against E. coli X90 cells (data not shown), but inhibited E. coli XL-1 cells more profoundly (Fig. 5B). This growth inhibition was blocked when cells express the imE5 gene (Fig. 5B), which encodes an immunity protein that specifically neutralizes ColE5-CT activity (Ogawa et al., 1999, Lin et al., 2005). Because ColE5-CT cleaves the anticodon loops of tRNAHis, tRNATyr, tRNAAsp and tRNAAsn molecules (Ogawa et al., 1999), we examined tRNATyr by northern blot to detect nuclease activity. Cleaved tRNATyr was detected in treated cells that either lack immunity or express cdiI536 (Fig. 5C). By contrast, there was less cleaved tRNATyr in cells that express imE5 (Fig. 5C), consistent with the ability of ImE5 to neutralize ColE5-CT activity. Together, these results indicate that the N-terminal domain of CdiA-CT536 is sufficient to translocate the colicin E5 nuclease domain into E. coli cells.
Figure 5. The N-terminal domain of CdiA-CT536 is sufficient for import.

A) Colicin E5 is composed of translocation, receptor-binding and nuclease domains. The C-terminal domain (ColE5-CT) has anticodon nuclease activity. The NT536-CTE5 protein contains residues Val3016 – Gly3097 of CdiA536 fused to the ColE5-CT nuclease domain. B) E. coli XL-1 cells were treated with purified ColE5-CT or NT536-CTE5 fusion at 30 min (indicated by the arrow) and growth monitored by OD600 measurements. Where indicated, cells carried plasmid-borne cdiI536 (cdiI536) or imE5 immunity genes. C) Northern blot analysis of toxin-treated cells. Total RNA was isolated from the cells in panel B and tRNATyr analyzed by northern blot. The migration positions of full-length and cleaved tRNATyr are indicated.
CdiA-CT536 import is independent of BamA, Tol and Ton translocation pathways
Cell mediated CDI requires specific receptors on target cells. Therefore, we asked whether CdiA-CT536 import requires the BamA receptor using bamA101 mutant cells, which have approximately five-fold less BamA on the cell surface and are significantly resistant to cell-mediated CDI (Aoki et al., 2008, Webb et al., 2013). E. coli bamA101 cells were inhibited by purified CdiA-CT536 to the same extent as bamA+ cells (Fig. 6A), suggesting the toxin is not imported through the usual CDI pathway. This result led us to consider other mechanisms for CdiA-CT536 import. Colicins exploit either the Tol or Ton systems to translocate their toxin domains into E. coli cells (Cascales et al., 2007). Therefore, we generated X90 ΔtolA and ΔtonB strains and tested these cells for resistance to group A (colicin E5) and group B (colicin D) colicins. As expected from previous studies, we found that ΔtolA cells were completely resistant to colicin E5, but inhibited by colicin D (Fig. S2). Reciprocally, ΔtonB cells were resistant to colicin D and inhibited by colicin E5 (Fig. S2). Moreover, X90 ΔtolA cells were resistant to phage M13, but not phage R17 (Table 1), consistent with the established phenotype of this mutant. We then treated the ΔtolA and ΔtonB strains with purified CdiA-CT536 and found that the mutants were as sensitive as tolA+ tonB+ cells (Figs. 6B & 6C). We also tested a ΔtolA ΔtonB strain to test for possible redundancy and found that the double mutant was also inhibited by CdiA-CT536, though it grew to slightly higher density than treated tolA+ tonB+ cells (Fig. 6D). Together, these findings suggest that purified CdiA-CT536 toxin enters E. coli cells through a novel pathway.
Figure 6. CdiA-CT536 is not translocated via known toxin-import pathways.

Wild-type E. coli X90 and the indicated mutants were treated with purified CdiA-CT536 where indicated (+CT), and cell growth was monitored by OD600 measurements. A) E. coli X90 (bamA+) and bamA101 mutants. B) E. coli X90 ΔtolA mutants. C) E. coli X90 ΔtonB mutants. D) E. coli X90 ΔtolA ΔtonB mutants.
Table 1.
Bacteriophage plating efficiencya
| Bacteriophage | ||
|---|---|---|
| Bacterial strain | R17 | M13 |
| X90 ΔtolA | 0.93 ± 0.12 | 0.0b |
| X90 ΔtonB | 1.2 ± 0.26 | 1.0 ± 0.20 |
| X90 ΔtolA ΔtonB | 1.1 ± 0.15 | 0.0b |
| X90 ΔtraA::cat pTrc | 0.0b | 0.0b |
| X90 ΔtraA::cat pTrc::traA | 1.2 ± 0.18 | 1.2 ± 0.15 |
| X90 ΔtraA::cat pTrc::traA(D74G) | 0.0b | 1.1 ± 0.09 |
| X90 ΔtraA::cat pTrc::traA(G120C) | 0.09 ± 0.005c | 0.77 ± 0.06c |
| X90 ΔtrbI::cat | 0.1 ± 0.04c | 0.1 ± 0.03c |
The indicated bacterial strains were infected with 10 and 100 plaque forming units (pfu). The number of plaques for each strain was divided by the plaques obtained with wild-type X90 cells. The mean plating efficiency ± SEM is reported (n = 4).
No plaques were detected when bacteria were incubated with 103 pfu of phage.
Plaques were turbid.
CdiA-CT536 import requires the F pilus
Although E. coli X90 and XL-1 strains are inhibited by purified CdiA-CT536, E. coli strain MC4100 is resistant to the toxin (data not shown). There are several genetic differences between these strains, but both sensitive strains carry F′ episomes whereas the resistant stain does not. Therefore, we tested E. coli X90 cells that had been cured of F′ and found that this isolate is resistant to purified CdiA-CT536 (Fig. 7A). Reintroduction of a different F′ episome (from E. coli XL-1) into the cured X90 cells restored toxin sensitivity (Fig. 7A). Moreover, E. coli MC4100 cells that carry F′ were also sensitive to purified CdiA-CT536 (data not shown). Together, these results indicate that one or more genes on F are required for CdiA-CT536 import. As a first step to identify F-encoded import gene(s), we tested whether cells carrying a subset of F genes are sensitive to CdiA-CT536. Plasmid pOX38::gent is derivative of F that contains the tra genes required for conjugation and a selectable gentamicin-resistance (gent) marker (Guyer et al., 1981, Johnson & Reznikoff, 1984). We introduced pOX38::gent into E. coli X90 F− and found that the resulting cells were sensitive to CdiA-CT536 (Fig. 7A), suggesting that tra gene(s) could mediate toxin import. We took a candidate approach and disrupted two genes, traA and traT, that encode abundant cell-surface proteins. The traA gene encodes F pilin, which polymerizes to form the conjugative pilus; and traT encodes an outer membrane β-barrel protein that functions to prevent mating between F+ cells (Achtman et al., 1977, Manning et al., 1980). The ΔtraT mutant was still inhibited by CdiA-CT536, but ΔtraA cells were completely resistant to purified toxin (Fig. 7B). To exclude possible polar effects from the ΔtraA mutation, we complemented the mutant with plasmid-borne traA and restored sensitivity to the toxin (Fig. 7C). These results suggest that the F pilus is required for the import of CdiA-CT536 toxin.
