

Abstract
Objective
The ability of high density lipoprotein (HDL) particles to accept cholesterol from peripheral cells such as lipid-laden macrophages and to transport cholesterol to the liver for catabolism and excretion in a process termed reverse cholesterol transport (RCT) is believed to underlie the beneficial cardiovascular effects of elevated HDL. The liver X receptors (LXRα and LXRβ) regulate RCT by controlling the efflux of cholesterol from macrophages to HDL and the excretion, catabolism and absorption of cholesterol in the liver and intestine. Importantly, treatment with LXR agonists increases RCT and decreases atherosclerosis in animal models. Nevertheless, LXRs are expressed in multiple tissues involved in RCT and their tissue specific contributions to RCT are still not well defined.
Approach and Results
Utilizing tissue-specific LXR deletions together with in vitro and in vivo assays of cholesterol efflux and fecal cholesterol excretion we demonstrate that macrophage LXR activity is neither necessary nor sufficient for LXR agonist-stimulated RCT. In contrast, the ability of LXR agonists primarily acting in the intestine to increase HDL mass and HDL function appears to underlie the ability of LXR agonists to stimulate RCT in vivo.
Conclusions
We demonstrate that activation of LXR in macrophages makes little or no contribution to LXR agonist-stimulated RCT. Unexpectedly our studies suggest that the ability of macrophages to efflux cholesterol to HDL in vivo is not regulated by macrophage activity but is primarily determined by the quantity and functional activity of HDL.
Keywords: Reverse cholesterol transport, Liver X receptors, Macrophage, HDL, Atherosclerosis
INTRODUCTION
Cardiovascular disease (CVD) is a leading cause of death globally and it is well established that elevated levels of cholesterol in the blood is a major contributor to disease development1. Excess plasma cholesterol accumulates in macrophages lodged in blood vessel walls which along with an associated inflammatory response initiate the formation of atherosclerotic lesions2. Statin therapy is highly effective for lowering disease-causing low-density lipoprotein (LDL) cholesterol thereby reducing morbidity and mortality associated with CVD3. Nevertheless, the residual risk for major cardiac events remains high for patients receiving LDL lowering therapies prompting the search for complementary therapeutic approaches4. Epidemiological studies have demonstrated that levels of high density lipoprotein particle (HDL) cholesterol are inversely associated with CVD suggesting the potential therapeutic benefit of raising HDL5. Recent clinical trials with cholesteryl ester transfer protein (CETP) inhibitors and niacin, however, have failed to demonstrate clinical benefits of increasing HDL cholesterol6, 7. The clinical trial results have led to the suggestion that HDL functionality, rather than the absolute mass of HDL cholesterol may be a more accurate indicator for CVD risk8, 9. The ability of HDL to promote cholesterol efflux from macrophage foam cells within atherosclerotic lesions was one of its earliest recognized functions10, 11. Importantly, cholesterol efflux from foam cells has been shown to increase macrophage egression and to reduce lesion burden in animal models of cardiovascular disease12–14. Measuring the dynamic rate of macrophage cholesterol efflux, therefore, may be a better predictor of the anti-atherogenic effects of novel HDL-targeted therapies15.
The movement of cholesterol from peripheral cells such as macrophages to HDL constitutes the first step in a process termed reverse cholesterol transport (RCT). HDL-derived cholesterol is then trafficked to the liver where it is catabolized or excreted to the bile16, 17. Recent studies have also described hepatic-independent pathways for cholesterol secretion18. Studies in animal models indicate that measurements of RCT can strongly predict the effect of genetic and pharmacological manipulations on atherosclerosis19. Similarly, in humans an inverse relationship has been uncovered between the ability of patient sera to accept cholesterol from macrophages in vitro and measurements of carotid intima media thickness with cholesterol acceptor capacity being a strong predictor of coronary disease status15. The utility of in vitro measurements of plasma cholesterol acceptor activity for predicting CVD as well as the proteins/particles in human sera responsible for accepting cholesterol, however, remain controversial20, 21.
