

Abstract
There is an emerging paradigm shift in oncology that seeks to emphasize molecularly targeted approaches for cancer prevention and therapy. Chalcones (1,3-diphenyl-2-propen-1-ones), naturally-occurring compounds with widespread distribution in spices, tea, beer, fruits and vegetables, consist of open-chain flavonoids in which the two aromatic rings are joined by a three-carbon α, β-unsaturated carbonyl system. Due to their structural diversity, relative ease of chemical manipulation and reaction of α, β-unsaturated carbonyl moiety with cysteine residues in proteins, some lead chalcones from both natural products and synthesis have been identified in a variety of screening assays for modulating important pathways or molecular targets in cancers. These pathways and targets that are affected by chalcones include MDM2/p53, tubulin, proteasome, NF-kappa B, TRIAL/death receptors and mitochondria mediated apoptotic pathways, cell cycle, STAT3, AP-1, NRF2, AR, ER, PPAR-γ and β-catenin/Wnt. Compared to current cancer targeted therapeutic drugs, chalcones have the advantages of being inexpensive, easily available and less toxic; the ease of synthesis of chalcones from substituted benzaldehydes and acetophenones also makes them an attractive drug scaffold. Therefore, this review is focused on molecular targets of chalcones and their potential implications in cancer prevention and therapy.
Keywords: Chalcones, molecular targets, bioactive dietary compounds, chemoprevention
1. INTRODUCTION
Chalcones are widely occurring natural plant products and presented in spices, tea, beer, fruits and vegetables [1–3]. Chalcones belong to the flavonoid family and act as intermediates in the biosynthesis of flavonoids. Chalcones are pharmacologically active and have demonstrated a wide range of biological activities, including anti-oxidative, anti-cancer, anti-mutagenic, anti-microbial, anti-protozoal, anti-histaminic, anti-inflammatory, analgesic and immune-modulator properties [1–3]. More importantly, chalcones have been used in clinics for treatment of gastric ulcers, duodenal ulcers, bronchial asthma, Addison’s disease, skin disorders, diabetes, cardiac disease and helicobacter pylori infection [1–3].
Chalcones contain a common 1, 3-diphenyl, propenone template that can be easily modified to alter the biological potential of these molecules. Through the addition of a wide variety of functional groups (aryls, halogens, hydroxlys, carboxylic groups, benzenes, etc.) [4], the biologic activities of chalcones are altered probably due to their interaction with different molecular targets. Even minor structural modification can results distinct cellular and molecular alterations. The diversity of the chalcone family also lends itself to broad-spectrum biologic applications in oncology, particularly for the development of novel targeted therapies.
Molecular targeted and “biologic” agents in cancer therapy are the result of a rapidly growing understanding of the molecular events which are involved in carcinogenesis, including crucial aberrations in the regulation of apoptosis, cell-cycle control, metastasis and tumor angiogenesis. Currently, small molecule tyrosine-kinase inhibitors and monoclonal antibodies are targeted approaches used to selectively inhibit these key pathways in tumor growth and progression [5]. Indeed, a variety of these agents, have gained widespread use in the clinical treatment of a number of malignancies, such as carcinoma of the colon, breast, ovary, or lung and leukemia [5]. Unlike conventional chemotherapy, targeted agents have a relatively wide therapeutic window and have non-overlapping toxicity profiles [5]. Phytochemicals, like chalcones, have similarly been shown to be relatively nontoxic, and certain chalcone moieties can target key molecular events that may lead to carcinogenesis. Population-based studies in countries with high dietary intake of certain types of chalcones have also shown a correlation with lower incidences of cancer in these areas, which provides support for the further investigation of such natural derivatives for cancer prevention and therapy. Chalcones and their synthetic derivatives thus present a unique opportunity to utilize these well-tolerated, highly modifiable molecules as either chemopreventive drugs or as components in cancer treatment.
2. CHALCONES TARGET THE P53 PATHWAY
The role of p53, the ‘guardian of the genome,’ has been extensively studied over the last 30 years due to its role as an important innate tumor suppressor. In response to DNA damage, oncogene activation, or other cellular stress signals, p53 can mediate a number of varied biologic effects that lead to cell senescence, cell cycle arrest, or apoptosis. Thus, p53 functions to maintain cellular and genomic integrity and prevent proliferation of incipient cancer cells. Loss of p53 function occurs in most human tumors by either mutation of TP53 itself or by inactivation of the p53 signal transduction pathway. Mutations in p53 have been identified across a number of different human cancers. In addition, p53 is inactivated by the overexpression of negative regulators, most notably MDM2. The p53 pathway in these tumors can be reactivated by small molecules that inhibit p53/MDM2 interaction, thus preventing p53 proteasomal degradation. Currently, there are at least two compounds of MDM2 inhibitors (JNJ-26854165, Johnson & Johnson and RG7112, F. Hoffmann–La Roche) being tested in phase I clinical trials in patients with hematologic neoplasms and advanced solid tumors [6, 7].
Chalcones were among the first class of compounds that demonstrated activity in modulating the p53/MDM2 interaction. Stoll and colleagues using multidimensional NMR spectroscopy, an ELISA assay that employed a p53 peptide and a gel shift assay, were the first to report that chalcones bound to MDM2 at the p53 transactivation domain and resulted in a release of p53 from both the p53/MDM2 complex as well as the DNA-bound p53/MDM2 complexes [8]. Based on thisNMR data, Kumar et al [9] hypothesized that the carboxylic acid group of the chalcones could be placed near the base of K51 lysine to form a salt bridge and then break the salt bridge interacting with Glut25 in the p53/MDM2 complex. Therefore, Kumar et al [9] designed and synthesized a series of boronic acid chalcone analogues that might form a stronger salt bridge with K51 lysine than the carboxylic acid analogues. They showed that these boronic chalcones were 5–10 fold more toxic to human breast cancer cell lines at low µM range compared to normal breast epithelial cell lines [9]. Modzelewska et al [10] from the same group further synthesized a new class of chalcones (bischalcones) that contain a pair of α, β-unsaturated groups. Some of these chalcones were even more potent in preferentially inhibiting the growth of breast cancer cell lines. One bis-chalcone exhibited differential cytotoxicity to an isogenic pair of colon cancer HCT116 cells; p53 +/+ cells were more sensitive to the bis-chalcone compared with the p53 −/− cells [10]. Hsu et al [11] also reported that the effect of isoliquiritigenin (4, 2',4'-trihydroxychalcone) on the growth of Hep G2 can be attenuated by a dominant negative p53 that blocks p53 transcriptional activity. However, these studies did not specifically investigate the effect of these chalcones on p53/MDM2 interaction. Achanta et al [12] reported that 3, 5-bis-(4-boronic acid-benzylidene)-1-methyl-piperidin-4-one (AM114), a bischalcone, selectively inhibited the growth of HCT116 p53 +/+ versus p53 −/− cells. The mechanism of action of AM114 was shown to be associated with inhibiting the chymotrypsin-like activity of the 20S proteasome in vitro and then leading to p53 accumulation but not with p53/MDM2 disruption. In another study, Chen et al [13] found that trans-4-Iodo, 4′-boranyl-chalcone (TIBC) inhibited MG132, a proteasome inhibitor-caused accumulation of ubiquitinated p53 in human lung cells, suggesting that TIBC may inhibit the interaction of MDM2/p53. Further study by Sasayama et al [14] showed that TIBC effectively inhibited the growth of human glioma cell lines irrespective of their p53 status and even decreased the expression of p53 in some glioma cell lines. Using a NMR chemical shift perturbation method for studying lead compounds in ligand-protein interaction, D'Silva L et al [15] tested three lead compounds: nutlin-3, a sulfonamide compound (NSC 279287), and a boronic chalcone, with recently reported activity to block the p53-MDM2 interaction. Only nutlin-3 was found to effectively release p53 from the p53/MDM2 complex, whereas NSC279287and the boronic chalcone either precipitated the protein or acted as a much weaker MDM2 inhibitor. Cumulatively, these results suggest that inhibition of the MDM2/p53 interaction may not be the primary anticancer mechanism of chalcones.
In general, most of chalcones demonstrated selectivity against the growth of cancer cells versus normal cells. Some chalcones (e.g. Hydroxysafflor yellow AHSYA and imidazole-chalcone) [16–18] have been shown to increase the expression of p53 and selectively inhibited the growth of cancer cells with wild-type p53, whereas other chalcones [e.g. toluenesulfonylamido-chalcone, 4′-(p-toluene sulfonyl amino)-3, 4-dihydroxy chalcone (TSHDC)] was reported to reduce p53 expression [19]. We have demonstrated that flavokawain A, a chalcone from Kava extracts, preferentially inhibited the growth of p53 mutant bladder cancer lines and its effect on cell cycle arrest differed in p53 wild and mutant type bladder cancer cell lines [20]. Flavokawain A induced a G1 arrest in RT4 cells harboring wild-type p53 and G2M arrest in bladder cancer cell lines with mutant p53 [20]. We further have demonstrated that dietary flavokawain A significantly inhibited urothelial tumorigenesis in vivo in the UPII-SV40T transgenic model that resembles human urothelial cell carcinoma (UCC) with defects in the p53 and the retinoblastoma (RB) protein pathways [21]. Together, these studies suggest that the diverse chemical structures of chalcones may contribute to their differential effects related to the p53 pathway. Mechanisms of Chalcones’ effect on the p53 pathway in cancer cells are summarized as Table 1. There is a potential opportunity to identify those chalcones that re-activate the p53 tumor suppressing pathway through a systematic study of their structure-relationship with mutant p53 cells.
Table 1.
Chalcones and the p53 pathway
| Lead Compounds | Chemical structures | Mechanisms of Action | Reference |
|---|---|---|---|
| Chalcone carboxylic acid | 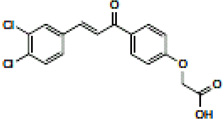 |
Disrupts p53/MDM2 interaction to restore p53 activity. IC50 = 49 µM (ELISA) Kd = 90 µM (NMR) |
Stoll et al 2001 [8] |
| Boronic chalcones | 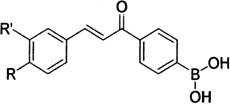 |
Preferentially inhibit the growth of human breast cancer cells (MCF-7, MDA-MB-231, MDA-MB-435) versus non-malignant breast epithelial cell lines (MCF-10A and MCF-12A). | Kumar et al 2003 [9] |
| Trans-4-lodo, 4’-boranyl-chalcone (TLBC) | 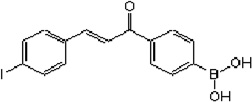 |
Decreases p53 ubiquitination in lung cells. | Chen et al 2007 [13] |
| 3,5-Bis-(4-boronic acid-benzylidene)-1-methyl-piperidin-4-one (AM114) | 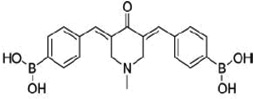 |
Inhibits the chymotrypsin-like activity of the 20S proteasome, induces p53 accumulation, and preferentially inhibit the growth of HCT116, p53+/+ versus p53 −/−. | Achanta et al 2006 [12] |
| Flavokawain A | 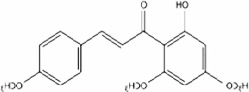 |
Effects on cell cycle arrest vary in p53 wild-type (RT4) versus mutant human bladder cancer cell lines (T24, UMUC3, TCCSUP, 5637, HT1376, HT1197) | Tang et al 2008 [20] |
| Imidazothiazole chalcones | 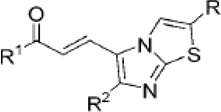 |
Induces p53 expression and G1 phase arrest. | Kamal et al 2010 [17]. |
| 3, 2',3',4'-Tetrahydroxychalcone | 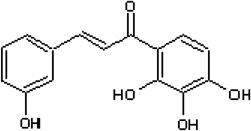 |
Inhibits the SIRT1-mediated deacetylation of a p53 acetylated peptide and recombinant protein in vitro, and induces SIRT1-mediated hyperacetylation of p53 in cells | Kahyo et al 2008 [22] |
3. CHALCONES TARGET TUBULIN POLYMERIZATION
The use of chalcones as antimitotic agents was first reported nearly 20 years ago, and represents some of the earliest investigations of chalcones as therapeutic agents [23]. Building on the knowledge of colchicine-tubulin structure-activity relationships, chalcone derivatives were modeled as colchicine analogues to achieve a similar reactivity in the tubulin binding site, leading to destabilization of microtubules. Structure activity relationships of a large number of chalcone derivatives were then studied with colchicine and vinblastine (known tubulin-binding antimitotic agents) as controls. This study paved the way for the multiple studies that have followed looking at the effects of various substitutions on the antimitotic activity of chalcones that will be reviewed here [24–38] (Table 2).
Table 2.