Figure 7. The F pilus is required for CdiA-CT536 import.

E. coli X90 and its derivatives were treated with purified CdiA-CT536 where indicated (+CT), and cell growth was monitored by OD600 measurements. A) Wild-type X90 is indicated as F′ and cured cells as F−. The F′::Tn10 episome and pOX38::gent (pOX38) were introduced into cured X90 and the resulting cells treated with CdiA-CT536. B) E. coli X90 ΔtraA::cat and ΔtraT::cat mutants. C) E. coli X90 ΔtraA::cat cells complemented with plasmid-borne traA. D) and E) E. coli X90 ΔtraA::cat cells complemented with plasmid-borne traA(D74G) and traA(G120C). Error bars correspond to the standard error of the mean for three independent experiments. F) E. coli X90 ΔtrbI::cat mutant. Wild-type (trbI+) cells expressing trbI from a plasmid (pTrbI) were also tested for toxin-resistance.
CdiA-CT536 blocks F-mediated conjugation
We hypothesized that the N-terminal domain of CdiA-CT536 binds directly to F pili and exploits the organelle to enter cells. This import pathway could be analogous to that used by some F-dependent bacteriophages to infect E. coli F+ cells. Therefore, we examined traA mutations that confer resistance to phage R17, reasoning that these alleles may also protect cells from purified CdiA-CT536. We focused on the Asp74Gly and Gly120Cys mutations, because they produce functional conjugative pili, but interfere with phage R17 binding (Manchak et al., 2002, Daehnel et al., 2005). We introduced each mutation into plasmid-borne traA and used the constructs to complement X90 ΔtraA::cat mutants. Each pilin variant supported conjugation, though the mating efficiency was somewhat reduced by the Asp74Gly mutation (Table 2). The mutant pilins also provided resistance to phage R17, but not phage M13 (Table 1). Cells expressing the Asp74Gly pilin were still inhibited by purified CdiA-CT536, but the Gly120Cys mutation provided partial protection against the toxin (Figs. 7D & 7E). These results suggest that CdiA-CT536 binds F pili at a site that overlaps with the phage R17 binding site.
Table 2.
Mating efficiencya
| F+ donor strain | Percent exconjugants (%) |
|---|---|
| CH11717 | 79 ± 2.8 |
| CH11717 ΔtraA::cat | 0.0 |
| CH11717 ΔtraA::cat pTrc | 0.0 |
| CH11717 ΔtraA::cat pTrc::traA | 75 ± 5.4 |
| CH11717 ΔtraA::cat pTrc::traA(D74G) | 58 ± 5.7 |
| CH11717 ΔtraA::cat pTrc::traA(G120C) | 72 ± 0.5 |
| CH11717 ΔtrbI::cat | 13 ± 3.6 |
Donor strains were derived from strain CH11717, which is X90 transduced with the Tn10 marker from E. coli XL-1. Donors were cultured at a 1:10 ratio with StrR recipient cells as described in Methods. Mating efficiency was determined by dividing the number of TetR StrR colonies by the number of total TetR colonies and expressed as a percent. The mean plating efficiency ± SEM is reported for three independent experiments.
Because F-dependent bacteriophage bind directly to F pili, they can interfere with conjugation (Lin et al., 2011). We reasoned that if CdiA-CT536 binds directly to the F pilus, then it may also reduce mating efficiency. We first tested the effects of M13 and R17 phages on mating efficiency. The phages were inactivated with ultraviolet radiation (to prevent infection and cell lysis), then added to conjugation co-cultures. The phage particles reduced mating efficiency to ~50% from about 88% for mock-treated cultures (Fig. 8). We next tested purified CdiA-CT536 carrying the His178Ala mutation so that the F+ donor cells were not inhibited during culture. Purified CdiA-CT536 at 1 μM reduced mating efficiency approximately 2.5-fold compared to mock-treated cells (Fig. 8). By contrast, the purified tRNase domain (which lacks the N-terminal domain of CdiA-CT536) had little effect on mating efficiency (Fig. 8). Similarly, the Cys-less and DUF638-containing toxins (see Fig. 2A) also had no substantive effect on conjugation (Fig. 8). These observations suggest that the tRNase domain and the latter CdiA-CT536 variants do not inhibit cell growth because they do not bind to F pili. Taken together with the protective effect of the traA(G120C) mutation, these results support a model in which the N-terminal domain of CdiA-CT536 binds to the F pilus.
Figure 8. Purified CdiA-CT536 interferes with F-mediated conjugation.

The efficiency of conjugation was determined using the F′::Tn10 episome as described in methods. Cell suspensions were treated with UV-inactivated phage particles or the indicated CdiA-CT536 constructs (see Fig. 2A). The CdiA-CT536 construct contains the His178Ala mutation to ablate tRNase activity.
trbI mutants are resistance to F-dependent bacteriophage and CdiA-CT536
F pili are dynamic and undergo cycles of extension and retraction (Clarke et al., 2008). Pilus retraction is critical for F-dependent phage infection and provides a possible mechanism to internalize CdiA-CT536 toxin. The trbI gene is thought to be required for pilus retraction (Maneewannakul et al., 1992), so we generated an in-frame trbI deletion strain for analysis. We found that ΔtrbI cells are resistant to both R17 and M13 phage (Table 1), but still capable of conjugation, albeit at lower efficiency than wild-type cells (Table 2). These observations indicate that the ΔtrbI mutant produces functional F pili as reported previously (Maneewannakul et al., 1992). We next treated ΔtrbI cells with purified CdiA-CT536 and found that the mutant was fully resistant to the toxin (Fig. 7F). We attempted to complement the mutant with plasmid-borne trbI, but were unable to do so with both IPTG- and arabinose-inducible trbI constructs (data not shown). We note that Maneewannakul et al. could not complement trbI mutants with multi-copy plasmids. Moreover, they reported that trbI over-expression in wild-type cells phenocopies the trbI null mutation (Maneewannakul et al., 1992). We also found that trbI over-expression in the trbI+ background conferred resistance to CdiA-CT536, similar to the ΔtrbI phenotype (Fig. 7F). Therefore, pili dynamics appear to be adversely affected by under- and over-expression of trbI (Maneewannakul et al., 1992). These results are consistent with a model in which pilus-bound toxin is internalized during retraction.