Integral to the regulation of RCT are the liver X receptors, LXRα (NR1H3) and LXRβ (NR1H2), which are members of the nuclear hormone receptor superfamily of ligand-activated transcription factors. Studies using genetic knockouts and synthetic agonists have defined important roles for LXRs in the control of cholesterol homeostasis and fatty acid metabolism22–24. Treatment of animals with LXR agonists results in changes in gene expression promoting the efflux of cholesterol from peripheral cells such as macrophages, the secretion of cholesterol from the liver, and the inhibition of cholesterol absorption in the intestine22. Importantly, the endogenous ligands for LXRs are oxidized forms of cholesterol (oxysterols) that increase coordinately with intracellular cholesterol levels, thus allowing these receptors to act as sensors to maintain appropriate cholesterol levels throughout the body25, 26. At the molecular level, LXRs control macrophage cholesterol efflux by regulating expression of genes encoding the ATP-binding cassette (ABC) transporters ABCA1 and ABCG1 as well the gene encoding apolipoprotein E (APOE)22. Up-regulation of ABCA1 and ABCG1 results in increased transfer of intracellular cholesterol to HDL particles, and genome-wide association studies have linked both transporters to HDL cholesterol levels in humans27, 28. Mutations in the human ABCA1 gene results in a genetic syndrome referred to as Tangier disease. Tangier disease patients characteristically present with little or no HDL, massive accumulation of cholesterol in lymph tissues and are at increased risk for atherosclerosis19, 29, 30. LXR also regulates expression of ABCG5 and ABCG8, two half-transporters that dimerize to form an additional cholesterol transporter31, 32. Expression of ABCG5/ABCG8 is largely restricted to the liver and intestine, where these proteins function to promote the excretion of cholesterol (liver) and limit cholesterol absorption (intestine)33. Genetic deletion of ABCG5/G8 or deletion of LXRα in the liver largely blocks the ability of LXR agonists to stimulate fecal excretion of cholesterol34, 35. Thus activation of LXRs promotes a net movement of cholesterol from the periphery out of the body. Not surprisingly, LXR agonists decrease atherosclerosis in animal models of CVD34, 36–38.
Treatment with LXR agonists also increases plasma HDL cholesterol34, 39 suggesting LXRs can regulate RCT in both a cell autonomous fashion, by controlling the transporters required to mobilize intracellular cholesterol, as well as in a non-autonomous fashion by regulating the amount of cholesterol acceptor in plasma. Interestingly, the ability of LXR agonists to increase HDL cholesterol levels is largely mediated by the induction of ABCA1 expression in the intestine34, 40. Not unexpected then is the observation that an intestinal-specific LXR agonist increases RCT41. Although LXR agonists appear to act in macrophages, the liver and the intestines to stimulate RCT, studies utilizing genetic knockouts indicate that macrophages are the major site of LXR agonist-dependent anti-atherogenic activity38, 42, 43. The atherosclerosis studies therefore led us to question the tissue-specific contributions of LXRs to the regulation of RCT. Combining in vivo measurements with tissue-selective knockouts we show that the ability of LXRs to regulate HDL quantity and activity is a major driver of RCT. In contrast, macrophage LXR activity is neither necessary nor sufficient. Furthermore, our studies suggest that the ability of macrophages to efflux cholesterol to HDL in vivo is primarily determined by the quantity and functional activity of HDL in the surrounding environment.
MATERAILS AND METHODS
Materials and Methods are available in the online-only Supplement.