Chalcones affect tubulin polymerization
| Lead Compound | Chemical structure | Mechanisms of Action | Reference |
|---|---|---|---|
| Trans-1-(2,5-dimethoxyphenyl)-3-[4-(dimethylamino)phenyl]-2-methyl-2-propen-1-one], MDL 27048 |  |
Binds to tubulin at the colchicine-binding site, and inhibits microtubule assembly. IC50 1 µM | Peyrot et al 1989 [23] |
| The crude extract of Calythropsis aurea (Myrtaceae). Calythropsin and dihydrocalythropsin are active compounds from the extract | 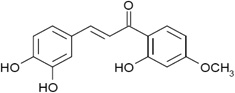 |
Produce a pattern of differential cytotoxicity in the NCI 60 cell line assay which was similar to those of known tubulin-interactive compounds | Boumendjel et al 2008 [24] |
| Combrestastin-like, alpha-methyl chalcones SD400 | 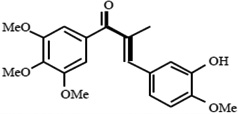 |
Combretastatin-like analogues populate the colchicine-binding site of beta-tubulin | Ducki et al 2009 [25] |
| A series of Combretastatin-like alpha-aryl chalcones | 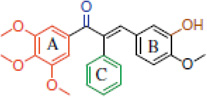 |
Inhibits tubulin assembly/antimitotic activity | Ducki et al 2009 [26] |
| Thiophene analogues of chalcones; (3,4,5-Trimethoxyphenyl) (5-(2-methoxyphenyl) thiophen-2-yl)methanone | 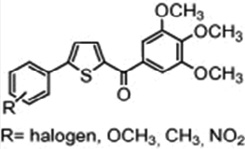 |
Inhibits tubulin assembly/antimitotic activity | Romagnoli et al 2008 [27] |
| Triazole or tetrazole analogues of Chalcones | 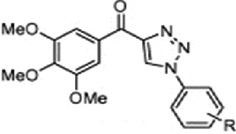 |
Inhibits tubulin assembly/antimitotic activity | Mesenzani et al 2011 [37] |
| Resveratrol derivatives with a chalcone moiety | 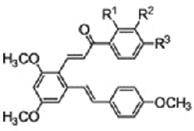 |
Inhibits tubulin assembly/antimitotic activity | Ruan et al 2011 [36] |
| Dihalogenated chalcones and structurally related dienones | 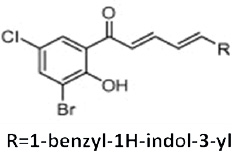 |
Stabilizes tubulin assembly/antimitotic activity | Dyrager et al 2011 [38] |
A number of chalcones that are analogues of combretastatin 4A, such as SD400 and MDL, have demonstrated a similar capacity to bind β-tubulin and thus destabilize microtubule polymers. Ducki et al [25, 26] presented a thorough review of alpha-aryl chalcone derivatives, whose structure-activity relationship studies demonstrated both antimitotic properties (IC50s in the low micromolar range) in K562 leukemia cells with arrest at G2/M phase, as well as antivascular activity (discussed below, see Angiogenesis). Boumendjel et al [24] similarly synthesized a library of chalcones and identified 3 candidates demonstrating significant antimitotic activity, again with IC50s in the low micromolar range. As demonstrated by Ducki et al, antimitotic activity appeared to be highly correlated to the degree of methoxylation (25, 26). These candidates were further tested in vivo and found to be nontoxic in animal models. Zoldakova et al [29] also reported on combretastatin-like chalcones and identified a candidate demonstrating marked microtubule disruption and cell-cycle arrest in melanoma 512A cells and neural cells. Presumably a similar tubulin-binding mechanism is also at play. Further, this same group of investigators also added a functional platinum complex onto the parent chalcone, rendering further cytotoxic potential by adding DNA adduct effects to the tubulin polymerization effects [35]. Ruan et al [36] synthesized a series of resveratrol derivatives with a chalcone moiety and found that these compounds reduced the growth of cancer cell lines through their binding to the tubulin-colchicine binding site, inhibiting tubulin polymerization and inducing G2M arrest during cell cycle progression.
Romagnoli R et al [27] reported that the replacement of the double bond of enone system of chalcones with a thiophene moiety enhanced the growth inhibitory and anti-tubulin polymerization effects of the chalcones. Similarly, Mesenzani O et al [37] replaced the double bond of antitubulin chalcones with triazoles and tetrazoles. His study suggested that the double bond in the chalcone scaffold may not be essential for the interaction with tubulin, and that these new analogues may have the advantage of being metabolically stable due to the introduction of a chemically inert heterocyclic ring (i.e. triazole or tetrazole). Dyrager C et al [38] identified one compound from a series of dihalogenated chalcones and structurally related dienones, which binds to the paclitaxel binding site of tubulin and stabilizes tubulin to the same extent as the docetaxel.
Based on the similarity of the chemical structure of chalcones to combretastatin and their ease of chemical modification, chalcones represent a very attractive starting material for synthesis of novel agents targeting tubulin for treatment of cancers.
4. CHALCONES TARGET PROTEASOMAL ACTIVITY
Chalcones have been shown to exhibit anti-cancer activity based on their ability to affect the proteasome via several different mechanisms. The Ubiquitin-Proteasome System (UPS) plays a major role in the regulation of cancer cell growth and proliferation, being responsible for the majority of protein degradation within the cell [39]. Proteasomal inhibitors, such as bortezomib and carfilzomib have already been used effectively in the clinical setting as targeted therapeutic agents [40]. A novel non-boronic chalcone based derivative has recently been synthesized and characterized by Bazzaro et al [41] which targets the catalytic 20s subunit of the proteasome via the interaction of its α, β-unsaturated carbonyl system with the N-terminal threonine residues in the catalytic sites of the proteasome. Significantly, this novel compound exhibited synergistic killing of HPV+ cervical cancer cells when used in combination with the clinically approved proteasome inhibitor Bortezomib. However, chalcone based compound RAMB1 has been shown to inhibit cervical cancer cell growth in a non-20s proteasome dependent manner, by stabilizing p53 levels from preventing its degradation, leading to a compensatory increase in lysosomal protein degradation [42]. This study demonstrated that chalcones can prevent HPV E6 mediated p53 degradation. Importantly, this allows co-treatment with RAMB1 and Chloroquine (a lysosome inhibitor) to synergistically reduce cervical cancer cell viability [42].
Chalcone derivatives can also affect proteasomal activity by targeting deubiquitinating enzymes without inhibiting the activity of the 20s proteasomal subunit itself. Three recently described chalcones act as inhibitors of deubiquitinating enzymes, depleting free ubiquitin in the cell and causing an accumulation of polyubiquitinated proteins ultimately resulting in p53 stabilization, tumor suppressor upregulation, and oncogene suppression [43]. This provides a promising alternate means of targeting proteasomal activity in a highly effective manner without having to directly target proteasomal subunits.
5. CHALCONES MODULATE CELL CYCLE PROGRESSION
The use of chalcones as antimitotic agents that can induce G2M arrest in cell cycle progression was first reported almost 20 years ago as described previously [23]. In addition to cell cycle interruption by targeting tubulin and microtubule polymerization, there have been a number of other chalcone targets that also interrupt cell cycle progression and subsequently signal apoptosis [44–47]. The proposed mechanisms of action are varied but centered primarily around their impact on various cyclins and cyclin-dependent kinases (CDKs). Cell cycle arrest and cytotoxicity with these chalcone derivatives is mediated by downregulation of cyclin expression [45, 46], inhibition of topoisomerase II [47], or enhanced expression of p21(CIP1/WAF1), which is a universal inhibitor of cyclin-dependent kinases [46, 47]. All of these pathways eventually trigger apoptotic pathways and subsequent dose-dependent growth inhibition [46–51]. The previously discussed chalcone-based inhibitors of deubiquitinating enzymes similarly resulted in downregulation of cyclin D1 and upregulation of several tumor suppressor genes, most notably p53, resulting once again in G2M arrest [43]. Interestingly, the chalcone known as Cardamonin has been shown by Park et al to suppress colon cancer cell proliferation by depleting β-catenin levels in a proteasome-dependent fashion, thereby also suppressing cyclin D1 and c-myc expression [51].
6. MCHANISMS OF CHALCONES INDUCED CELL DEATH
Many chalcones have demonstrated the ability to induce apoptosis through the targeting of the mitochondrial pathway. The specific molecular components targeted by chalcones that affect the mitochondrial pathway are varied, as are their chemical structures. There does not seem to be an overall requisite set of substitutions to render apoptotic effects of chalcones. A number of authors have demonstrated effects of their respective chalcones on the induction of pro-apoptotic proteins Bak [52, 53] and Bax [52–54], and decrease in anti-apoptotic proteins Bcl-2 [11, 52, 53, 55] and Bcl-X [11, 52, 53, 56], resulting in the cumulative response of caspase-9 activation. In line with these observations, two studies reported that apoptotic cell death marked by caspase-9 activation [52, 55, 57] could be inhibited with the pretreatment of cells with a caspase inhibitor [11]. An additional line of indirect evidence regarding mitochondrial uncoupling was described by Navarini et al [58], whereby mitochondrial ATP and GSH were reduced in melanoma cells, potentially destabilizing the mitochondrial membrane. Finally, direct effects of chalcone treatment on cell mitochondrial membrane depolarization have been reported in multiple studies, leading to activation of apoptotic pathways [59–63].
In addition, a number of studies have demonstrated that chalcones can induce cancer cell apoptosis through effects on the extrinsic pathway. Tang et al [64] demonstrated that the chalcone flavokawain B (FKB) led to upregulation of proapoptotic proteins death-receptor 5 (DR5/TRAILR2), Puma and Bim, while decreasing expression of apoptotic inhibitors XIAP and survivin in androgen receptor negative prostate cancer cells. Further, synergy with TRAIL was demonstrated. This was accompanied by an increase in cytotoxicity in vitro, as well as decreased tumor growth in vivo. The effects of FKB on cell growth inhibition could additionally be negated with shRNA silencing of Bim expression. Notably, there was minimal toxic effect on normal prostate epithelial cells. Upregulation of death receptors was similarly demonstrated by Yun et al [54] who found that Pandaturin A increased both Fas and TRAIL expression leading to increased apoptosis in prostate cancer cells. Similarly, several other investigators noted increased DR5 expression in both leukemia and colon cancer cells, and in once case was able to demonstrate that chalcone-induced death receptor overexpression was able to restore sensitivity to TRAIL-mediated apoptosis in TRAIL-resistant cells [65–68].
Szliszka et al [69] also studied a panel of five chalcones in prostate cancer cell lines, finding that all five compounds markedly increased TRAIL-mediated apoptosis and cytotoxicity in cells previously demonstrated to be TRAIL resistant. The same investigators then studied another panel of molecules including 2 chalcones and 3 dihydrochalcones and again demonstrated markedly increased TRAIL-mediated apoptosis in the same resistant prostate cancer cell line [70]. Thus, several independent reports suggest that there is some synergy between chalcones and TRAIL which can overcome aspects of TRAIL resistance in cancer cells [64, 69, 70). Further, given that the broad spectrum of chalcones structures appears to have similar effects on the death receptor pathway, combined with the clear activity in prostate cancer cell lines and animal models, suggests that chalcones have significant potential as chemopreventive drugs or therapeutic adjuncts in cancer therapy.
Recently, Champelovier et al [71] reported that two chalcone derivatives, JAI-51 and MBL-II-58, with minor structural differences induce cell death in glioblastoma cell lines via a significantly different mechanism. JAI-51 induces apoptosis through the activation of caspase-3, -8 and -9, whereas, MBL-II-58 treatment causes autophagic cell death in a ROS, but not caspase and protein synthesis, dependent manner. JAI-51also selectively inhibited high proliferative cancer cells versus normal human nucleated blood cells and normal human skin fibroblast cells [71]. These results indicated that even minor changes in chalcone structures can lead to significantly different molecular mechanisms for chalcone induced cell death. He et al [72] also showed that Chalcone-24 did not activate caspase cascade and induce apoptosis. Instead, Chalcone-24 treatment of lung cancer (A549 and H1299) and bladder cancer (UM-UC-3) cell lines caused a non-apoptotic death, autophagy-mediated and Receptor-Interacting Protein (RIP) 1- and RIP3-dependent necroptosis. Robinson et al [73] described that a chalcone derivative 3-(2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (i.e., MIPP) induced massive accumulation of vacuoles by dysregulation of macropinocytosis in glioblastoma cells leading to a novel caspase-independent cell death, named Methuosis. Mechanisms of chalcones induced cell death are summarized in Figure 1 and Table 3.
Fig. (1).

Chalcones with minor structural changes result in distinct processes of cell death. Flavokawain B and Isobavachalcone induce apoptosis by activation of both mitochondria and death receptor mediated pathways. Chalcone-24 induces autophagic cell death via activation of the JNK pathway and down-regulation of cIAPs.
Table 3.