Discussion
The experiments presented here reveal an unexpected import pathway for CDI toxins. Purified CdiA-CT536 and CdiA-CTECL enter E. coli F+ cells and inhibit growth. These toxins inhibit growth using different C-terminal nuclease domains, but they share a common N-terminal domain. The N-terminal domain is not necessary for nuclease activity in vitro (Diner et al., 2012), but is required for toxin translocation into F+ cells. These results indicate that the N-terminal domain has autonomous import activity and carries tethered passenger domains into the cell. Indeed, the translocation domain is capable of transporting a heterologous nuclease domain from colicin E5 into F+ cells. Several observations suggest that toxin import requires a binding interaction between the N-terminal domain and F pili. First, purified CdiA-CT536 interferes with mating when added to conjugation co-cultures. The same phenomenon is observed with phage R17 and M13 particles, both of which bind directly to F pili. Presumably, the binding of either phage or toxin to F pili disrupts the formation of conjugation bridges. This effect is specific because the C-terminal tRNase domain of CdiA-CT536 has little to no effect on mating efficiency, and mutations within the N-terminal domain that block cell-import also abrogate the toxin’s effect on conjugation. Furthermore, cells expressing F pilin with the Gly120Cys mutation are significantly resistant to purified CdiA-CT536. This mutation also disrupts the interaction between F pili and phage R17 (Daehnel et al., 2005), suggesting that phage and CdiA-CT536 share overlapping binding sites. Taken together, these results indicate that CdiA-CT536 and CdiA-CTECL contain a pilus-binding domain that facilitates entry into F+ cells.
F pili are dynamic structures that extend in search of potential mating partners and retract to bring donor and recipient cells together to establish conjugation bridges (Novotny & Fives-Taylor, 1974, Clarke et al., 2008). This cycle of extension and retraction also provides a possible mechanism to import CdiA-CT536 toxin across the cell envelope. Retraction is driven by pilus disassembly into pilin monomers within the inner membrane (Moore et al., 1981). Therefore, pilus-bound CdiA-CT536 could be carried directly to the inner membrane during retraction. If this model is correct, then the toxin must remain stably associated with the pilus as it retracts through the lumen of the type IV secretion assembly. Type IV secretion assemblies are large, barrel-shaped structures that span the entire cell envelope (Chandran et al., 2009, Waksman & Fronzes, 2010). The F-pilus assembly is composed of a hetero-oligomeric complex of TraV, TraK and TraB, with a tetradecameric ring of TraV forming an aperture in the outer membrane through which the pilus emerges (Silverman & Clarke, 2010). Presumably, TraV forms a tight seal around the pilus to prevent the loss of periplasmic contents to the media. Therefore, the associated CdiA-CT536 must pass through the TraV aperture and not be stripped from the pilus surface. Pilus retraction is also critical for F-dependent phage infection, and a number of phage-resistance mutations appear to disrupt pilus dynamics (Burke et al., 1979, Willetts et al., 1980). These resistance mutations are unmapped, and to our knowledge, trbI is the only F gene to be specifically linked to defects in pilus retraction (Maneewannakul et al., 1992). As predicted by the retraction-import model, in-frame deletion of trbI protects F+ cells from CdiA-CT536, just as it provides resistance to F-dependent phages (Maneewannakul et al., 1992). However, we note that pili dynamics have not been directly examined in ΔtrbI mutants. The retraction-import model would be strengthened if ΔtrbI retraction defects could be confirmed through real-time video microscopy as described by Silverman and colleagues (Clarke et al., 2008).
As outlined above, there are parallels between CdiA-CT536 import and infection by F-dependent phages. Two bacteriophage families exploit conjugative pili as host-cell receptors. The Inoviridae are filamentous, single-strand DNA viruses (e.g. phages fd and M13) that bind to the tip of the pilus using the g3p capsid protein (Daehnel et al., 2005, Manchak et al., 2002). These phages also require TolA as a co-receptor (Riechmann & Holliger, 1997). However, CdiA-CT536 import does not require TolA, suggesting that the toxin is internalized through another pathway. Moreover, CdiA-CT536 appears to bind F pili in a manner similar to phage R17 and other leviviruses. The Leviviridae are icosahedral, single-strand RNA viruses that attach to the side of conjugative pili using a single copy of the maturation (or assembly) protein present in the phage capsid (Roberts & Steitz, 1967, Steitz, 1968, Dent et al., 2013). Upon binding the pilus, the maturation protein is released from the capsid and proteolytically processed into two fragments (Krahn et al., 1972, Paranchych et al., 1971, Paranchych et al., 1970). These peptide fragments remain associated with the phage genome and are internalized with viral RNA during infection (Oriel, 1969, Osborn et al., 1970, Krahn et al., 1972). The internalization process also requires TraD motor function and other plasmid-transfer initiation proteins (Lang et al., 2014, Lang et al., 2011, Schoulaker & Engelberg-Kulka, 1978, Willetts & Achtman, 1972). Together, these observations suggest that the maturation protein guides the tethered genome into the cytoplasm. A similar mechanism may underlie CdiA-CT536 import into F+ cells. Although the N-terminal domain of CdiA-CT536 lacks homology with known maturation proteins, we note that these proteins are quite diverse between different phage (Rumnieks & Tars, 2012, Friedman et al., 2009). For example, the maturation proteins from MS2, GA and Qβ phages are only 26 – 49% identical to one another, yet are all thought to bind along the shafts of F pili. Given this sequence diversity, it seems reasonable to posit that the N-terminal domain of CdiA-CT536 mimics a leviviral maturation protein.