RESULTS
Macrophage LXR is not necessary for LXR agonist-dependent RCT
LXR activity in the liver and the macrophage is thought to contribute to RCT44 but the relative contribution of LXR at these sites has not been well defined. To determine the contribution of macrophage LXR to RCT, we injected bone marrow derived macrophages (BMM) that had been loaded with 3H-cholesterol in vitro into the peritoneal space of mice and followed the movement of macrophage-derived cholesterol to the plasma and ultimately to the feces as described by Naik et al.45. For these studies we used C57BL/6J (LXR+) and Lxrα−/−/Lxrβ−/− (DKO) mice in the C57BL/6J background to generate three groups of animals: LXR+ macrophage introduced into LXR+ mice (referred to as MacLXR+/LXR+), LXR+ macrophage introduced into DKO mice (referred to as MacLXR+/DKO) and DKO macrophages into LXR+ mice (referred to as MacDKO/LXR+). For the RCT experiments age-matched male mice were treated with vehicle or the LXR agonist T0901317 (10mpk) daily by oral gavage for 3 days prior to injection. Following injection of radiolabeled macrophage, mice continued to be treated with vehicle or agonist for the duration of the experiment (for a total of 5 doses) and the appearance of 3H sterol was quantitated in the plasma at 6, 24 and 48 hours after injection. At completion of the experiment (48 hours) the amount of 3H-sterol in the feces and liver was determined. In preliminary experiments we found that LXR activation (e.g. rise in plasma triglycerides) can be observed following 3 doses of T0901317 at 10mpk and that the plasma concentrations of T0901317 are similar between C57BL/6J and Lxrα−/−/Lxrβ−/− mice and at least 10 times above the reported EC50 (data not shown).
As expected, agonist treatment of MacLXR+/LXR+ mice stimulates the appearance of macrophage-derived cholesterol in plasma over the time course and in the feces at 48 hours (Figure 1A–B). When LXR is present only in macrophages (MacLXR+/DKO), however, the amount of macrophage-derived cholesterol in the plasma and feces is significantly decreased (Figure 1A–B). Similarly, the ability of T0901317 to increase the accumulation of macrophage-derived cholesterol in the plasma of MacLXR+/DKO mice is decreased by 70% (Figure 1A) and agonist-stimulated fecal excretion is completely blocked in these animals (Figure 1B). Quantification of ABCA1 mRNA levels in macrophage re-extracted from the peritoneal space at completion of the experiment demonstrates that placing LXR+ macrophages into DKO mice does not impair macrophage LXR transcriptional activity (Figure 1C). In contrast to the decreased RCT observed in the MacLXR+/DKO mice, selective deletion of LXR in macrophages (MacDKO/LXR+) has little or no effect on either the accumulation of 3H-cholesterol in the plasma or the feces (Figure 1A–B). Little or no differences among the groups are seen when hepatic levels of 3H-sterols were examined (Supplemental Figure I). To further address the contribution of macrophage LXR activity to the ability of LXR agonists to increase the accumulation of macrophage-derived cholesterol in the plasma we examined 3H-cholesterol levels in vehicle and T0901317 treated MacLXR+/LXR+ and MacDKO/LXR+ mice at 30, 60 and 90 minutes after introducing radiolabeled macrophage into the peritoneal space. As shown in Figure 1D, pretreatment of mice with T0901317 significantly increases 3H-cholesterol in the plasma by 60 minutes. Even at these short time points, however, the LXR genotype of the macrophages has no effect on the response to agonist treatment. The observation that LXR macrophage activity does not appear to play a role in the accumulation of 3H-cholesterol in the plasma in vivo is consistent with studies in vitro demonstrating that ABCA1 expression and cholesterol efflux is actually slightly increased in Lxrα−/−/Lxrβ−/− macrophages46. In the absence of agonists LXRs repress transcription by interacting with corepressors and this activity is lost upon genetic deletion46. A similar up-regulation of ABCA1 expression is observed in DKO macrophages recovered from the peritoneal space of LXR+ mice after in vivo RCT experiments (Figure 1C).
Figure 1. Macrophage LXRs are not required for RCT.