Mechanisms of chalcone induced Cell death
| Lead Compound | Chemical Structure | Mechanisms of Action | Reference |
|---|---|---|---|
| Isobavachalcone | 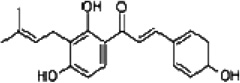 |
Increases Bak and Bax expression and decreases Bcl2 expression, as well as activates caspase-9 and -3 | Nishimura et al [55] |
| JAI-51 | 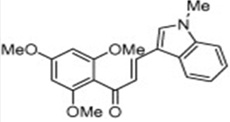 |
Increases Bax/Bcl2 ratio and activates caspases-8, -9 and -3, leading to apoptosis in glioblastoma cell lines | Champelovier et al [71] |
| MBL-II-58 | 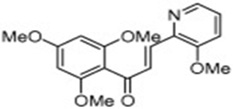 |
Induces ROS production and autophagic cell death | Champelovier et al [71] |
| Chalcone-24 (Chal-24)Chal-24 | 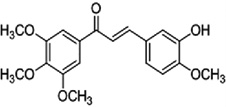 |
Inducing autophagy-mediated necroptosis and c-IAP1 and c-IAP2 degradation and ripoptosome formation | He et al 2013 [72] |
| 3-(5-methoxy, 2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (i.e., MOMIPP) | 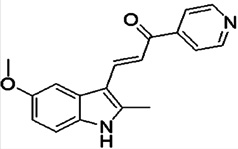 |
An inducer of methuosis | Robinson et al 2012 [73] |
| Flavokawain B | 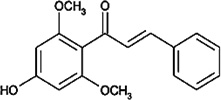 |
Increases the expression of death receptor-5, Bim and Bax and down-regulates the expression of survivin and XIAP, leading to apoptosis. | Tang et al [64] |
7. CHALCONES AND THE NF-κB PATHWAY
NF-κB is a critical transcription factor with widespread cellular effects that plays an important role in carcinogenesis, although, the oncogenic factors that result in the constitutive activation of NF-κB seen in many cancers have yet to be clearly elucidated. Nevertheless, NF-κB downstream effects are well recognized in cancer, including upregulation of tumor promoting cytokines and survival genes (e.g., Bcl-2), inhibition of apoptosis, and promotion of angiogenic factors, as well as a migratory and invasive phenotype that is associated with tumor progression. Thus, aberrant regulation of NF-κB is involved in cancer development and progression as well as in drug resistance. Inhibitors of NF-κB mediate effects potentially leading to antitumor responses or greater sensitivity to the action of antitumor agents. With regard to chalcones as inhibitors, it would appear that structural complexity is not a requisite for NF-κB inhibitory activities, as reported in structural activity relationship studies [74, 75].
NF-κB activity is negatively regulated primarily by IĸBα, which essentially keeps NF-κB in an inactive state. IĸBα in turn is regulated by IĸBα kinase β (IKK), which phosphorylates IĸBα in the presence of cellular stress signals and causes the dissociation if the IĸBα/NF-κB complex, leaving NF-κB free to translocate to the nucleus and affect gene expression. Inhibition of IKK, therefore, can lead to inhibition of NF-κB by keeping it locked to IĸBα in an inactive state. This mechanism of NF-κB regulation is the target of a number of agents, including chalcones. Chalcones contain a highly electrophilic α, β-unsaturated carbonyl moiety that can react with free sulfhydryl groups of thioredoxin and cysteine residues in proteins [76, 77]. Indeed, Pandey et al [78] demonstrated that butein (3,4,2',4'-tetrahydroxy chalcone) inhibited NF-κB and NF-κB regulated gene expression through its conjugation of cysteine 179 residue of the IKKβ and then stabilization of IĸBα protein. Similarly to this result, the following multiple naturally-occurring chalcones were found to prevent the degradation of IκBα and in turn block NF-κB activation: broussochalcone A from Broussonetia papyrifera Vent, Isoliquiritigenin present in Glycyrrhiza and Dalbergia, 4-hydroxylonchocarpin obtained from Psoralea corylifolia, Cardamonin isolated from Alpinia rafflesiana, flavokawain A and B from the kava plant, Xanthoangelol D isolated from Angelica keiskei, Xanthohumol present in hops (Humulus lupus L.), Hydroxysafflor yellow A (HSYA) from Carthamus tinctorius L, panduratin A from Boesenbergia rotunda stercurensin, etc. [1, 2, 79–85]. However, Orlikova et al [86] reported that 4′-hydroxychalcone inhibited NF-κB activation via an IKK-independent mechanism, which was suggested to be involved in its mediated inhibition of proteasome function and then stabilization of IĸBα. 4′-Hydroxychalcone has no direct effect on IKK but nevertheless may attenuate its activity indirectly through inhibition of IĸBα. Kim et al [87] found that stercurensin, a chalcone isolated from the leaves of Syzygium samarangense, could inhibit the NF-κB activation via preventing the formation of the transforming growth factor-β-activated kinase 1 (TAK1)/ TAK1-binding protein 1 (TAB1) complex.
In addition to conjugated double bonds, chalcones have a completely delocalized Π-electron system on both benzene rings, providing chalcones a greater probability of undergoing electron transfer reactions. The B-ring has a flexible ring structure and can easily convert cis-chalcone to trans-chalcone or vice versa. Trimethoxy chalcone at the A-ring with fluoro, chloro, bromo substitution in on the B-Ring, like 2'-hydroxy-3-bromo-6'-methoxychalcone, 2'-methoxy-3,4-dichlorochalcone, flavokawain A, or flavokawain B, were demonstrated to be better inhibitors of NF-κB [83, 84, 86]. Reddy et al [88] synthesized a series of bichalcones and identified that some significantly blocked the nuclear translocation of NF-κB p65. The differential effects of chalcones on different components of the NF-κB pathways are shown in Table 4 and Figure 2.
Table 4.
Mechanisms of chalcones inhibiting the NF-κB pathway
| Lead Compound | Chemical structure | Mechanisms of Action | Reference |
|---|---|---|---|
| Butein | 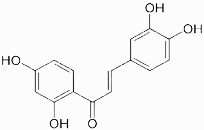 |
Conjugation of cysteine 179 residue of the IKKβ and then stabilization of IĸBα protein | Pandey et al 2007 [78] |
| Xanthohumol | 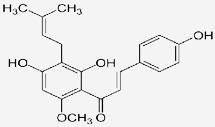 |
Prevents the degradation of IκBα and in turn blocks NF-κB activation | Harikumar et al 2009 [81] |
| 4′-Hydroxychalcone | 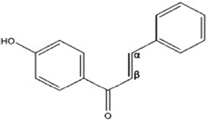 |
Inhibits proteasome function and then stabilizes IĸBα. via an IKK-independent mechanism | Orlikova et al 2006 [86] |
| 2',4'-Dihydroxy-6'-methoxy-3'-methylchalcone (stercurensin) | 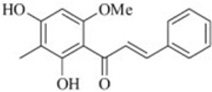 |
Inhibits TAK1-TAB1 complex formation | Kim et al 2011 [87] |
| Bichalcones | 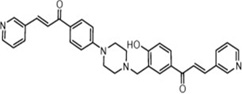 |
Blocks the nuclear translocation of NF-κB p65 | Reddy et al 2011 [88] |
Fig. (2).

Different structural types of chalcones primarily target different components of canonical NF-κB pathway. (a) Stercurensin inhibits the formation of TAK1/TAK1 complex; (b) 3-Hydroxy-4,3′,4′,5′-Tetramethoxychalcone conjugates the lysine residuals of IKK β and block IKK β activity; (c) 4′-hydroxy chalcone inhibits the proteasomal activity and stabilize IκBα; (d) Bichalcones inhibits NF-κB subunits p50/p65 nuclear translocation
8. ANDROGEN AND ESROGEN RECEPTOR PATHWAYS
The androgen receptor (AR) is an important mediator of prostate cancer development and progression and consequently a therapeutic opportunity for chalcones therapy. Shirota et al [89] isolated five antiandrogenic diels-alder-type Adducts from Brosimum rubescens consisting of chalcone derivatives and a prenylcoumarin. Prostate cancer progression is frequently accompanied by mutations in the AR, which render an antagonist-to-agonist conversion. Thus, previously hormone-sensitive cancers become insensitive to androgen ablation therapy, while the AR signal pathway remains active. As a result, ARs can inappropriately activate transcription in androgen-independent prostate cancer cells via mechanisms that are resistant to castration and AR antagonism. Zhou et al [90] screened a series of ionone-based chalcones and discovered one pan-antagonist of the AR in a panel of prostate cancer cell lines with various AR mutations. This chalcone represents a novel antiandrogen that is simultaneously effective against multiple AR mutants that confer resistance to anti-androgens currently used in the clinics. Chen et al [91] similarly investigated isoliquiritigenin in hormone-resistant prostate cancer cells and demonstrated a downregulation of AR and an AR-related product (prostate specific antigen), which was coupled with cytotoxicity in 2 prostate cancer cell lines with IC50s in the low micromolar range. Kim et al [92] identified that chalcones with an o-methoxy group on A ring can inhibit AR nuclear translocation and AR mediated gene expression by increasing the formation of the AR-Hsp90 complex in the cytoplasm.
Anti-estrogen targeted therapies, such as those commonly employed in breast cancer treatment, are mediated though a number of different molecules in the steroid pathway. The use of chalcones to target the estrogen pathway has focused on modulation of either the estrogen receptor (ER) or aromatase activity responsible for conversion of androgens to estrogens. One of the early reports of targeting the estrogen receptor with chalcones was demonstrated by Satomi et al [93], who reported that 3’-methyl-3hydroxychalcone, apart from cell cycle arrest effects, blocked the binding of estradiol to type-II estrogen binding sites yielding downstream pathway effects and inhibiting cell proliferation. Similarly, De Vincenzo et al [49] investigated a series of both natural and synthetic chalcones in established and primary ovarian cancer cell lines, showing inhibition of estradiol binding to the type-II estrogen receptor and inhibiting cell proliferation.
Anti-estrogen activity mediated by aromatase inhibition with the chalcones butein and isoliquiritigenin (ISL) have been demonstrated in breast cancer models [94, 95]. Both butein and isoliquiritigenin showed inhibitory K(i) values of 0.32 and 3 µM range, respectively. ISL was further studied in vivo in a xenograft model using MCF-7 breast cancer cells transfected with aromatase and showed a significant decrease in tumor growth. These effects were correlated with a demonstration of decreased mRNA expression of aromatase in the presence of ISL. In contrast to the anti-estrogenic and antiproliferative effects reported by Ye et al [95], Maggiolini and colleagues [96] reported the paradoxical action of ISL as an estrogenic agonist of both estrogen receptor isoforms in the same breast cancer cell model. The authors found that at lower concentrations (10 nM to 1 µM), ISL stimulated the estrogen receptor and downstream transcriptional activity leading to cell proliferation which could be blocked with known anti-estrogens. At higher concentrations (1 to 10 µM), however, ISL was cytotoxic to both hormone dependent and hormone independent cell lines, suggesting that ISL cytotoxic effect may be independent of the estrogen receptor at these concentrations. These studies highlight the importance of the need for appropriate quantification of chalcone concentrations in dietary supplements in order to derive the specific chemopreventive benefits. Differential effects that are independent of estrogen are also suggested by the work of Rafi et al [97]. Licochalcone-A was demonstrated to be a phytoestrogen with paradoxical cytotoxic effect in both breast cancer and leukemia cell lines [97, 98]. The authors attributed the cytotoxicity of this estrogenic compound to the modulation of Bcl-2 in favor of apoptosis. Thus, while chalcones can have estrogenic agonist properties, the balance between hormone dependent cancer cell proliferation versus hormone independent cytotoxicity depends on the specificity of these compounds to other cellular targets, like Bcl-2, which can be simultaneously targeted.
We present the summary results of mechanisms of chacones’ action on androgen receptor and estrogen receptor pathways in Table 5 and Figure 3.
Table 5.
Chalcones affect androgen receptor and estrogen receptor signaling
| Lead Compound | Chemical structure | Mechanisms of Action | Reference |
|---|---|---|---|
| Ionone-based chalcones | 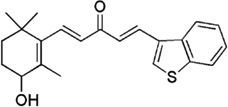 |
A pan-inhibitor against the wild type and the clinically relevant T877A, W741C and H874Y mutated AR activities | Zhou et al 2010 [90] |
| Methoxychalcones | 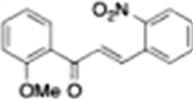 |
Inhibits androgen receptor nuclear translocation and AR mediated gene expression. | Kim et al 2012 [92] |
| 3’-Methyl-3hydroxychalcone | 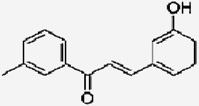 |
Blocks the binding of estradiol to type-II estrogen binding sites | Satomi et al 1993[93] |
| Isoliquiritigenin | 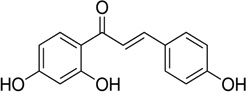 |
Stimulates the estrogen receptor and downstream transcriptional activity at low concentrations (10 nM to 1 µM), and cytotoxic effect at higher concentrations (1 to 10 µM) | Maggiolini et al 2002 [97]. |
Fig. (3).

The simplified mechanisms of chalcones and their analogs’ action on androgen receptor signaling. o-Methoxy chalcones increase the formation of the AR/Hsp90 complex and block the nuclear translocation of AR. Flavokawain B down-regulates the expression of Sp1 and inhibits the Sp1 transcriptional regulated expression of AR and AR splicing variants. Ionone-based chalcone analogs inhibit both wild-type and mutant types of ARs mediated gene transcription.
9. CHALCONES TARGET MULTIDRUG RESISTANCE TRANSPORTERS
A number of authors have investigated the ability of chalcones to mediate drug resistance to conventional chemotherapeutics by modulating multidrug efflux transporters (MDRs) known to be important components in cellular accumulation of drugs. The majority of studies have studied the ability of chalcones to inhibit two well-characterized members of the ABC transporter family, P-glycoprotein (P-gp or ABCB1) and BCRP (breast cancer resistance protein/ABCG2). Indeed, many cancers have been shown to overexpress MDR transport proteins, and expression of MDR proteins has been associated with poor prognosis in a number of cancers.