In principle, pilus-mediated import provides an additional mechanism to deliver CDI toxins into target bacteria. However, it is not clear that this mode of delivery occurs naturally. We have previously reported that E. coli EC93 cells release CdiA fragments into the extracellular milieu, but these fragments are not inhibitory (Aoki et al., 2005). Similarly, CdiA536 fragments from UPEC 536 culture supernatants do not inhibit the growth of F+ cells (C.M.B & C.S.H., unpublished data). Another possibility is that the binding of CdiA536 to F pili augments cell-cell interactions to potentiate CDI. We have found that F+ cells are generally better CDI targets than F− cells, but this is also true for CdiA effector proteins that lack the pilus-interaction domain (C.M.B & C.S.H, unpublished data). Thus, there is no evidence that full-length CdiA536 on the surface of inhibitor cells binds to F pili on target cells. If the interaction with F pili plays no role in cell-mediated CDI, then what is the biological significance of these findings? We hypothesize that these phenomena reflect a critical function for the N-terminal domain in transport across the target-cell inner membrane during CDI. In this model, the unusual pilus-binding activity of CdiA-CT536 allows the toxin to by-pass the CDI receptor and gain entry into the periplasm. Once in the periplasm, the toxin is predicted to resume its normal translocation pathway, using the N-terminal domain to cross the inner membrane and enter the cytoplasm. Though this model has not been tested directly, there is evidence that the N-terminal domain of CdiA-CT536 is critical for CDI. Mutation of residues Cys3028/Cys3034 within full-length CdiA536 abrogates CDI, but still allows transfer of CdiA-CT536 toxin antigen to the surface of target bacteria (Julia S. Webb & D.A.L., unpublished data). These observations indicate that CDI is disrupted at a later stage in the pathway, as expected for a defect in toxin translocation. If this model is correct, then our results suggest that CDI toxins and leviviral genomes may use the same basic mechanism to cross the inner membrane of bacteria.
Experimental Procedures
Bacterial strains and growth conditions
Bacterial strains used in this study are listed in Table 3. E. coli cells were grown in LB medium supplemented with antibiotics at the following concentrations: ampicillin (Amp), 150 μg mL−1; chloramphenicol (Cm), 33 μg mL−1; kanamycin (Kan), 50 μg mL−1; rifampicin (Rif), 250 μg mL−1; streptomycin (Str), 50 μg mL−1; and tetracycline (Tet), 10 μg mL−1. Bacteria were grown in baffled flasks at 37 °C with shaking (215 rpm) unless otherwise indicated. The F− derivative of E. coli X90 (strain KW1070) was a generous gift from Dr. Kelly P. Williams. The ΔtolA::kan and ΔtonB::kan gene disruptions were obtained from the Keio collection (Baba et al., 2006) and introduced into E. coli X90 by bacteriophage P1-mediated transduction. The ΔtraA::cat, ΔtraT::cat and ΔtrbI::cat disruptions were generated by phage λ Red-mediated recombination as described (Datsenko & Wanner, 2000). The chloramphencol acetyltransferase (cat) open reading frame was amplified with oligonucleotides (listed in Table S1) containing homology to regions flanking traA (traA-cat-for/traA-cat-rev) and traT (traT-cat-for/traT-cat-rev). A similar procedure was used for the trbI gene, but the cat gene was amplified by two sequential reactions with trbI-cat-for1/trbI-cat-rev1 and trbI-cat-for2/trbI-cat-rev2 primer pairs. The resulting PCR products were electroporated into E. coli X90 cells expressing the Red proteins from plasmid pSIM6 (Datta et al., 2006). The F′::Tn10 (from strain XL-1) and pOX38::gent plasmids were transferred into E. coli strain KW1070 by conjugation. Donor and recipient cells were mixed and spotted onto LB-agar for 4 hr at 37 °C. Exconjugants were selected on LB-agar supplemented with: Tet/Rif for F′::Tn10 transfer into E. coli KW1070, Gent/Rif for pOX38::gent transfer into KW1070, and Tet/Str for F′::Tn10 transfer into E. coli MC4100.
Table 3.
Bacterial strains and plasmids
| Strains or plasmids | Descriptiona | Reference |
|---|---|---|
| Strain | ||
| X90 | F′ lacIq lac′ pro′/araΔ(lac-pro) nal1 argE(amb) rifr thi-1, RifR | (Beckwith & Signer, 1966) |
| XL-1 | F′::Tn10 proA+B+ lacIq Δ(lacZ)M15/recA1 endA1 gyrA96 thi hsdR17 (rK− rK+) glnV44 relA1 lac. TetR, NalR | Stratagene |
| MC4100 | F− araD139 Δ(argF-lac)U169 rpsL150 relA1 deoC1 rbsR fthD5301 fruA25 λ−. StrR | (Casadaban, 1976) |
| KW1070 | X90 F− | Kelly P. Williams |
| CH2016 | X90 (DE3) Δrna ΔslyD::kan, KanR | (Garza-Sánchez et al., 2006) |
| CH6479 | X90 ΔtolA::kan, KanR | This study |
| CH6480 | X90 ΔtonB::kan, KanR | This study |
| CH6680 | X90 bamA101::kan, KanR | (Aoki et al., 2008) & this study |
| CH6866 | KW1070 pOX38::gent, GentR | This study |
| CH6939 | X90 ΔtraA::cat, CmR | This study |
| CH7035 | X90 ΔtraT::cat, CmR | This study |
| CH9027 | KW1070 F′::Tn10, TetR | This study |
| CH9354 | X90 ΔtonB tolA::kan, KanR | This study |
| CH11476 | X90 ΔtrbI::cat, CmR | This study |
| CH11703 | CH11717 ΔtrbI::cat, TetR CmR | This study |
| CH11717 | X90 Tn10, TetR | This study |
| CH11718 | CH11717 ΔtraA::cat, TetR CmR | This study |
| Plasmids | ||
| pBR322 | Cloning vector, AmpR TetR | (Bolivar et al., 1977) |
| pACYC184 | Cloning vector, CmR, TetR | (Chang & Cohen, 1978) |
| pTrc99A | IPTG-inducible expression vector, AmpR | GE Healthcare |