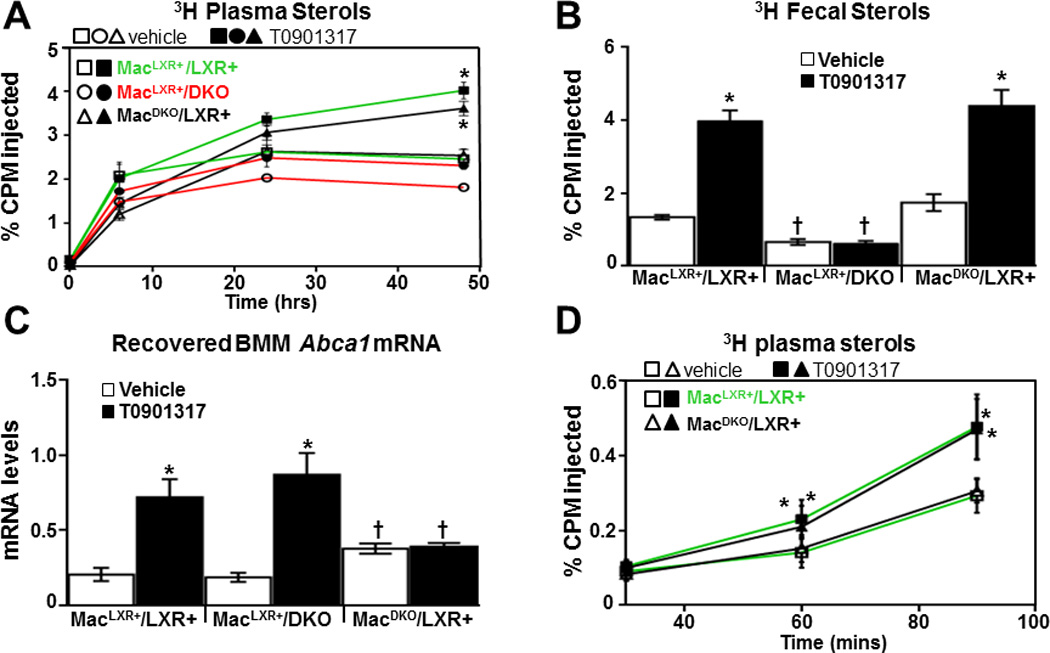
3H-cholesterol and acetylated LDL-loaded C57BL6/J or DKO BMMs were injected into C57BL6/J or DKO mice to generate MacLXR+/LXR+, MacLXR+/DKO, and MacDKO/LXR+ mice (see text). Animals were treated for 3 days with or without 10 mg/kg T0901317 (n=6/group), and the amount of 3H sterol in plasma (A and D) and feces (B) were determined as described in the Materials and Methods. Mice continued to receive vehicle or T0901317 treatment for the duration of the experiment. C) Total RNA was isolated from BMM that were recovered from the peritoneal space and the mRNA levels of Abca1 were measured by quantitative real-time PCR. Data are mean ± SEM. *Statistically significant difference between vehicle and T0901317-treated animals of the same genotype (p ≤ 0.05%). † Statistically significant difference between MacLXR+/LXR+ and MacLXR+/DKO or between MacLXR+/LXR+ and MacDKO/LXR+ mice with the same treatment (p ≤ 0.05%).
HDL levels and adipose activity drive LXR-agonist-dependent RCT
LXR agonists are known to increase HDL cholesterol predominately by increasing expression of ABCA1 in the intestine40. Consistent with an LXR agonist-dependent increase in HDL cholesterol (Table 1), plasma from T0901317 treated C57BL/6J (LXR+) mice has increased cholesterol acceptor activity in vitro when 3H-cholesterol loaded RAW264.7 cells are used as donor macrophages. The effect of agonist, however, is lost when plasma from DKO animals is used (Figure 2A). To further address the contribution of HDL to macrophage efflux, a similar series of in vitro efflux experiments were carried out using FPLC-purified HDL particles (Figure 2B). For experiments with FPLC-purified HDL, peak HDL fractions were pooled (Supplemental Figure II) and normalized by the amount of apolipoprotein AI (APOAI) as determined by Western blotting (Supplemental Figure IIIA). Using APOA1 as a relative measure for particle number, HDL from agonist treated C57BL/6J accept greater amounts of macrophage cholesterol compared to DKO mice (Figure 2B). Together these experiments show that LXR agonist treatment increases both HDL mass and HDL function.