In the late 1990’s, Bois and colleagues explored the ability of chalcones to bind recombinant P-gp, found that certain moieties including a trihyrdoxy-4-iodochalcone and a 4’-octyloxy-trihydoxychalcone demonstrated the highest affinities for P-gp [99, 100]. These moieties also bound the ATP-binding region, increasing the likelihood that these compounds would inhibit P-gp transport function, though this was not specifically tested in their series. Other authors have looked at the ability of their chalcone derivatives to modulate increased drug accumulation via effects on P-gp. Ivanova et al [101] demonstrated a 100-fold increase in drug accumulation via inhibition of P-gp in lymphoma cells, most effective in a chalcone with a π-chloro group on Ring B, with IC50 concentrations in the low-micromolar range. Similarly, Liu et al [102] demonstrated that chalcones with basic moieties on Ring A increased drug accumulation in breast cancer cells by inhibition of P-gp using the calcein assay. However, these same basic chalcone molecules were ineffective in modulation BCRP-mediated drug efflux of mitoxantrone. Non-basic Ring A moieties, by contrast, were highly effective in inhibiting BCRP and increasing mitoxantrone uptake, up to 300%. These findings are in agreement with Han et al [103], who also found that non-basic chalcones, specifically dimethoxy- or dihydroxyl- substitutions on Ring A, increased both drug uptake of mitoxantrone and cytotoxicity by 2 to 5-fold in breast cancer cells. Qian et al [104] further corroborated such findings, similarly utilizing a dihydroxy-substituted dimethylchalcone and showing a 3.9-fold increase in cytotoxicity in combination with doxorubicin in a drug resistant cell line. They further demonstrated an in vivo increase in chemosensitivity to doxorubicin showing decreased tumor weights in a xenograft model using the same drug-resistant cell line [104]. Finally, Boumendjel et al [105] looked at their compound, JAI-51, noted to be structurally similar to that of Liu et al, and found a similar restoration of chemosensitivity to both daunorubicin and mitoxantrone in paired sensitive and resistant cell lines, indication that their chalcone inhibited both P-gp and BCRP. Further, their compound did not show any differential cytotoxicity in cell lines overexpressing either P-gp or BCRP compared to parental controls, implying that JAI-51 was not a substrate for these pumps. Unique to this study was the additional investigation of tubulin binding, which showed that in addition to efflux drug pump inhibition, JAI-51 also destabilized microtubules by binding in the colchicine-tubulin site. Antitumor activity was substantiated in an in vivo glioblastoma model. Since one of the limiting phenomena for the use of common microtubule destabilizers like paclitaxel or vinblastine is drug resistance mediated by P-gp or BCRP, these compounds may be active agents in taxane-resistant cancers [105]. De Vincenzo et al [49] by contrast, looked for activity of their 3-methyl-2-butenyl Ring A substituted chalcones against P-gp, but did not find significant inhibition. Gu et al [106] synthesized a series of bifendate-chalcone hybrids without stimulation on the P-gp ATPase activity and found that these chalcone hybrids are more potent than known P-gp inhibitors bifendate and verapamil. Thus, varying substitutions can clearly affect the utility of chalcones to bind and inhibit P-glycoprotein or BCRP, and the ideal chalcone would address both efflux pathways. The results described above are summarized in Table 6.
Table 6.
Chalcones target multidrug resistance transporters
| Lead Compound | Chemical structure | Mechanisms of Action | Reference |
|---|---|---|---|
| 3’, 4’, 5’-Trimethoxy-4-chlorochalcone | 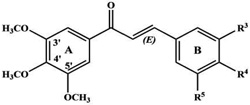 |
Inhibits the transport activity of P-glycoprotein. | Ivanova et al 2008 [101] |
| 3-(2,4-Dimethoxyphenyl)-1-(4-(piperazin-1-yl)phenyl)prop-2-en-1-one | 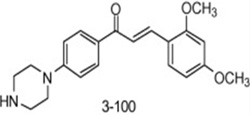 |
inhibits both P-glycoprotein and BCRP | Liu et al 2008 [102] |
| JAI-51 | 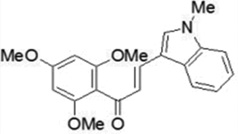 |
An inhibitor of P-glycoprotein and BCRP in in-vitro and in-vivo glioblastoma models. | Boumendjel et al 2009 [105] |
| Bifendate-chalcone hybrids | 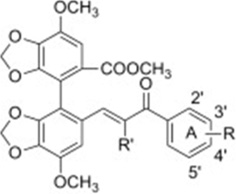 |
P-glycoprotein inhibitors with minimal cytotoxicity and no stimulation on the P-gp ATPase activity | Gu et al 2012 [106] |
Other drug resistance pathways or modulators, such as inhibition of glutathione S-transferase P1-1 (GSTP1-1) are less well studied, but Wang et al [107] has reported that carboxylic acid Ring A substitutions confer inhibition of the enzyme. The authors speculate that these compounds might conjugate with glutathione through Michael addition to act like suicide inhibitors of GSTP1-1. In a screening assay, trans-chalcones were found to activate the kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor 2 (NRF2) pathway suggesting their usefulness for detoxifying oxidative/electrophilic stress [108].
10. CHALCONES TARGET THE TUMOR VASCULATURE
Both targeting tumor neovascularization and existing tumor blood vessels using chalcones have been reported. With regard to targeting neovascularization, a number of authors have investigated chalcone derivatives’ efficacy in targeting endothelial cell invasion, migration and tube formation using in vitro assays [109–113]. Both Chang et al [114] and Albini et al [109] reported on the inhibition of NFκB and Akt pathways after chalcone exposure which was associated with subsequent endothelial cell apoptosis. This was accompanied by decreased endothelial migration, invasion, and formation of tube-like structures. Madan et al [115] similarly noted inhibition of NFκB in endothelial cells after chalcone exposure, noting that relatively high concentrations of their hydroxychalcone (up to 60 µM) were nontoxic to cells. Given that NFκB is a key transcriptional factor for cellular adhesion molecules (CAMs), the authors also investigated the expression of CAMs in endothelial cells and found that chalcone treatment decreased levels of various CAMs including VCAM and ICAM [116]. Since cellular adhesion molecules are important proangiogenic mediators of endothelial cell interaction and adherence in the tumor extracellular matrix, this data suggests that certain chalcones may induce antiangiogenic effects through multiple mechanisms, both intracellular and extracellular. Zhu and colleagues [117] demonstrated inhibition of VEGF-R2 (KDR) signaling as well as upstream Akt activation. This was correlated with decreased endothelial growth as well as VEGF-stimulated tumor growth in two xenograft models, due in part to decreased vessel density. In a structure-based virtual screening, Rizvi et al [118] identified a series of quinolyl-thienyl chalcones as potent VEGFR-2 kinase inhibitors with IC50 of 73.41 nM for the lead compound. Interestingly, several authors have reported that the effect of chalcones on endothelial cells, including decreased proliferation and apoptosis, seem to occur at very low concentrations when compared to those needed to reach IC50 levels in tumor cells. Nam and colleagues found their compound, 2-chloro-2',5'-dihydroxychalcone, to have an IC50 value up to 66-fold more potent in HUVEC cells compared to tumor cells [112]. Similarly, Pilatova et al [113] found that in concentrations non-toxic to tumor cells, their compound inhibited VEGF-induced migration of HUVECs, and decreased MMP-9 and VEGF secretion. Thus, these findings would suggest that given the proper chemical modifications, certain chalcones appear to selectively and potently target endothelial cells, but can also have cytotoxic effects on tumor cells in high enough concentrations. Further, several xenograft models have confirmed the activity of chalcones in significantly slowing tumor growth, indicating that these compounds likely effect both tumor cells as well as tumor endothelium. Thus, chalcones may offer a viable strategy to target multiple cells within the tumor compartment. The mechanisms of chalcones’ action are listed in Table 7.
Table 7.
Chalcones affect the tumor vasculature
| Lead Compound | Chemical structure | Mechanisms of Action | Reference |
|---|---|---|---|
| Boronic acid chalcone | 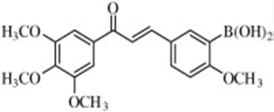 |
Inhibits angiogenesis in HUVEC tube formation and aortic ring assays. | Kong et al 2009 [110] |
| 2-Chloro-2',5'-dihydroxychalcone | 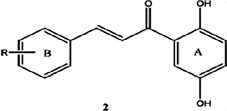 |
Exhibit the highest 66 fold selective toxicity toward HUVECs versus HCT116 cells | Nam et al 2003 [112] |
| 2'-Hydroxychalcone | 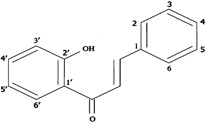 |
Inhibiting the expression of ICAM-1, VCAM-1, and E-selectin and the adhesion of peripheral neutrophils to the endothelial cell monolayers | Madan et al 2000 [115]. |
| Quinolyl-thienyl chalcones | 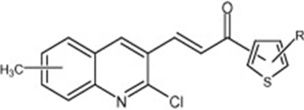 |
Inhibition of VEGF-R2 (KDR) kinase activity | Rizvi et al 2012 [118] |
Targeting existing tumor vasculature, as opposed to the neovasculature discussed above, has also been reported using chalcone derivatives. By and large, a significant proportion of the compounds which produce this effect are combretastatin analogues. Combretastatins, such as CA-4P, have been extensively studied and are currently in advanced stages of investigation in cancer clinical trials. Although it was first recognized that combretastatins functioned by anti-tubulin and antimitotic mechanisms, like colchicine they were also found to have significant effects in tumor blood vessels. The classic histopathologic finding of combretastatins is a central tumor necrosis, which ensues as a result of disruption of the tumor vessels which feed the tumor. This has not yet been fully investigated with chalcone-based combretastatin analogues, though similar properties, affecting both the cell cycle as well as tumor vessels have been demonstrated [25, 26, 110].
11. OTHER MOLECULAR CANCER TARGETS ARE MODULATED BY CHALCONES
Signal-transducer-and-activator-of-transcription 3 (STAT3) is a latent cytoplasmic transcription factor that plays pivotal roles in malignant transformation, cell growth, tumorigenesis, inflammation and angiogenesis by increasing the expression of various genes such as MMP2, MMP9, Bcl-xL, Bcl-2, cyclin D1, Mcl-1and VEGF. STAT3 is therefore considered as a suitable anti-cancer drug target. Funakoshi-Tago et al [119] have shown that Licochalcone A potently induce apoptosis of TEL-Jak2-transformed cells by inhibiting the phosphorylation and nuclear localization of STAT3 in human leukemia cells. Pandey et al [120] demonstrated that Butein inhibited both constitutive and inducible STAT 3 activation by induction of a protein tyrosine phosphatase SHP-1 in multiple myeloma (MM) cells. Cardamonin also exhibited an in vivo anti-inflammatory activity in lipopolysaccharide (LPS)-challenged peritoneal macrophages of ICR mice by suppressing IFN-γ induction and STATs-1, 2, 3 and 4 activation [121].
Human epidermal growth factor receptor kinases, such as EGFR, ErbB-2, and ErbB-3, have demonstrated their clinical values as cancer drug targets. Li et al [122] reported that ON-III (2', 4'-dihydroxy-6'-methoxy-3', 5'-dimethylchalcone) inhibits ErbB-2 tyrosine kinase phosphorylation and its mediated activation of AKT and MAPK, leading to induction of Bim expression and apoptosis in ErbB-2 overexpressing human breast cancer cells. Yang et al [68] have shown that Butein inhibits EGF-stimulated auto phosphotyrosine level and tyrosine-specific protein kinase activities of EGF receptor and p60c-src in vitro in Human hepatoma cells (HepG2) cells. Molecular modeling suggested that butein may be docked into the ATP binding pocket of EGFR. Jung et al [123] also showed that Isoliquiritigenin inhibited cell proliferation of human (DU145) and rat (MatLyLu) prostate cancer cells via inhibition of ErbB3 signaling and PI3K/Akt pathway.
The wingless-type (Wnt) pathway plays a central role in embryonic development and aberrant activation of the Wnt pathway contributes to the progression of several major human cancers [124]. Therefore, chalcones with Wnt inhibitor activity have major therapeutic potential. Cho et al [125] demonstrated that cardamonin the Wnt/beta-catenin signaling pathway through promoting the degradation of intracellular beta-catenin in HEK293 reporter cells and human normal melanocytes.
Agents that target the mammalian target of rapamycin (mTOR) signaling pathway are of significant clinical translational values. Sun et al [126] reported that the WJ9708011 (a methoxychalcone derivative) inhibited the mTOR pathway and protein synthesis via an Akt- and AMPK-independent fashion. Similarly, naturally-occurring chalcones, including licochalcone-A, isoliquiritigenin, cardamonin and xanthohumol were also shown to modulate the mTOR activity under different experimental conditions [127, 128]. Other important molecular targets, including c-Myc, Nrf2, mutant BRAF and aurora kinases are found to be modulated by chalcones (see Table 8).