| pCH450 | pACYC184 derivative containing E. coli araC and the L-arabinose-inducible ParaBAD promoter, TetR | (Hayes & Sauer, 2003) |
| pET21::cdiA-CT/cdiI3937 | Over-produces CdiA-CT3937 and CdiI3937-His6, AmpR | (Aoki et al., 2010) |
| pET21::cdiA-CT/cdiI536 | Over-produces CdiA-CT536 and CdiI536-His6, AmpR | (Aoki et al., 2010) |
| pET21::DUF-CT/cdiI536 | Over-produces DUF638-CdiA-CT536 and CdiI536-His6, AmpR | This study |
| pET21::VENN-less-CT/cdiI536 | Over-produces VENNless-CdiA-CT536 and CdiI536-His6. AmpR | This study |
| pET21::Cys-less-CT/cdiI536 | Over-produces CdiA-CT536 containing Cys13Ser and Cys19Ser mutations together with CdiI536-His6, AmpR | This study |
| pET21::tRNase/cdiI536 | Over-produces the C-terminal tRNase domain of CdiA-CT536 together with CdiI536-His6, AmpR | (Diner et al., 2012) |
| pET21::cdiA-CT(H178A)/cdiI536 | Over-produces catalytically inactive His178Ala variant of CdiA-CT536 toxin and CdiI536-His6, AmpR | (Diner et al., 2012) |
| pET21K::cdiA-CT/cdiIECL | Over-produces CdiA-CTECL and CdiIECL-His6 from Enterobacter cloacae ATCC 13047, AmpR | This study |
| pCH450::cdiA-CT536 | Expresses cdiA-CT536 under control of PBAD promoter, TetR | This study |
| pCH450::DUF-CT536 | Expresses DUF638-cdiA-CT536 under control of PBAD promoter, TetR | This study |
| pCH450::VENN-less-CT536 | Produces VENNless-CdiA-CT536, TetR | This study |
| pCH450::Cys-less-CT536 | Produes CdiA-CT536 containing Cys13Ser and Cys19Ser mutations, TetR | This study |
| pCH450::tRNase536 | C-terminal tRNase domain of CdiA-CT536, TetR | This study |
| pCH450::cdiA-CT(H178A)536 | catalytically inactive His178Ala variant of CdiA-CT536, TetR | This study |
| pDAL776 | pBR322 derivative that constitutively expresses cdiI536, AmpR | (Aoki et al., 2010) |
| pDAL852 | pBR322 derivative that constitutively expresses cdiI3937 from Dickeya dadantii 3937, AmpR | (Aoki et al., 2010) |
| pOX38::gent | pOX38 with integrated gentamicin-resistance cassette, GentR | (Johnson & Reznikoff, 1984) |
| pTrc99A::cdiI536 | Expresses cdiI536 under control of the Ptrc promoter, AmpR | This study |
| pTrc99A::cdiIECL | Expresses cdiIECL from Enterobacter cloacae ATCC 13047 under control of the Ptrc promoter, AmpR | This study |
| pTrc99A::imE5 | Expresses imE5 under control of the Ptrc promoter, AmpR | This study |
| pET21::colE5-CT/imE5 | Over-produces ColE5-CT (residues Met429 - Gln556) and together with ImE5-His6, AmpR | This study |
| pET21::NT536-colE5-CT/imE5 | Over-produces a protein containing residues Val1 – Tyr82 of CdiA-CT536 fused to the nuclease domain of colicin E5 (colE5-CT) together with ImE5-His6, AmpR | This study |
| pTrc99A::traA | Expresses wild-type F-pilin under the control of the Ptrc promoter, AmpR | This study |
| pTrc99A::traA(D74G) | Expresses the Asp74Gly variant of F-pilin under control of the Ptrc promoter, AmpR | This study |
| pTrc99A::traA(G120C) | Expresses the Gly120Cys variant of F-pilin under control of the Ptrc promoter, AmpR | This study |
| pTrc99A::trbI | Expresses trbI under the control of the Ptrc promoter, AmpR | This study |
| pCH450::trbI | Expresses trbI under the control of the PBAD promoter, TetR | This study |
Abbreviations: AmpR, ampicillin resistant; CmR, chloramphenicol resistant; GentR, gentamicin resistant; KanR, kanamycin resistant; NalR, nalidixic acid resistant; RifR, rifampicin resistant; StrR, streptomycin resistant; TetR, tetracycline resistant
Plasmids
Plasmids used in this study are listed in Table 3. Plasmids pET21::cdiA-CT/cdiI536 and pET21::cdiA-CT/cdiI3937 were used to over-produce CdiA-CT/CdiI536-His6 and CdiA-CT/CdiI3937-His6 complexes (respectively) (Aoki et al., 2010). All other CdiA-CT/CdiI536-His6 over-expression constructs were generated by PCR and ligated into plasmid pET21P using NcoI/XhoI restriction sites. The DUF-CT, VENN-less and tRNase domain constructs were amplified using primers DUF-536-Nco-for, VENN-less-Nco-for and tRNase-Nco-for (respectively) in conjunction with primer 536-Xho-rev (Table S1). Residues Cys13 and Cys19 of CdiA-CT536 were mutated to serine by mega-primer PCR (Aiyar & Leis, 1993). A fragment of plasmid pET21::cdiA-CT/cdiI536 was amplified using primers pET-Sph/Pst and 536-Cys(mut). This product was purified and used as a mega-primer in a second PCR with primer 536-Xho-rev. The final product was digested with NcoI/XhoI and ligated to plasmid pET21P. The colE5-CT and imE5 coding sequences were amplified from plasmid ColE5-099 using primers colE5-M429-Nco and imE5-Spe, and the product digested with NcoI/SpeI and ligated to plasmid pET21P. The CdiA-CT/ColE5-CT fusion construct was generated by sequential ligation of cdiA-CT536 and colE5-imE5 fragments into plasmid pET21P. The colE5-CT/imE5 sequence was amplified with primers ColE5-Bam-for and ImE5-Xho-rev and ligated to plasmid pET21P. The coding sequence for Val1 – Tyr82 of CdiA-CT536 was amplified with primers 536-Nco-for and 536-Bam-rev and ligated to plasmid pET21::colE5-CT/imE5 using NcoI and BamHI restriction sites. The CdiA-CT/CdiIECL-His6 expression construct was generated in two steps. First, the T7 promoter region of pET21P was amplified with primers pET-Sph/Pst and pET-Kpn/Nco, and the fragment ligated to PstI/NcoI-digested pET21::cdiA-CT/cdiI3937. The resulting pET21K::cdiA-CT/cdiI3937 plasmid contains a unique KpnI site upstream of NcoI. The cdiA-CT/cdiIECL region was then amplified with ECL-CT-Kpn-for and ECL-cdiI-Nhe-rev and ligated to KpnI/SpeI-digested plasmid pET21K::cdiA-CT/cdiI3937.