Table 1.
Plasma Lipid Levels.
| Diet | Strain | Drug treatment |
Total Cholesterol (mg/dL) |
HDL- Cholesterol (mg/dL) |
Triglycerides (mg/dL) |
|---|---|---|---|---|---|
| Chow | C57bl6/J | Vehicle | 122.2 ± 5.4 | 65.9 ± 1.2 | 55.3 ± 3.6 |
| T0901317 | 155.4± 3.9* | 100.0 ± 4.8* | 90.5 ± 7.2* | ||
| Chow | Lxrα−/− Lxrβ−/− | Vehicle | 113.6 ± 3.9 | 46.1 ± 1.7** | 40.2 ± 2.7 |
| T0901317 | 112.5 ± 3.6 | 54.5 ± 1.8** | 55.5 ± 5.2 | ||
| Chow | Floxed | Vehicle | 109.0 ± 8.1 | 65.3 ± 3.6 | 64.2 ± 7.8 |
| T0901317 | 163.1 ± 8.3* | 121.5 ± 10.9* | 113.4 ± 10.5* | ||
| Chow | LivKO | Vehicle | 115.2 ± 9.4 | 44.9 ± 5.2** | 45.1 ± 3.9 |
| T0901317 | 166.4 ± 9.9* | 86.4 ± 6.7* | 47.9 ± 3.1 | ||
| 0.2% cholesterol | Floxed | Vehicle | 159.6 ± 12.5† | 73.9 ± 5.3 | 59.9 ± 3.4 |
| T0901317 | 216.9 ± 16.0*† | 93.8 ± 5.4*† | 197.2 ± 17.6*† | ||
| 0.2% cholesterol | LivKO | Vehicle | 166.7 ± 11.0† | 54.1 ± 2.5** | 47.7 ± 7.2 |
| T0901317 | 167.9 ± 6.2 | 56.0 ± 5.6 | 35.4 ± 3.2 | ||
| Chow | CETP− | Vehicle | 116.9 ± 2.6 | 68.2 ± 3.3 | 52.6 ± 3.4 |
| T0901317 | 193.6 ± 6.8* | 92.4 ± 1.7* | 85.2 ± 7.5* | ||
| Chow | CETP+ | Vehicle | 105.3 ± 4.6 | 52.4 ± .9 | 57.0 ± 6.2 |
| T0901317 | 78.4 ± 4.6* | 38.0 ± 1.9* | 92.2 ± 9.2* |
Statistically significant difference between vehicle and T0901317-treated animals of the same genotype (n = 6, p ≤ 0.05%).
Statistically significant difference between chow and 0.2% cholesterol diet fed mice of the same genotype and treatment (n = 6, p ≤ 0.05%).
Statistically significant difference between C57bl6/J and Lxrα−/− Lxrβ−/− or floxed and LivKO mice with the same treatment. Data are mean ± SEM.
Figure 2. HDL function and adipose tissue drives LXR-dependent RCT.