Table 8.
Miscellaneous targets
| Lead compound | Chemical structure | Mechanisms of action | Reference |
|---|---|---|---|
| Licochalcone A | 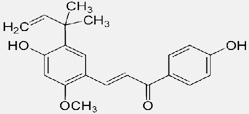 |
Prevents TEL-Jak2-mediated transformation through significant inhibition of the phosphorylation and nuclear localization of Stat3 | Funakoshi-Tago M et al 2008 [119] |
| ON-III (2',4'-dihydroxy-6'-methoxy-3',5'-dimethylchalcone) | 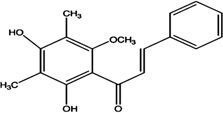 |
Inhibits ErbB-2 tyrosine kinase phosphorylation, disables AKT, MAPK, and downstream pathway, and induces apoptosis via induction of Bim | Li et al 2009 [122] |
| Isoliquiritigenin | 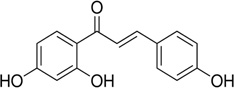 |
Inhibits cell proliferation via inhibition of ErbB3 signaling and PI3K/Akt pathway | Jung et al 2006 [123] |
| Cardamonin | 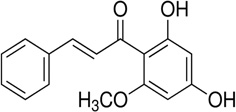 |
Promoting β-catenin degradation | Cho et al 2010 [125] |
| WJ9708011 (a methoxy chalcone derivative) | 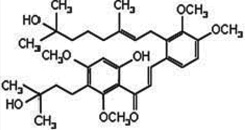 |
Inhibits mTOR signaling pathway | Sun et al 2010 [126] |
| Toluenesulfonylamido-chalcone, 4'-(p-toluene sulfonyl amino)-3,4-dihydroxy chalcone (TSHDC) | 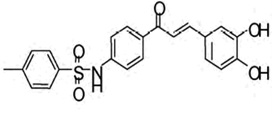 |
Increasing c-Myc-mediated reactive oxygen species production | Kim et al 2010 [129] |
| 2-trifluoromethyl-2'-methoxychalone | 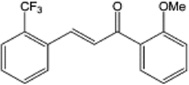 |
A potent activator of Nrf2, both in vitro and in vivo and independent of reactive oxygen species or redox changes. | Kumar et al 2011 [130] |
| A (E)-α-benzylsulfonyl chalcone derivative |
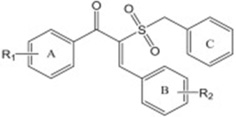 R1= 4-Cl; R2= 4-NO2 |
Exhibiting inhibitory activity with an IC50 value of 0.17 µM for BRAFV600E and GI50 value of 0.52 µM for mutant BRAF-dependent cells. | Li et al 2012 [131] |
| 1-(5-(2,4-Dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)naphthalen-2-ol | 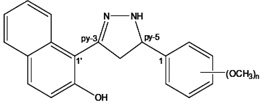 |
A selective inhibitor of aurora kinases A and B | Shin et al 2013 [132] |
12. SUMMARY AND PERSPECTIVES
Chalcones play a central role in the flavonoid synthesis pathway and is ubiquitously present in many kinds of natural products, including many dietary products like spices, tea, beer, fruits and vegetables. In different screening assays, chalcones have been able to target multiple cellular molecules, such as MDM2/p53, tubulin, proteasome, NF-kappa B, TRIAL/death receptors and mitochondria mediated apoptotic pathways, cell cycle, STAT3, AP-1, NRF2, AR, ER, PPAR-γ, β-catenin/Wnt and others (Figure 4). However, it remains unclear whether these concentrations of chalcones that were used in vitro can also be reached in in vivo conditions. Further experiments are necessary to examine the pharmacologically reachable concentrations of these chalcones. Nevertheless, chalcones are easy to chemically modify and synthesis to generate compounds with a wide variety of structural diversities. This property of chalcones makes chalcones to be very attractive as one of basic building blocks for synthesis of molecularly targeting agents. In the coming era of molecularly targeted therapy and personalized medicine, both naturally-occurring and synthetic chalcones may be very useful tools for studying basic mechanisms of cancer treatment, prevention and for developing novel agents for targeted cancer therapies.
Fig. (4).

Schematic presentation of different sturctural types of chalcones on a wide variety of molecular targets for their anti-cancer mechanisms.
Chalcones offer a very large repository of bioactive compounds with diverse molecular targets. Chalcones with even minor structural changes can result in targeting distinct cellular processes [71, 72, 110, 111]. The chemical structures of chalcones appear to play a critical role in determining their molecular targets. Some chalcones can have multiple targets and then affect various steps of carcinogenesis from tumor initiation to metastasis. Limited pilot clinical trials also have reported that some chalcones are well tolerated and non-toxic to humans and have reasonable pharmacokinetic properties [133]. Therefore, some lead chalcones are in the process of further characterization and optimization for investigative clinical trials for treatment of cancer, viral and cardiovascular disorders, and some have been in human use as cosmetic formulation ingredients and food additives [134–136]. These chalcones, either as nutraceuticals or as novel therapeutic agents, would provide a promising approach to cancer chemoprevention.
ACKNOWLEDGMENTS
We apologize for not being able to cite all of the publications in the field due to the limitations of the length of the review. XZ is supported by NIH grants R01CA122558 and DJ is supported in part by 3R01CA122558-03S1.
ABBREVIATIONS
- ABCB1
P-glycoprotein
- AM114
3, 5-bis-(4-boronic acid-benzylidene)-1-methyl-piperidin-4-one
- AP-1
the activator protein 1
- AR
androgen receptor
- ATP
Adenosine triphosphate
- BCRP or ABCG2
breast cancer resistance protein
- CDKs
cyclin-dependent kinases
- DR5/TRAILR2
death-receptor 5/ TNF-related apoptosis-inducing ligand receptor 2
- ER
estrogen receptor
- ELISA
The enzyme-linked immunosorbent assay
- FKB
flavokawain B
- GSH
Glutathione
- HSYA
Hydroxysafflor yellow A (HSYA)
- ICAM-1
Intercellular Adhesion Molecule 1
- ISL
isoliquiritigenin
- IKK
IĸBα kinase β
- Keap1
kelch-like ECH-associated protein 1
- LPS
lipopolysaccharide
- MDRs
multidrug efflux transporters
- MDM2
Mouse double minute 2 homolog
- MM
multiple myeloma
- NF-κB
nuclear factor-kappa B
- NRF2
nuclear factor erythroid 2-related factor 2
- RIP
Receptor-Interacting Protein
- NMR
Nuclear magnetic resonance
- NRF2
nuclear factor erythroid 2 [NF-E2]-related factor 2
- P-gp
P-glycoprotein
- PPAR-γ
peroxisome proliferator-activated receptor-γ
- RB
retinoblastoma
- SIRT1
sirtuin-1
- STAT3
Signal transducer and activator of transcription 3
- TAB1
TAK1-binding protein 1
- TAK1
the formation of the transforming growth factor-β-activated kinase 1 ()/
- TIBC
trans-4-Iodo, 4′-boranyl-chalcone
- TRAIL
TNF-related apoptosis-inducing ligand
- TSHDC
toluenesulfonylamido-chalcone, 4′-(p-toluene sulfonyl amino)-3, 4-dihydroxy chalcone
- UCC
urothelial cell carcinoma
- UPS
Ubiquitin-Proteasome System
- VCAM-1
Vascular cell adhesion protein 1
- VEGF-R2
vascular endothelial growth factor receptor 2
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.Orlikova B, Tasdemir D, Golais F, Dicato M, Diederich M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011;6:125–147. doi: 10.1007/s12263-011-0210-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yadav VR, Prasad S, Sung B, Aggarwal BB. The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int Immunopharmacol. 2011;11:295–309. doi: 10.1016/j.intimp.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimmock JR, Elias DW, Beazely MA, Kandepu NM. Bioactivities of chalcones. Curr Med Chem. 1999;6:1125–1149. [PubMed] [Google Scholar]
- 4.Bukhari SN, Jasamai M, Jantan I. Synthesis and biological evaluation of chalcone derivatives (mini review) Mini Rev Med Chem. 2012;12:1394–1403. doi: 10.2174/13895575112091394. [DOI] [PubMed] [Google Scholar]
- 5.Dy GK, Adjei AA. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA Cancer J Clin. 2013;63:249–279. doi: 10.3322/caac.21184. [DOI] [PubMed] [Google Scholar]
- 6.Tabernero J, Dirix L, Schöffski P, Cervantes A, Lopez-Martin JA, Capdevila J, van Beijsterveldt L, Platero S, Hall B, Yuan Z, Knoblauch R, Zhuang SH. A phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin Cancer Res. 2011;17:6313–6321. doi: 10.1158/1078-0432.CCR-11-1101. [DOI] [PubMed] [Google Scholar]
- 7.Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski F, Janson C, Tovar C, Filipovic ZM, Higgins B, Glenn K, Packman K, Vassilev LT, Graves B. Discovery of RG7388, a Potent and Selective p53-MDM2 Inhibitor in Clinical Development. J Med Chem. 2013 doi: 10.1021/jm400487c. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 8.Stoll R, Renner C, Hansen S, Palme S, Klein C, Belling A, Zeslawski W, Kamionka M, Rehm T, Mühlhahn P, Schumacher R, Hesse F, Kaluza B, Voelter W, Engh RA, Holak TA. Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry. 2001;40:336–344. doi: 10.1021/bi000930v. [DOI] [PubMed] [Google Scholar]
- 9.Kumar SK, Hager E, Pettit C, Gurulingappa H, Davidson NE, Khan SR. Design, synthesis, and evaluation of novel boronic-chalcone derivatives as antitumor agents. J Med Chem. 2003;46:2813–2825. doi: 10.1021/jm030213+. [DOI] [PubMed] [Google Scholar]
- 10.Modzelewska A, Pettit C, Achanta G, Davidson NE, Huang P, Khan SR. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg Med Chem. 2006;14:3491–3495. doi: 10.1016/j.bmc.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Hsu YL, Kuo PL, Lin CC. Isoliquiritigenin induces apoptosis and cell cycle arrest through p53-dependent pathway in Hep G2 cells. Life Sci. 2005;77:279–292. doi: 10.1016/j.lfs.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 12.Achanta G, Modzelewska A, Feng L, Khan SR, Huang P. A boronic-chalcone derivative exhibits potent anticancer activity through inhibition of the proteasome. Mol Pharmacol. 2006;70:426–433. doi: 10.1124/mol.105.021311. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z, Knutson E, Wang S, Martinez LA, Albrecht T. Stabilization of p53 in human cytomegalovirus-initiated cells is associated with sequestration of HDM2 and decreased p53 ubiquitination. Biol Chem. 2007;282:29284–29295. doi: 10.1074/jbc.M705349200. [DOI] [PubMed] [Google Scholar]
- 14.Sasayama T, Tanaka K, Mizukawa K, Kawamura A, Kondoh T, Hosoda K, Kohmura E. Trans-4-lodo,4'-boranyl-chalcone induces antitumor activity against malignant glioma cell lines in vitro and in vivo. J Neurooncol. 2007;85:123–132. doi: 10.1007/s11060-007-9395-2. [DOI] [PubMed] [Google Scholar]
- 15.D'Silva L, Ozdowy P, Krajewski M, Rothweiler U, Singh M, Holak TA. Monitoring the effects of antagonists on protein-protein interactions with NMR spectroscopy. J Am Chem Soc. 2005;127:13220–13226. doi: 10.1021/ja052143x. [DOI] [PubMed] [Google Scholar]
- 16.Lian ZQ, Zhao DL, Zhu HB. Hydroxysafflor yellow A up-regulates HIF-1alpha via inhibition of VHL and p53 in Eahy 926 cell line exposed to hypoxia] Yao Xue Xue Bao. 2008;43:484–489. In Chinese. [PubMed] [Google Scholar]
- 17.Kamal A, Dastagiri D, Ramaiah MJ, Reddy JS, Bharathi EV, Srinivas C, Pushpavalli SN, Pal D, Pal-Bhadra M. Synthesis of imidazothiazole-chalcone derivatives as anticancer and apoptosis inducing agents. Chem Med Chem. 2010;5:1937–1947. doi: 10.1002/cmdc.201000346. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Srinivasan B, Xing C, Lü J. A new chalcone derivative (E)-3-(4-methoxyphenyl)-2-methyl-1-(3, 4, 5-trimethoxyphenyl) prop-2-en-1-one suppresses prostate cancer involving p53-mediated cell cycle arrests and apoptosis. Anticancer Res. 2012;32:3689–3698. [PubMed] [Google Scholar]