The cdiI536, cdiIECL and imE5 immunity genes were amplified from pET21 expression plasmids using 536-cdiI-Eco-for, ECL-cdiI-Eco-for and imE5-Eco-for primers (respectively) in conjunction with the pET-Pst reverse primer. All PCR products were digested with EcoRI and PstI and ligated to plasmid pTrc99A. The traA coding sequence from the F plasmid was amplified with primers traA-Nco-for and traA-Spe-rev and the product ligated to NcoI/SpeI-digested plasmid pTrc99A::rhsIB (Poole et al., 2011). The Gly120Cys mutation in traA was generated by PCR using primers traA-Nco-for and traA-G120C-Spe. The Asp74Gly mutation in traA was generated by megaprimer PCR using primer traA-D74G in conjunction with the traA-Nco-for/traA-Spe-rev primer pair. The trbI gene was amplified with primers trbI-Kpn-for and trbI-Xho-rev and the product ligated to KpnI/XhoI-digested pTrc99KX and pCH450K to generate trbI expression constructs for complementation analysis.
Protein purification
All CdiA-CT/CdiI-His6 complexes were over-produced in E. coli strain CH2016. Cultures were grown to OD600 ~ 0.7 and protein production induced with 1.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for three hr. Cells were harvested over ice and collected by centrifugation at 6,000 rpm for 10 min. Cells were resuspended in 10 mL of extraction buffer [20 mM sodium phosphate (pH 7.0), 150 mM sodium chloride, 0.05% Triton X-100, 10 mM β-mercaptoethanol (β-ME), 1 mM PMSF] and broken by french press passage at 20,000 psi. Cell lysates were clarified by two consecutive centrifugations at 14,000 rpm for 10 min (SS-34 rotor) and supernatants were incubated with Ni2+-nitrilotriacetic acid (Ni2+-NTA) resin for 1.5 hr at 4 °C. The resin was washed with 20 mM sodium phosphate (pH 7.0), 150 mM sodium chloride, 0.05% Triton X-100, 10 mM β-ME, 20 mM imidazole and loaded onto a Poly-Prep column (Bio-Rad) at 4 °C. CdiA-CT/CdiI-His6 complexes were eluted from the resin with native elution buffer [20 mM sodium phosphate (pH 7.0), 150 mM sodium chloride, 10 mM β-ME, 250 mM imidazole], followed by dialysis into storage buffer [20 mM sodium phosphate (pH 7.0), 150 mM NaCl, 10 mM β-ME]. CdiA-CT toxins were isolated from CdiI-His6 immunity proteins by Ni2+-affinity chromatography under denaturing conditions [6 M guanidine-HCl, 20 mM sodium phosphate (pH 7.0), 10 mM β-ME] at room temperature. Purified CdiA-CT toxins were dialyzed against storage buffer and quantified by absorbance at 280 nm.
CdiA-CT inhibition assays
Mid-log phage cells were diluted to OD600 = 0.05 in fresh LB medium and incubated at 37 °C with shaking. After growth to OD600 = 0.1 – 0.15, purified CdiA-CT, ColE5-CT or CdiA-CT/CdiI complex was added to the culture at a final concentration of 100 nM. Cell growth was monitored by measuring OD600 every 30 min. Samples were collected periodically and total RNA isolated by guanidine isothiocyanate-phenol extraction as described (Garza-Sánchez et al., 2006). Northern blot analysis was performed as described (Hayes & Sauer, 2003) using 5′-radiolabeled oligonucleotide probes to tRNAICGArg and tRNAGUATyr (Table S1). Expression of plasmid-borne traA alleles was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) prior to the addition of purified CdiA-CT536. The stability of CdiA-CT536 in shaking broth cultures was determined by immunoblot analysis. Culture samples were removed at 0, 1, 3 and 5 hr and the media clarified by centrifugation at 14,000 μg for 10 min. The supernatants (10 μL) were run on SDS-polyacrylamide gels and blotted onto PVDF membrane. Proteins were detected using polyclonal antibodies raised against CdiA-CT536 (Webb et al., 2013).
Bacteriophage plating and mating efficiency
Bacteriophage resistance was determined by measuring the efficiency of plating. X90 and derivatives were incubated with 10 and 100 plaque forming units (pfu) and then plated in soft agar. The number of plaques was divided by the number of infecting particles. Phage-resistant strains were also challenged with 103 pfu, but no plaques were detected. The traA and trbI mutants were tested for mating efficiency under the same conditions as CdiA-CT inhibition assays. F′::Tn10 donor cells (TetR) were co-cultured with MC4100 recipient cells (StrR) at a 1:10 ratio in LB for 3 hr at 37 °C. Samples were collected and plated onto LB-agar supplemented with Tet to enumerate F′::Tn10 donor cells and Tet/Str quantify exconjugants. Mating efficiency was calculated by dividing the number of exconjugants by total donor cells and expressed as a percentage. The effect of purified CdiA-CT536 proteins on conjugation was determined using MC4100 F′::Tn10 donors and MC4100 pTrc recipients. CdiA-CT proteins were added at 1 μM final concentration to conjugation co-cultures and mating efficiency determined as described above. Phage were inactivated with UV irradiation (64 mJ m−2) in a Stratalinker. This dose resulted in a 106-fold reduction in R17 and M13 plaque-forming units (pfu). UV-inactivated phage (~105 particles mL−1) were added to conjugation co-cultures to determine the effect on mating efficiency.
Supplementary Material
Acknowledgments
We thank Beth Traxler for providing bacteriophages M13 and R17, and Brian Janssen for sequence analysis to determine the translation start site for colicin E5 (Genbank accession: KF925332). This work was supported by grants GM078634 (C.S.H.) and GM102318 (D.A.L & C.S.H.) from the National Institutes of Health.