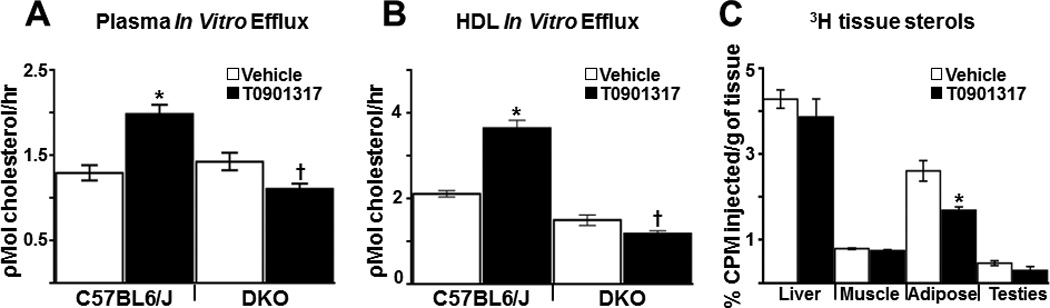
C57BL6/J and DKO mice (n = 5–6/group) were treated for 5 days with vehicle or 10 mg/kgT0901317 and In vitro macrophage cholesterol efflux was measured as described in Materials and Methods using 3H-cholesterol labeled Raw264.7 cells that were incubated with 0.03% pooled plasma (A) or FPLC purified HDL (B) Efflux data are representative of 3 independent experiments. C) 3H-cholesterol and acetylated LDL-loaded C57BL6/J BMM were injected into C57bl6/J mice treated for 3 days with or without 10 mg/kg T0901317 (n=6/group), and the amount of 3H sterol in tissues were determined as described in the Materials and Methods. Mice continued to receive vehicle or T0901317 treatment for the duration of the experiment. All data are expressed as mean ± SEM. *Statistically significant difference between vehicle and T0901317-treated animals of the same genotype (p ≤ 0.05%). † Statistically significant difference between C57BL6/J and DKO with the same treatment (p ≤ 0.05%).
Over the course of in vivo RCT experiments it is likely that macrophage-derived 3H-cholsterol incorporates into cells and tissues throughout the body. Thus along with increasing the cholesterol acceptor activity of HDL, LXR agonists may also increase the amount of cholesterol in plasma by promoting efflux from other tissues via transcriptional up-regulation of ABCA1, ABCG1 and APOE. To address the possible contributions of different tissues to LXR agonist-stimulated RCT, radiolabeled LXR+ macrophages were introduced into vehicle and T0901317 treated LXR+ mice (MacLXR+/LXR+) and multiple tissues were harvested at 48 hours post injection to determine if agonist treatment promotes a net loss in tissue-associated 3H-sterols. As shown in Figure 2C, a significant agonist-dependent decrease is observed in white adipose (gonadal fat pad) suggesting that fat tissue may make an important contribution to LXR-stimulated accumulation of cholesterol in the plasma and feces. T0901317-dependent changes in 3H-sterol levels were not observed in other tissues (Figure 2C). Importantly, the decrease in adipose 3H-sterol levels could result from increased LXR transcriptional activity in fat cells, the improved acceptor activity of HDL or both.
Diet-dependent regulation of Liver LXR activity and RCT