- 19.Kim TH, Seo WD, Ryu HW, Seo HR, Jin YB, Lee M, Ji YH, Park KH, Lee YS. Anti-tumor effects by a synthetic chalcone compound is mediated by c-Myc-mediated reactive oxygen species production. Chem Biol Interact. 2010;188:111–118. doi: 10.1016/j.cbi.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 20.Tang Y, Simoneau AR, Xie J, Shahandeh B, Zi X. Effects of the kava chalcone flavokawain A differ in bladder cancer cells with wild-type versus mutant p53. Cancer Prev Res (Phila Pa) 2008;1:439–451. doi: 10.1158/1940-6207.CAPR-08-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, Xu X, Li X, Liu S, Simoneau AR, He F, Wu XR, Zi X. KAVA chalcone, Flavokawain A, inhibits urothelial tumorigenesis in the UPII-SV40T transgenic mouse model. Cancer Prev Res (Phila) 2013;6:1365–1375. doi: 10.1158/1940-6207.CAPR-13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kahyo T, Ichikawa S, Hatanaka T, Yamada MK, Setou M. A novel chalcone polyphenol inhibits the deacetylase activity of SIRT1 and cell growth in HEK293T cells. J Pharmacol Sci. 2008;108:364–371. doi: 10.1254/jphs.08203fp. [DOI] [PubMed] [Google Scholar]
- 23.Peyrot V, Leynadier D, Sarrazin M, Briand C, Rodriquez A, Nieto JM, Andreu JM. Interaction of tubulin and cellular microtubules with the new antitumor drug MDL 27048. A powerful and reversible microtubule inhibitor. J Biol Chem. 1989;264:21296–21301. [PubMed] [Google Scholar]
- 24.Boumendjel A, Boccard J, Carrupt PA, Nicolle E, Blanc M, Geze A, Choisnard L, Wouessidjewe D, Matera EL, Dumontet C. Antimitotic and antiproliferative activities of chalcones: forward structure-activity relationship. J Med Chem. 2008;51:2307–2310. doi: 10.1021/jm0708331. [DOI] [PubMed] [Google Scholar]
- 25.Ducki S, Mackenzie G, Greedy B, Armitage S, Chabert JF, Bennett E, Nettles J, Snyder JP, Lawrence NJ. Combretastatin-like chalcones as inhibitors of microtubule polymerisation. Part 2: Structure-based discovery of alpha-aryl chalcones. Bioorg Med Chem. 2009;17:7711–7722. doi: 10.1016/j.bmc.2009.09.044. [DOI] [PubMed] [Google Scholar]
- 26.Ducki S, Rennison D, Woo M, Kendall A, Chabert JF, McGown AT, Lawrence NJ. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: synthesis and biological evaluation of antivascular activity. Bioorg Med Chem. 2009;17:7698–7710. doi: 10.1016/j.bmc.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 27.Romagnoli R, Baraldi PG, Carrion MD, Cara CL, Cruz-Lopez O, Preti D, Tolomeo M, Grimaudo S, Di Cristina A, Zonta N, Balzarini J, Brancale A, Sarkar T, Hamel E. Design, synthesis, and biological evaluation of thiophene analogues of chalcones. Bioorg Med Chem. 2008;16:5367–5376. doi: 10.1016/j.bmc.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 28.ter Haar E, Hamel E, Balachandran R, Day BW. Cellular targets of the anti-breast cancer agent Z-1,1-dichloro-2,3-diphenylcyclopropane: type II estrogen binding sites and tubulin. Anticancer Res. 1997;17:1861–1869. [PubMed] [Google Scholar]
- 29.Zoldakova M, Kornyei Z, Brown A, Biersack B, Madarasz E, Schobert R. Effects of a combretastatin A4 analogous chalcone and its Pt-complex on cancer cells: A comparative study of uptake, cell cycle and damage to cellular compartments. Biochem Pharmacol. Pharmacol. 2010;80:1487–1496. doi: 10.1016/j.bcp.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 30.Tu HY, Huang AM, Hour TC, Yang SC, Pu YS, Lin CN. Synthesis and biological evaluation of 2',5'-dimethoxychalcone derivatives as microtubule-targeted anticancer agents. Bioorg Med Chem. 2010;18:2089–2098. doi: 10.1016/j.bmc.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 31.Ducki S, Forrest R, Hadfield JA, Kendall A, Lawrence NJ, McGown AT, Rennison D. Potent antimitotic and cell growth inhibitory properties of substituted chalcones. Bioorg Med Chem Lett. 1998;8:1051–1056. doi: 10.1016/s0960-894x(98)00162-0. [DOI] [PubMed] [Google Scholar]
- 32.Quintin J, Desrivot J, Thoret S, Le Menez P, Cresteil T, Lewin G. Synthesis and biological evaluation of a series of tangeretin-derived chalcones. Bioorg Med Chem Lett. 2009;19:167–169. doi: 10.1016/j.bmcl.2008.10.126. [DOI] [PubMed] [Google Scholar]
- 33.Utsintong M, Massarotti A, Caldarelli A, Theeramunkong S. Parallel synthesis of "click" chalcones as antitubulin agents. Med Chem. 2013;9:510–516. doi: 10.2174/1573406411309040004. [DOI] [PubMed] [Google Scholar]
- 34.Vitorović-Todorović MD, Erić-Nikolić A, Kolundžija B, Hamel E, Ristić S, Juranić IO, Drakulić BJ. (E)-4-aryl-4-oxo-2-butenoic acid amides, chalcone-aroylacrylic acid chimeras: design, antiproliferative activity and inhibition of tubulin polymerization. Eur J Med Chem. 2013;62:40. doi: 10.1016/j.ejmech.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schobert R, Biersack B, Dietrich A, Knauer S, Zoldakova M, Fruehauf A, Mueller T. Pt(II) complexes of a combretastatin A-4 analogous chalcone: effects of conjugation on cytotoxicity, tumor specificity, and long-term tumor growth suppression. J Med Chem. 2009;52:241–246. doi: 10.1021/jm801001d. [DOI] [PubMed] [Google Scholar]
- 36.Ruan BF, Lu X, Tang JF, Wei Y, Wang XL, Zhang YB, Wang LS, Zhu HL. Synthesis, biological evaluation, and molecular docking studies of resveratrol derivatives possessing chalcone moiety as potential antitubulin agents. Bioorg Med Chem. 2011;19:2688–2695. doi: 10.1016/j.bmc.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Mesenzani O, Massarotti A, Giustiniano M, Pirali T, Bevilacqua V, Caldarelli A, Canonico P, Sorba G, Novellino E, Genazzani AA, Tron GC. Replacement of the double bond of antitubulin chalcones with triazoles and tetrazoles: Synthesis and biological evaluation. Bioorg Med Chem Lett. 2011;21:764–768. doi: 10.1016/j.bmcl.2010.11.113. [DOI] [PubMed] [Google Scholar]
- 38.Dyrager C, Wickström M, Fridén-Saxin M, Friberg A, Dahlén K, Wallén EA, Gullbo J, Grøtli M, Luthman K. Inhibitors and promoters of tubulin polymerization: synthesis and biological evaluation of chalcones and related dienones as potential anticancer agents. Bioorg Med Chem. 2011;19:2659–2665. doi: 10.1016/j.bmc.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Crawford LJ, Walker B, Irvine AE. Proteasome inhibitors in cancer therapy. Journal of Cell Communication and Signaling. 2011;5:101–110. doi: 10.1007/s12079-011-0121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. 2008;12:164–179. doi: 10.1016/j.drup.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Bazzaro M, Anchoori RK, Mudiam MK, Issaenko O, Kumar S, Karanam B, Lin Z, Isaksson Vogel R, Gavioli R, Destro F, Ferretti V, Roden RB, Khan SR. α, β-Unsaturated carbonyl system of chalcone-based derivatives is responsible for broad inhibition of proteasomal activity and preferential killing of human papilloma virus (HPV) positive cervical cancer cells. J Med Chem. 2011;54:449–456. doi: 10.1021/jm100589p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anchoori RK, Khan SR, Sueblinvong T, Felthauser A, Iizuka Y, Gavioli R, Destro F, Isaksson Vogel R, Peng S, Roden RB, Bazzaro M. Stressing the ubiquitin-proteasome system without 20S proteolytic inhibition selectively kills cervical cancer cells. PLoS One. 2011;6:e23888. doi: 10.1371/journal.pone.0023888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Issaenko OA, Amerik AY. Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes. Cell Cycle. 2012;11:1804–1817. doi: 10.4161/cc.20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eskander RN, Randall LM, Sakai T, Guo Y, Hoang B, Zi X. Flavokawain B, a novel, naturally occurring chalcone, exhibits robust apoptotic effects and induces G2/M arrest of a uterine leiomyosarcoma cell line. J Obstet Gynaecol Res. 2012;38:1086–1094. doi: 10.1111/j.1447-0756.2011.01841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ji T, Lin C, Krill LS, Eskander R, Guo Y, Zi X, Hoang BH. Flavokawain B, a kava chalcone, inhibits growth of human osteosarcoma cells through G2/M cell cycle arrest and apoptosis. Mol Cancer. 2013;12:55. doi: 10.1186/1476-4598-12-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsieh TC, Guo J, Kunicki J, Lee MY, Darzynkiewicz Z, Wu JM. Licochalcone-A, a novel flavonoid isolated from licorice root (Glycyrrhiza glabra), causes G2 and late-G1 arrests in androgen-independent PC-3 prostate cancer cells. Biochem Biophys Res Commun. 2004;322:263–270. doi: 10.1016/j.bbrc.2004.07.094. [DOI] [PubMed] [Google Scholar]
- 47.Lee YM, Lim DY, Choi HJ, Jung JI, Chung WY, Park JH. Induction of cell cycle arrest in prostate cancer cells by the dietary compound isoliquiritigenin. J Med Food. 2009;12:8–14. doi: 10.1089/jmf.2008.0039. [DOI] [PubMed] [Google Scholar]
- 48.Park I, Park KK, Park JH, Chung WY. Isoliquiritigenin induces G2 and M phase arrest by inducing DNA damage and by inhibiting the metaphase/anaphase transition. Cancer Lett. 2009;277:174–181. doi: 10.1016/j.canlet.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 49.De Vincenzo R, Ferlini C, Distefano M, Gaggini C, Riva A, Bombardelli E, Morazzoni P, Valenti P, Belluti F, Ranelletti FO, Mancuso S, Scambia G. In vitro evaluation of newly developed chalcone analogues in human cancer cells. Cancer Chemother Pharmacol. 2000;46:305–312. doi: 10.1007/s002800000160. [DOI] [PubMed] [Google Scholar]
- 50.Ii T, Satomi Y, Katoh D, Shimada J, Baba M, Okuyama T, Nishino H, Kitamura N. Induction of cell cycle arrest and p21(CIP1/WAF1) expression in human lung cancer cells by isoliquiritigenin. Cancer Lett. 2004;207:27–35. doi: 10.1016/j.canlet.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 51.Park S, Gwak J, Han SJ, Oh S. Cardamonin Suppresses the Proliferation of Colon Cancer Cells by Promoting β-Catenin Degradation. Biological and Pharmaceutical Bulletin. 2013;6:1040–1044. doi: 10.1248/bpb.b13-00158. [DOI] [PubMed] [Google Scholar]
- 52.Hsu YL, Chia CC, Chen PJ, Huang SE, Huang SC, Kuo PL. Shallot and licorice constituent isoliquiritigenin arrests cell cycle progression and induces apoptosis through the induction of ATM/p53 and initiation of the mitochondrial system in human cervical carcinoma HeLa cells. Mol Nutr Food Res. 2009;53:826–835. doi: 10.1002/mnfr.200800288. [DOI] [PubMed] [Google Scholar]
- 53.Shen KH, Chang JK, Hsu YL, Kuo PL. Chalcone arrests cell cycle progression and induces apoptosis through induction of mitochondrial pathway and inhibition of nuclear factor kappa B signalling in human bladder cancer cells. Basic Clin Pharmacol Toxicol. 2007;101:254–261. doi: 10.1111/j.1742-7843.2007.00120.x. [DOI] [PubMed] [Google Scholar]
- 54.Yun JM, Kweon MH, Kwon H, Hwang JK, Mukhtar H. Induction of apoptosis and cell cycle arrest by a chalcone panduratin A isolated from Kaempferia pandurata in androgen-independent human prostate cancer cells PC3 and DU145. Carcinogenesis. 2006;27:1454–1464. doi: 10.1093/carcin/bgi348. [DOI] [PubMed] [Google Scholar]
- 55.Nishimura R, Tabata K, Arakawa M, Ito Y, Kimura Y, Akihisa T, Nagai H, Sakuma A, Kohno H, Suzuki T. Isobavachalcone, a chalcone constituent of Angelica keiskei, induces apoptosis in neuroblastoma. Biol Pharm Bull. 2007;30:1878–1883. doi: 10.1248/bpb.30.1878. [DOI] [PubMed] [Google Scholar]
- 56.Sasayama T, Tanaka K, Mizukawa K, Kawamura A, Kondoh T, Hosoda K, Kohmura E. Trans-4-lodo,4'-boranyl-chalcone induces antitumor activity against malignant glioma cell lines in vitro and in vivo. J Neurooncol. 2007;85:123–132. doi: 10.1007/s11060-007-9395-2. [DOI] [PubMed] [Google Scholar]