References
- Achtman M, Kennedy N, Skurray R. Cell–cell interactions in conjugating Escherichia coli: role of traT protein in surface exclusion. Proc Natl Acad Sci U S A. 1977;74:5104–5108. doi: 10.1073/pnas.74.11.5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiyar A, Leis J. Modification of the megaprimer method of PCR mutagenesis: improved amplification of the final product. Biotechniques. 1993;14:366–369. [PubMed] [Google Scholar]
- Anderson MS, Garcia EC, Cotter PA. The Burkholderia bcpAIOB genes define unique classes of two-partner secretion and contact dependent growth inhibition systems. PLoS Genet. 2012;8:e1002877. doi: 10.1371/journal.pgen.1002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, Jones AM, Webb JS, Hayes CS, Cotter PA, Low DA. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature. 2010;468:439–442. doi: 10.1038/nature09490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki SK, Malinverni JC, Jacoby K, Thomas B, Pamma R, Trinh BN, Remers S, Webb J, Braaten BA, Silhavy TJ, Low DA. Contact-dependent growth inhibition requires the essential outer membrane protein BamA (YaeT) as the receptor and the inner membrane transport protein AcrB. Mol Microbiol. 2008;70:323–340. doi: 10.1111/j.1365-2958.2008.06404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. Contact-dependent inhibition of growth in Escherichia coli. Science. 2005;309:1245–1248. doi: 10.1126/science.1115109. [DOI] [PubMed] [Google Scholar]
- Aoki SK, Poole SJ, Hayes CS, Low DA. Toxin on a stick: modular CDI toxin delivery systems play roles in bacterial competition. Virulence. 2011;2:356–359. doi: 10.4161/viru.2.4.16463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki SK, Webb JS, Braaten BA, Low DA. Contact-dependent growth inhibition causes reversible metabolic downregulation in Escherichia coli. J Bacteriol. 2009;191:1777–1786. doi: 10.1128/JB.01437-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CM, Morse RP, Cunningham DA, Iniguez A, Low DA, Goulding CW, Hayes CS. CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure. 2014;22:707–718. doi: 10.1016/j.str.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwith JR, Signer ER. Transposition of the lac region of Escherichia coli. I. Inversion of the lac operon and transduction of lac by phi80. J Mol Biol. 1966;19:254–265. doi: 10.1016/s0022-2836(66)80003-7. [DOI] [PubMed] [Google Scholar]
- Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, Crosa JH, Falkow S. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene. 1977;2:95–113. [PubMed] [Google Scholar]
- Burke JM, Novotny CP, Fives-Taylor P. Defective F pili and other characteristics of Flac and Hfr Escherichia coli mutants resistant to bacteriophage R17. J Bacteriol. 1979;140:525–531. doi: 10.1128/jb.140.2.525-531.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban MJ. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, Postle K, Riley M, Slatin S, Cavard D. Colicin biology. Microbiol Mol Biol Rev. 2007;71:158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J, Waksman G. Structure of the outer membrane complex of a type IV secretion system. Nature. 2009;462:1011–1015. doi: 10.1038/nature08588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AC, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke M, Maddera L, Harris RL, Silverman PM. F-pili dynamics by live-cell imaging. Proc Natl Acad Sci U S A. 2008;105:17978–17981. doi: 10.1073/pnas.0806786105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daehnel K, Harris R, Maddera L, Silverman P. Fluorescence assays for F-pili and their application. Microbiology. 2005;151:3541–3548. doi: 10.1099/mic.0.28159-0. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Costantino N, Court DL. A set of recombineering plasmids for gram-negative bacteria. Gene. 2006;379:109–115. doi: 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Dautin N, Barnard TJ, Anderson DE, Bernstein HD. Cleavage of a bacterial autotransporter by an evolutionarily convergent autocatalytic mechanism. EMBO J. 2007;26:1942–1952. doi: 10.1038/sj.emboj.7601638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent KC, Thompson R, Barker AM, Hiscox JA, Barr JN, Stockley PG, Ranson NA. The asymmetric structure of an icosahedral virus bound to its receptor suggests a mechanism for genome release. Structure. 2013;21:1225–1234. doi: 10.1016/j.str.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Beck CM, Webb JS, Low DA, Hayes CS. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI) Genes Dev. 2012;26:515–525. doi: 10.1101/gad.182345.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SD, Genthner FJ, Gentry J, Sobsey MD, Vinje J. Gene mapping and phylogenetic analysis of the complete genome from 30 single-stranded RNA male-specific coliphages (family Leviviridae) J Virol. 2009;83:11233–11243. doi: 10.1128/JVI.01308-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza-Sánchez F, Janssen BD, Hayes CS. Prolyl-tRNA(Pro) in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J Biol Chem. 2006;281:34258–34268. doi: 10.1074/jbc.M608052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer MS, Reed RR, Steitz JA, Low KB. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb Symp Quant Biol. 1981;45(Pt 1):135–140. doi: 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- Hayes CS, Sauer RT. Cleavage of the A site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol Cell. 2003;12:903–911. doi: 10.1016/s1097-2765(03)00385-x. [DOI] [PubMed] [Google Scholar]
- Johnson RC, Reznikoff WS. Copy number control of Tn5 transposition. Genetics. 1984;107:9–18. doi: 10.1093/genetics/107.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn PM, O’Callaghan RJ, Paranchych W. Stages in phage R17 infection. VI. Injection of A protein and RNA into the host cell. Virology. 1972;47:628–637. doi: 10.1016/0042-6822(72)90552-1. [DOI] [PubMed] [Google Scholar]