We have previously determined under severe hyperlipidemic conditions (Ldlr−/− mice on Western diet) that liver-specific deletion of LXRα impairs the accumulation of macrophage-derived cholesterol in both the plasma and in the feces34. To further investigate the contribution of liver LXR activity to RCT, liver-specific knockout LXRα (LivKO) mice34 and floxed littermate controls (carrying the floxed LXRα allele without albumin CRE) were placed on a standard chow diet with or without 0.2% cholesterol. LXRα is the major LXR subtype expressed in the liver47 and the ability of T0901317 to increase plasma triglycerides and to induce expression of hepatic ABCG5, ABCG8 and ABCA1 is significantly impaired in LivKO mice34 (Table 1 and Supplemental Figure IV). After 4 weeks on diet, plasma total cholesterol increases 30–50% in both LivKO and littermate control groups fed the 0.2% cholesterol diet (Table 1). Consistent with published data, the 0.2% cholesterol diet also significantly increases hepatic cholesterol in LivKO mice due to impaired fecal excretion and decreased bile acid synthesis34, 47 (Supplemental Figure VA). Hepatic triglycerides, however, are not increased (Supplemental Figure VB) and the increase in hepatic cholesterol measured in LivKO mice does not result in a significant increase in liver damage (Supplemental Figure VC–D), markers of inflammation or markers of endoplasmic reticulum stress (data not shown). For the final week of the diet treatment (week 4) mice were treated with vehicle or T0901317 and RCT was measured in vivo as in previous experiments by introducing radiolabeled LXR+ macrophages. On a standard chow diet the appearance of 3H-cholesterol in the plasma of T0901317 treated LivKO and littermate controls is significantly increased at 24 and 48 hours (Figure 3A) indicating that liver LXRα activity is not required for agonists to increase the accumulation of 3H-cholesterol in the plasma. On the other hand, the ability of LXR agonists to increase fecal sterol excretion is completely lost in LivKO mice (Figure 3B) a result consistent with decreased agonist-dependent regulation of ABCG5 and ABCG8 in the livers of these animals (Supplemental Figure IV). Interestingly, exposure to the 0.2% cholesterol diet impairs both LXR agonist-dependent plasma and fecal cholesterol accumulation in LivKO mice relative to controls (Figure 3C–D). Thus dietary cholesterol uncovers a critical role for hepatic LXR activity in controlling the accumulation of macrophage-derived cholesterol in