- 57.Echeverria C, Santibanez JF, Donoso-Tauda O, Escobar CA, Ramirez-Tagle R. Structural antitumoral activity relationships of synthetic chalcones. Int J Mol Sci. 2009;10:221–231. doi: 10.3390/ijms10010221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Navarini AL, Chiaradia LD, Mascarello A, Fritzen M, Nunes RJ, Yunes RA, Creczynski-Pasa TB. Hydroxychalcones induce apoptosis in B16-F10 melanoma cells via GSH and ATP depletion. Eur J Med Chem. 2009;44:1630–1637. doi: 10.1016/j.ejmech.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 59.Jung JI, Lim SS, Choi HJ, Cho HJ, Shin HK, Kim EJ, Chung WY, Park KK, Park JH. Isoliquiritigenin induces apoptosis by depolarizing mitochondrial membranes in prostate cancer cells. J Nutr Biochem. 2006;17:689–696. doi: 10.1016/j.jnutbio.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 60.Sabzevari O, Galati G, Moridani MY, Siraki A, O'Brien PJ. Molecular cytotoxic mechanisms of anticancer hydroxychalcones. Chem Biol Interact. 2004;148:57–67. doi: 10.1016/j.cbi.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 61.Sun ZJ, Chen G, Zhang W, Hu X, Huang CF, Wang YF, Jia J, Zhao YF. Mammalian target of rapamycin pathway promotes tumor-induced angiogenesis in adenoid cystic carcinoma: its suppression by isoliquiritigenin through dual activation of c-Jun NH2-terminal kinase and inhibition of extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2010;334:500–512. doi: 10.1124/jpet.110.167692. [DOI] [PubMed] [Google Scholar]
- 62.Sakai T, Eskander RN, Guo Y, Kim KJ, Mefford J, Hopkins J, Bhatia NN, Zi X, Hoang BH. Flavokawain B, a kava chalcone, induces apoptosis in synovial sarcoma cell lines. J Orthop Res. 2012;30:1045–1050. doi: 10.1002/jor.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zi X, Simoneau AR. Flavokawain A, a novel chalcone from kava extract, induces apoptosis in bladder cancer cells by involvement of Bax protein-dependent and mitochondria-dependent apoptotic pathway and suppresses tumor growth in mice. Cancer Res. 2005;65:3479–3486. doi: 10.1158/0008-5472.CAN-04-3803. [DOI] [PubMed] [Google Scholar]
- 64.Tang Y, Li X, Liu Z, Simoneau AR, Xie J, Zi X. Flavokawain B, a kava chalcone, induces apoptosis via up-regulation of death-receptor 5 and Bim expression in androgen receptor negative, hormonal refractory prostate cancer cell lines and reduces tumor growth. Int J Cancer. 2010;127:1758–1768. doi: 10.1002/ijc.25210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim N. Butein sensitizes human leukemia cells to apoptosis induced by tumor necrosis factor-related apoptosis inducing ligand (TRAIL) Arch Pharm Res. 2008;31:1179–1186. doi: 10.1007/s12272-001-1286-2. [DOI] [PubMed] [Google Scholar]
- 66.Ohtsuki T, Kikuchi H, Koyano T, Kowithayakorn T, Sakai T, Ishibashi M. Death receptor 5 promoter-enhancing compounds isolated from Catimbium speciosum and their enhancement effect on TRAIL-induced apoptosis. Bioorg Med Chem. 2009;17:6748–6754. doi: 10.1016/j.bmc.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 67.Yoshida T, Horinaka M, Takara M, Tsuchihashi M, Mukai N, Wakada M, Sakai T. Combination of isoliquiritigenin and tumor necrosis factor-related apoptosis-inducing ligand induces apoptosis in colon cancer HT29 cells. Environ Health Prev Med. 2008;13:281–287. doi: 10.1007/s12199-008-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang EB, Zhang K, Cheng LY, Mack P. Butein, a specific protein tyrosine kinase inhibitor. Biochem Biophys Res Commun. 1998;245:435–438. doi: 10.1006/bbrc.1998.8452. [DOI] [PubMed] [Google Scholar]
- 69.Szliszka E, Czuba ZP, Mazur B, Sedek L, Paradysz A, Krol W. Chalcones enhance TRAIL-induced apoptosis in prostate cancer cells. Int J Mol Sci. 2009;11:1–13. doi: 10.3390/ijms11010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Szliszka E, Czuba ZP, Mazur B, Paradysz A, Krol W. Chalcones and dihydrochalcones augment TRAIL-mediated apoptosis in prostate cancer cells. Molecules. 2010;15:5336–5353. doi: 10.3390/molecules15085336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Champelovier P, Chauchet X, Hazane-Puch F, Vergnaud S, Garrel C, Laporte F, Boutonnat J, Boumendjel A. Cellular and molecular mechanisms activating the cell death processes by chalcones: Critical structural effects. Toxicol In Vitro. 2013;27:2305–2315. doi: 10.1016/j.tiv.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 72.He W, Wang Q, Srinivasan B, Xu J, Padilla MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, Xing C, Lin Y. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene. 2013 doi: 10.1038/onc.2013.256. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson MW, Overmeyer JH, Young AM, Erhardt PW, Maltese WA. Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. J Med Chem. 2012;55:1940–1956. doi: 10.1021/jm201006x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Srinivasan B, Johnson TE, Lad R, Xing C. Structure-activity relationship studies of chalcone leading to 3-hydroxy-4,3',4',5'-tetramethoxychalcone and its analogues as potent nuclear factor kappaB inhibitors and their anticancer activities. J Med Chem. 2009;52:7228–7235. doi: 10.1021/jm901278z. [DOI] [PubMed] [Google Scholar]
- 75.Liu YC, Hsieh CW, Wu CC, Wung BS. Chalcone inhibits the activation of NF-kappaB and STAT3 in endothelial cells via endogenous electrophile. Life Sci. 2007;80:1420–1430. doi: 10.1016/j.lfs.2006.12.040. [DOI] [PubMed] [Google Scholar]
- 76.Gan FF, Kaminska KK, Yang H, Liew CY, Leow PC, So CL, Tu LN, Roy A, Yap CW, Kang TS, Chui WK, Chew EH. Identification of Michael acceptor-centric pharmacophores with substituents that yield strong thioredoxin reductase inhibitory character correlated to antiproliferative activity. Antioxid Redox Signal. 2013;19:1149–1165. doi: 10.1089/ars.2012.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Venkateswararao E, Sharma VK, Lee KC, Sharma N, Park SH, Kim Y, Jung SH. A SAR study on a series of synthetic lipophilic chalcones as inhibitor of transcription factor NF-κB. Eur J Med Chem. 2012;54:379–386. doi: 10.1016/j.ejmech.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 78.Pandey MK, Sandur SK, Sung B, Sethi G, Kunnumakkara AB, Aggarwal BB. Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene expression through direct inhibition of IkappaBalpha kinase beta on cysteine 179 residue. J Biol Chem. 2007;282:17340–17350. doi: 10.1074/jbc.M700890200. [DOI] [PubMed] [Google Scholar]
- 79.Zhang L, Chen W, Li X. A novel anticancer effect of butein: inhibition of invasion through the ERK1/2 and NF-kappa B signaling pathways in bladder cancer cells. FEBS Lett. 2008;582:1821–1828. doi: 10.1016/j.febslet.2008.04.046. [DOI] [PubMed] [Google Scholar]
- 80.Kumar S, Sharma A, Madan B, Singhal V, Ghosh B. Isoliquiritigenin inhibits IkappaB kinase activity and ROS generation to block TNF-alpha induced expression of cell adhesion molecules on human endothelial cells. Biochem Pharmacol. 2007;73:1602–1612. doi: 10.1016/j.bcp.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 81.Harikumar KB, Kunnumakkara AB, Ahn KS, Anand P, Krishnan S, Guha S, Aggarwal BB. Modification of the cysteine residues in IkappaBalpha kinase and NF-kappaB (p65) by xanthohumol leads to suppression of NF-kappaB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood. 2009;113:2003–2013. doi: 10.1182/blood-2008-04-151944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lorenzo P, Alvarez R, Ortiz MA, Alvarez S, Piedrafita FJ, de Lera AR. Inhibition of IkappaB kinase-beta and anticancer activities of novel chalcone adamantyl arotinoids. J Med Chem. 2008;51:5431–5440. doi: 10.1021/jm800285f. [DOI] [PubMed] [Google Scholar]
- 83.Zhou P, Gross S, Liu JH, Yu BY, Feng LL, Nolta J, Sharma V, Piwnica-Worms D, Qiu SX. Flavokawain B, the hepatotoxic constituent from kava root, induces GSH-sensitive oxidative stress through modulation of IKK/NF-{kappa}B and MAPK signaling pathways. FASEB J. 2010;24:4722–4732. doi: 10.1096/fj.10-163311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Folmer F, Blasius R, Morceau F, Tabudravu J, Dicato M, Jaspars M, Diederich M. Inhibition of TNFalpha-induced activation of nuclear factor kappaB by kava (Piper methysticum) derivatives. Biochem Pharmacol. 2006;71:1206–1218. doi: 10.1016/j.bcp.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 85.Orlikova B, Tasdemir D, Golais F, Dicato M, Diederich M. The aromatic ketone 4'-hydroxychalcone inhibits TNFα-induced NF-κB activation via proteasome inhibition. Biochem Pharmacol. 2011;82:620–631. doi: 10.1016/j.bcp.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 86.Hsu YL, Kuo PL, Tzeng WS, Lin CC. Chalcone inhibits the proliferation of human breast cancer cell by blocking cell cycle progression and inducing apoptosis. Food Chem Toxicol. 2006;44:704–713. doi: 10.1016/j.fct.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 87.Kim YJ, Kim HC, Ko H, Amor EC, Lee JW, Yang HO. Stercurensin inhibits nuclear factor-κB-dependent inflammatory signals through attenuation of TAK1-TAB1 complex formation. J Cell Biochem. 2011;112:548–558. doi: 10.1002/jcb.22945. [DOI] [PubMed] [Google Scholar]
- 88.Reddy MV, Shen YC, Yang JS, Hwang TL, Bastow KF, Qian K, Lee KH, Wu TS. New bichalcone analogs as NF-κB inhibitors and as cytotoxic agents inducing Fas/CD95-dependent apoptosis. Bioorg Med Chem. 2011;19:1895–1906. doi: 10.1016/j.bmc.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shirota O, Takizawa K, Sekita S, Satake M, Hirayama Y, Hakamata Y, Hayashi T, Yanagawa T. Antiandrogenic natural Diels--Alder-type adducts from Brosimum rubescens. J Nat Prod. 1997;60:997–1002. doi: 10.1021/np9607215. [DOI] [PubMed] [Google Scholar]
- 90.Zhou J, Geng G, Wu JH. Synthesis and in vitro characterization of ionone-based chalcones as novel antiandrogens effective against multiple clinically relevant androgen receptor mutants. Invest New Drug. 2010;28:291–298. doi: 10.1007/s10637-009-9251-7. [DOI] [PubMed] [Google Scholar]
- 91.Chen S, Gao J, Halicka HD, Traganos F, Darzynkiewicz Z. Down-regulation of androgen-receptor and PSA by phytochemicals. Int J Oncol. 2008;32:405–411. [PMC free article] [PubMed] [Google Scholar]
- 92.Kim YS, Kumar V, Lee S, Iwai A, Neckers L, Malhotra SV, Trepel JB. Methoxychalcone inhibitors of androgen receptor translocation and function. Bioorg Med Chem Lett. 2012;2:2105–2109. doi: 10.1016/j.bmcl.2011.12.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Satomi Y. Inhibitory effects of 3'-methyl-3-hydroxy-chalcone on proliferation of human malignant tumor cells and on skin carcinogenesis. Int J Cancer. 1993;55:506–514. doi: 10.1002/ijc.2910550330. [DOI] [PubMed] [Google Scholar]
- 94.Wang Y, Chan FL, Chen S, Leung LK. The plant polyphenol butein inhibits testosterone-induced proliferation in breast cancer cells expressing aromatase. Life Sci. 2005;77:39–51. doi: 10.1016/j.lfs.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 95.Ye L, Gho WM, Chan FL, Chen S, Leung LK. Dietary administration of the licorice flavonoid isoliquiritigenin deters the growth of MCF-7 cells overexpressing aromatase. Int J Cancer. 2009;124:1028–1036. doi: 10.1002/ijc.24046. [DOI] [PubMed] [Google Scholar]
- 96.Maggiolini M, Statti G, Vivacqua A, Gabriele S, Rago V, Loizzo M, Menichini F, Amdò S. Estrogenic and antiproliferative activities of isoliquiritigenin in MCF7 breast cancer cells. J Steroid Biochem Mol Biol. 2002;82:315–322. doi: 10.1016/s0960-0760(02)00230-3. [DOI] [PubMed] [Google Scholar]
- 97.Rafi MM, Rosen RT, Vassil A, Ho CT, Zhang H, Ghai G, Lambert G, DiPaola RS. Modulation of bcl-2 and cytotoxicity by licochalcone-A, a novel estrogenic flavonoid. Anticancer Res. 2000;20:2653–2658. [PubMed] [Google Scholar]