- Lang S, Gruber CJ, Raffl S, Reisner A, Zechner EL. Common Requirement for the Relaxosome of Plasmid R1 in Multiple Activities of the Conjugative Type IV Secretion System. J Bacteriol. 2014;196:2108–2121. doi: 10.1128/JB.00045-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang S, Kirchberger PC, Gruber CJ, Redzej A, Raffl S, Zellnig G, Zangger K, Zechner EL. An activation domain of plasmid R1 TraI protein delineates stages of gene transfer initiation. Mol Microbiol. 2011;82:1071–1085. doi: 10.1111/j.1365-2958.2011.07872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Jimenez J, Derr J, Vera P, Manapat ML, Esvelt KM, Villanueva L, Liu DR, Chen IA. Inhibition of bacterial conjugation by phage M13 and its protein g3p: quantitative analysis and model. PLoS One. 2011;6:e19991. doi: 10.1371/journal.pone.0019991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YL, Elias Y, Huang RH. Structural and mutational studies of the catalytic domain of colicin E5: a tRNA-specific ribonuclease. Biochemistry. 2005;44:10494–10500. doi: 10.1021/bi050749s. [DOI] [PubMed] [Google Scholar]
- Manchak J, Anthony KG, Frost LS. Mutational analysis of F-pilin reveals domains for pilus assembly, phage infection and DNA transfer. Mol Microbiol. 2002;43:195–205. doi: 10.1046/j.1365-2958.2002.02731.x. [DOI] [PubMed] [Google Scholar]
- Maneewannakul S, Maneewannakul K, Ippen-Ihler K. Characterization, localization, and sequence of F transfer region products: the pilus assembly gene product TraW and a new product, TrbI. J Bacteriol. 1992;174:5567–5574. doi: 10.1128/jb.174.17.5567-5574.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning PA, Beutin L, Achtman M. Outer membrane of Escherichia coli: properties of the F sex factor traT protein which is involved in surface exclusion. J Bacteriol. 1980;142:285–294. doi: 10.1128/jb.142.1.285-294.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D, Sowa BA, Ippen-Ihler K. Location of an F-pilin pool in the inner membrane. J Bacteriol. 1981;146:251–259. doi: 10.1128/jb.146.1.251-259.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse RP, Nikolakakis KC, Willett JL, Gerrick E, Low DA, Hayes CS, Goulding CW. Structural basis of toxicity and immunity in contact-dependent growth inhibition (CDI) systems. Proc Natl Acad Sci U S A. 2012;109:21480–21485. doi: 10.1073/pnas.1216238110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakakis K, Amber S, Wilbur JS, Diner EJ, Aoki SK, Poole SJ, Tuanyok A, Keim PS, Peacock S, Hayes CS, Low DA. The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol Microbiol. 2012;84:516–529. doi: 10.1111/j.1365-2958.2012.08039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny CP, Fives-Taylor P. Retraction of F pili. J Bacteriol. 1974;117:1306–1311. doi: 10.1128/jb.117.3.1306-1311.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa T, Tomita K, Ueda T, Watanabe K, Uozumi T, Masaki H. A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science. 1999;283:2097–2100. doi: 10.1126/science.283.5410.2097. [DOI] [PubMed] [Google Scholar]
- Oriel PJ. The thermal and alkaline degradiation of MS2 bacteriophage. Arch Biochem Biophys. 1969;132:8–15. doi: 10.1016/0003-9861(69)90333-6. [DOI] [PubMed] [Google Scholar]
- Osborn M, Weiner AM, Weber K. Large scale purification of A-protein from bacteriophage R17. Eur J Biochem. 1970;17:63–67. doi: 10.1111/j.1432-1033.1970.tb01134.x. [DOI] [PubMed] [Google Scholar]
- Paranchych W, Ainsworth SK, Dick AJ, Krahn PM. Stages in phage R17 infection. V. Phage eclipse and the role of F pili. Virology. 1971;45:615–628. doi: 10.1016/0042-6822(71)90176-0. [DOI] [PubMed] [Google Scholar]
- Paranchych W, Krahn PM, Bradley RD. Stages in phage R17 infection. Virology. 1970;41:465–473. doi: 10.1016/0042-6822(70)90168-6. [DOI] [PubMed] [Google Scholar]
- Poole SJ, Diner EJ, Aoki SK, Braaten BA, T’Kint de Roodenbeke C, Low DA, Hayes CS. Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet. 2011;7:e1002217. doi: 10.1371/journal.pgen.1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann L, Holliger P. The C-terminal domain of TolA is the coreceptor for filamentous phage infection of E. coli. Cell. 1997;90:351–360. doi: 10.1016/s0092-8674(00)80342-6. [DOI] [PubMed] [Google Scholar]
- Roberts JW, Steitz JE. The reconstitution of infective bacteriophage R17. Proc Natl Acad Sci U S A. 1967;58:1416–1421. doi: 10.1073/pnas.58.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhe ZC, Low DA, Hayes CS. Bacterial contact-dependent growth inhibition. Trends Microbiol. 2013a;21:230–237. doi: 10.1016/j.tim.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhe ZC, Wallace AB, Low DA, Hayes CS. Receptor polymorphism restricts contact-dependent growth inhibition to members of the same species. MBio. 2013b;4 doi: 10.1128/mBio.00480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumnieks J, Tars K. Diversity of pili-specific bacteriophages: genome sequence of IncM plasmid-dependent RNA phage M. BMC Microbiol. 2012;12:277. doi: 10.1186/1471-2180-12-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoulaker R, Engelberg-Kulka H. Escherichia coli mutant temperature sensitive for group I RNA bacteriophages. J Virol. 1978;25:433–435. doi: 10.1128/jvi.25.1.433-435.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman PM, Clarke MB. New insights into F-pilus structure, dynamics, and function. Integr Biol (Camb) 2010;2:25–31. doi: 10.1039/b917761b. [DOI] [PubMed] [Google Scholar]
- Steitz JA. Identification of the A protein as a structural component of bacteriophage R17. J Mol Biol. 1968;33:923–936. doi: 10.1016/0022-2836(68)90328-8. [DOI] [PubMed] [Google Scholar]
- Waksman G, Fronzes R. Molecular architecture of bacterial type IV secretion systems. Trends Biochem Sci. 2010;35:691–698. doi: 10.1016/j.tibs.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JS, Nikolakakis KC, Willett JLE, Aoki SK, Hayes CS, Low DA. Delivery of CdiA nuclease toxins into target cells during contact-dependent growth inhibition. PLoS ONE. 2013;8:e57609. doi: 10.1371/journal.pone.0057609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willetts N, Achtman M. Genetic analysis of transfer by the Escherichia coli sex factor F, using P1 transductional complementation. J Bacteriol. 1972;110:843–851. doi: 10.1128/jb.110.3.843-851.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willetts NS, Moore PM, Paranchych W. Variant pili produced by mutants of the Flac plasmid. J Gen Microbiol. 1980;117:455–464. doi: 10.1099/00221287-117-2-455. [DOI] [PubMed] [Google Scholar]
- Zarivach R, Deng W, Vuckovic M, Felise HB, Nguyen HV, Miller SI, Finlay BB, Strynadka NC. Structural analysis of the essential self-cleaving type III secretion proteins EscU and SpaS. Nature. 2008;453:124–127. doi: 10.1038/nature06832. [DOI] [PubMed] [Google Scholar]
- Zhang D, Iyer LM, Aravind L. A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 2011;39:4532–4552. doi: 10.1093/nar/gkr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.