- 98.Calliste CA, Le Bail JC, Trouillas P, Pouget C, Habrioux G, Chulia AJ, Duroux JL. Chalcones: structural requirements for antioxidant, estrogenic and antiproliferative activities. Anticancer Res. 2001;21:3949–3956. [PubMed] [Google Scholar]
- 99.Bois F, Beney C, Boumendjel A, Mariotte AM, Conseil G, Di Pietro A. Halogenated chalcones with high-affinity binding to P-glycoprotein: potential modulators of multidrug resistance. J Med Chem. 1998;41:4161–4164. doi: 10.1021/jm9810194. [DOI] [PubMed] [Google Scholar]
- 100.Bois F, Boumendjel A, Mariotte AM, Conseil G, Di Petro A. Synthesis and biological activity of 4-alkoxy chalcones: potential hydrophobic modulators of P-glycoprotein-mediated multidrug resistance. Bioorg Med Chem. 1999;7:2691–2695. doi: 10.1016/s0968-0896(99)00218-7. [DOI] [PubMed] [Google Scholar]
- 101.Ivanova A, Batovska D, Engi H, Parushev S, Ocsovszki I, Kostova I, Molnar J. MDR-reversal activity of chalcones. In Vivo. 2008;22:379–384. [PubMed] [Google Scholar]
- 102.Liu XL, Tee HW, Go ML. Functionalized chalcones as selective inhibitors of P-glycoprotein and breast cancer resistance protein. Bioorg Med Chem. 2008;16:171–180. doi: 10.1016/j.bmc.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 103.Han Y, Riwanto M, Go ML, Ee PL. Modulation of breast cancer resistance protein (BCRP/ABCG2) by non-basic chalcone analogues. Eur J Pharm Sci. 2008;35:30–41. doi: 10.1016/j.ejps.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 104.Qian F, Ye CL, Wei DZ, Lu YH, Yang SL. In vitro and in vivo reversal of cancer cell multidrug resistance by 2',4'-dihydroxy-6'-methoxy-3',5'-dimethylchalcone. J Chemother. 2005;17:309–314. doi: 10.1179/joc.2005.17.3.309. [DOI] [PubMed] [Google Scholar]
- 105.Boumendjel A, McLeer-Florin A, Champelovier P, Allegro D, Muhammad D, Souard F, Derouazi M, Peyrot V, Toussaint B, Boutonnat J. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models. BMC Cancer. 2009;9:242. doi: 10.1186/1471-2407-9-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gu X, Ren Z, Tang X, Peng H, Ma Y, Lai Y, Peng S, Zhang Y. Synthesis and biological evaluation of bifendate-chalcone hybrids as a new class of potential P-glycoprotein inhibitors. Bioorg Med Chem. 2012;20:2540–2548. doi: 10.1016/j.bmc.2012.02.050. [DOI] [PubMed] [Google Scholar]
- 107.Wang J, Wang S, Song D, Wang J, Wang S, Song D, Zhao D, Sha Y, Jiang Y, Jing Y, Cheng M, et al. Chalcone derivatives inhibit glutathione S-transferase P1-1 activity: insights into the interaction mode of alpha, beta-unsaturated carbonyl compounds. Chem Biol Drug Des. 2009;73:511–514. doi: 10.1111/j.1747-0285.2009.00807.x. [DOI] [PubMed] [Google Scholar]
- 108.Wu KC, McDonald PR, Liu J, Klaassen CD. Screening of Natural Compounds as Activators of the Keap1-Nrf2 Pathway. Planta Med. 2013 doi: 10.1055/s-0033-1351097. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Albini A, Dell'Eva R, Vene R, Ferrari N, Buhler DR, Noonan DM, Fassina G. Mechanisms of the antiangiogenic activity by the hop flavonoid xanthohumol: NF-kappaB and Akt as targets. FASEB J. 2006;20:527–529. doi: 10.1096/fj.05-5128fje. [DOI] [PubMed] [Google Scholar]
- 110.Kong Y, Wang K, Edler MC, Hamel E, Mooberry SL, Paige MA, Brown ML. A boronic acid chalcone analog of combretastatin A-4 as a potent anti-proliferation agent. Bioorg Med Chem. 2009;18:971–977. doi: 10.1016/j.bmc.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee YS, Lim SS, Shin KH, Kim YS, Ohuchi K, Jung SH. Anti-angiogenic and anti-tumor activities of 2'-hydroxy-4'-methoxychalcone. Biol Pharm Bull. 2006;29:1028–1031. doi: 10.1248/bpb.29.1028. [DOI] [PubMed] [Google Scholar]
- 112.Nam NH, Kim Y, You YJ, Hong DH, Kim HM, Ahn BZ. Cytotoxic 2',5'-dihydroxychalcones with unexpected antiangiogenic activity. Eur J Med Chem. 2003;38:179–187. doi: 10.1016/s0223-5234(02)01443-5. [DOI] [PubMed] [Google Scholar]
- 113.Pilatova M, Varinska L, Perjesi P, Sarissky M, Mirossay L, Solar P, Ostro A, Mojzis J. In vitro antiproliferative and antiangiogenic effects of synthetic chalcone analogues. Toxicol In Vitro. 2010;24:1347–1355. doi: 10.1016/j.tiv.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 114.Chang HJ, Yoon G, Park JS, Kim MH, Baek MK, Kim NH, Shin BA, Ahn BW, Cheon SH, Jung YD. Induction of apoptosis by the licochalcone E in endothelial cells via modulation of NF-kappaB and Bcl-2 Family. Biol Pharm Bull. 2007;30:2290–2293. doi: 10.1248/bpb.30.2290. [DOI] [PubMed] [Google Scholar]
- 115.Madan B, Batra S, Ghosh B. 2'-hydroxychalcone inhibits nuclear factor-kappaB and blocks tumor necrosis factor-alpha- and lipopolysaccharide-induced adhesion of neutrophils to human umbilical vein endothelial cells. Mol Pharmacol. 2000;58:526–534. doi: 10.1124/mol.58.3.526. [DOI] [PubMed] [Google Scholar]
- 116.Dell'Eva R, Ambrosini C, Vannini N, Piaggio G, Albini A, Ferrari N. AKT/NF-kappaB inhibitor xanthohumol targets cell growth and angiogenesis in hematologic malignancies. Cancer. 2007;110:2007–2011. doi: 10.1002/cncr.23017. [DOI] [PubMed] [Google Scholar]
- 117.Zhu XF, Xie BF, Zhou JM, Feng GK, Liu ZC, Wei XY, Zhang FX, Liu MF, Zeng YX. Blockade of vascular endothelial growth factor receptor signal pathway and antitumor activity of ON-III (2',4'-dihydroxy-6'-methoxy-3',5'-dimethylchalcone), a component from Chinese herbal medicine. Mol Pharmacol. 2005;67:1444–1450. doi: 10.1124/mol.104.009894. [DOI] [PubMed] [Google Scholar]
- 118.Rizvi SU, Siddiqui HL, Nisar M, Khan N, Khan I. Discovery and molecular docking of quinolylthienyl chalcones as anti-angiogenic agents targeting VEGFR-2 tyrosine kinase. Bioorg Med Chem Lett. 2012;22:942–944. doi: 10.1016/j.bmcl.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 119.Funakoshi-Tago M, Tago K, Nishizawa C, Takahashi K, Mashino T, Iwata S, Inoue H, Sonoda Y, Kasahara T. Licochalcone A is a potent inhibitor of TEL-Jak2-mediated transformation through the specific inhibition of Stat3 activation. Biochem Pharmacol. 2008;76:1681–1693. doi: 10.1016/j.bcp.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 120.Pandey MK, Sung B, Ahn KS, Aggarwal BB. Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT) 3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphatase SHP-1. Mol Pharmacol. 2009;75:525–533. doi: 10.1124/mol.108.052548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Takahashi A, Yamamoto N, Murakami A. Cardamonin suppresses nitric oxide production via blocking the IFN-γ/STAT pathway in endotoxin-challenged peritoneal macrophages of ICR mice. Life Sci. 2011;89:337–342. doi: 10.1016/j.lfs.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 122.Li DD, Wu XQ, Tang J, Wei XY, Zhu XF. ON-III inhibits erbB-2 tyrosine kinase receptor signal pathway and triggers apoptosis through induction of Bim in breast cancer cells. Cancer Biol Ther. 2009;8:739–743. doi: 10.4161/cbt.8.8.7917. [DOI] [PubMed] [Google Scholar]
- 123.Jung JI, Chung E, Seon MR, Shin HK, Kim EJ, Lim SS, Chung WY, Park KK, Park JH. Isoliquiritigenin (ISL) inhibits ErbB3 signaling in prostate cancer cells. Biofactors. 2006;28:159–168. doi: 10.1002/biof.5520280302. [DOI] [PubMed] [Google Scholar]
- 124.Zi X, Guo Y, Simoneau AR, Hope C, Xie J, Holcombe RF, Hoang BH. Expression of Frzb/secreted Frizzled-related protein 3, a secreted Wnt antagonist, in human androgen-independent prostate cancer PC-3 cells suppresses tumor growth and cellular invasiveness. Cancer Res. 2005;65:9762–9770. doi: 10.1158/0008-5472.CAN-05-0103. [DOI] [PubMed] [Google Scholar]
- 125.Cho M, Ryu M, Jeong Y, Chung YH, Kim DE, Cho HS, Kang S, Han JS, Chang MY, Lee CK, Jin M, Kim HJ, Oh S. Cardamonin suppresses melanogenesis by inhibition of Wnt/beta-catenin signaling. Biochem Biophys Res Commun. 2009;390:500–505. doi: 10.1016/j.bbrc.2009.09.124. [DOI] [PubMed] [Google Scholar]
- 126.Sun YW, Huang WJ, Hsiao CJ, Chen YC, Lu PH, Guh JH. Methoxychalcone induces cell-cycle arrest and apoptosis in human hormone-resistant prostate cancer cells through PI 3-kinase-independent inhibition of mTOR pathways. Prostate. 2010;70:1295–1306. doi: 10.1002/pros.21165. [DOI] [PubMed] [Google Scholar]
- 127.Niu P, Zhang Y, Shi D, Chen Y, Deng J. Cardamonin ameliorates insulin resistance induced by high insulin and high glucose through the mTOR and signal pathway. Planta Med. 2013;79:452–458. doi: 10.1055/s-0032-1328325. [DOI] [PubMed] [Google Scholar]
- 128.Chen G, Hu X, Zhang W, Xu N, Wang FQ, Jia J, Zhang WF, Sun ZJ, Zhao YF. Mammalian target of rapamycin regulates isoliquiritigenin-induced autophagic and apoptotic cell death in adenoid cystic carcinoma cells. Apoptosis. 2012;17:90–101. doi: 10.1007/s10495-011-0658-1. [DOI] [PubMed] [Google Scholar]
- 129.Kim TH, Seo WD, Ryu HW, Seo HR, Jin YB, Lee M, Ji YH, Park KH, Lee YS. Antitumor effects by a synthetic chalcone compound is mediated by c-Myc-mediated reactive oxygen species production. Chem Biol Interact. 2010;188:111–118. doi: 10.1016/j.cbi.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 130.Kumar V, Kumar S, Hassan M, Wu H, Thimmulappa RK, Kumar A, Sharma SK, Parmar VS, Biswal S, Malhotra SV. Novel chalcone derivatives as potent Nrf2 activators in mice and human lung epithelial cells. J Med Chem. 2011;54:4147–4159. doi: 10.1021/jm2002348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li QS, Li CY, Lu X, Zhang H, Zhu HL. Design, synthesis and biological evaluation of novel (E)-α-benzylsulfonyl chalcone derivatives as potential BRAF inhibitors. Eur J Med Chem. 2012;50:288–295. doi: 10.1016/j.ejmech.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 132.Shin SY, Yoon H, Hwang D, Ahn S, Kim DW, Koh D, Lee YH, Lim Y. Benzochalcones bearing pyrazoline moieties show anti-colorectal cancer activities and selective inhibitory effects on aurora kinases. Bioorg Med Chem. 2013;21:7018–7024. doi: 10.1016/j.bmc.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 133.Yang Z, Yang J, Jia Y, Tian Y, Wen A. Pharmacokinetic properties of hydroxysafflor yellow A in healthy Chinese female volunteers. J Ethnopharmacol. 2009;124:635–638. doi: 10.1016/j.jep.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 134.Weber TM, Ceilley RI, Buerger A, Kolbe L, Trookman NS, Rizer RL, Schoelermann A. Skin tolerance, efficacy, and quality of life of patients with red facial skin using a skin care regimen containing Licochalcone A. J Cosmet Dermatol. 2006;5:227–232. doi: 10.1111/j.1473-2165.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- 135.Higuchi K, Watanabe T, Tanigawa T, Tominaga K, Fujiwara Y, Arakawa T. Sofalcone, a gastroprotective drug, promotes gastric ulcer healing following eradication therapy for Helicobacter pylori: a randomized controlled comparative trial with cimetidine, an H2-receptor antagonist. J Gastroenterol Hepatol. 2010;25(Suppl 1):S155–S160. doi: 10.1111/j.1440-1746.2010.06232.x. [DOI] [PubMed] [Google Scholar]
- 136.Wananukul S, Chatproedprai S, Chunharas A, Limpongsanuruk W, Singalavanija S, Nitiyarom R, Wisuthsarewong W. Randomized, double-blind, split-side, comparison study of moisturizer containing licochalcone A and 1% hydrocortisone in the treatment of childhood atopic dermatitis. J Med Assoc Thai. 2013;96:1135–1142. [PubMed] [Google Scholar]