plasma. The ability of LXR agonists to increase HDL cholesterol levels in LivKO mice is also sensitive to dietary cholesterol (Figure 4A and Table 1) despite similar increases in the intestinal mRNA levels of ABCA1 (Supplemental Figure VI). Furthermore a dietary cholesterol-dependent decrease in cholesterol acceptor activity is also observed when FPLC-purified HDL particles isolated from T0901317 treated LivKO mice are compared to HDL particles from littermate controls in vitro (Figure 4B; see Supplemental Figures II and IIIC–D for FPLC profiles and APOA1 levels). The reason(s) why the cholesterol enriched diet impairs the ability of LXR agonist treatment to increase HDL mass and function remains to be determined. Nevertheless, the failure of T0901317 to modulate HDL levels and functional activity in cholesterol fed LivKO mice supports the hypothesis that the ability of LXR agonists to promote the accumulation of macrophage-derived cholesterol in plasma is largely derived from systemic effects on HDL and independent of macrophage LXR activity.
Figure 3. In vivo RCT in chow and 0.2% cholesterol diet fed LivKO mice.
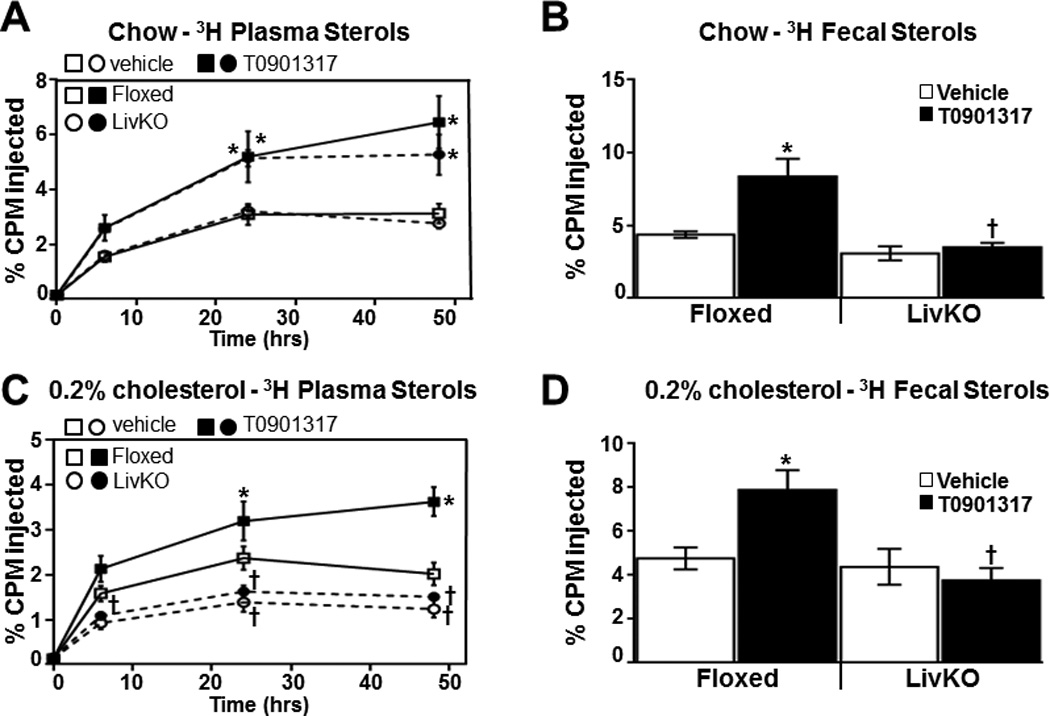
3H-cholesterol and acetylated LDL-loaded C57BL6/J BMMs were injected into Floxed or LivKO mice fed standard chow (A and B) or 0.2% cholesterol supplemented diet (C and D). Animals were treated for 3 days with or without 10 mg/kg T0901317 (n=6/group) prior to BMM injection, and the amount of 3H sterol in plasma (A and C) and feces (B and D) was determined as described in the Materials and Methods. Mice continued to receive vehicle or T0901317 treatment for the duration of the experiment. Data are mean ± SEM. *Statistically significant difference between vehicle and T0901317-treated animals of the same genotype (p ≤ 0.05%). † Statistically significant difference between Floxed and LivKO mice with the same treatment (p ≤ 0.05%).
Figure 4. LXR agonist dependent changes in HDL mass and function in LivKO mice.
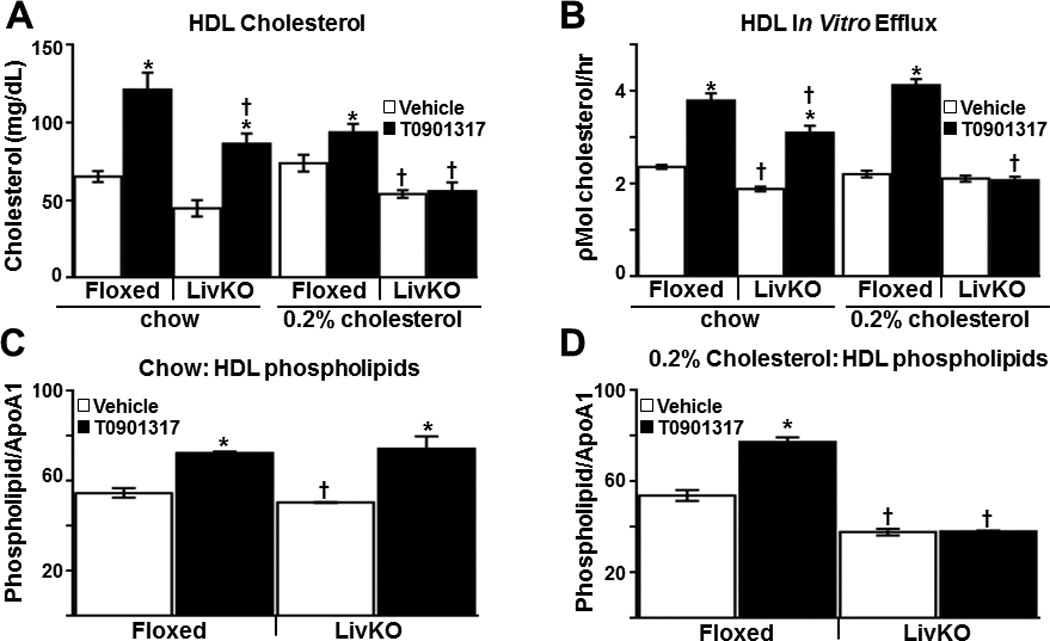
A) Plasma HDL cholesterol levels in chow and 0.2% cholesterol diet fed Floxed and LivKO mice (n = 6/group) treated for 5 days with vehicle or 10 mg/kg T0901317. B) In vitro macrophage cholesterol efflux was measured as described in Materials and Methods using 3H-cholesterol labeled Raw264.7 cells that were incubated with FPLC purified HDL from animals treated with vehicle or T0901317 (10 mpk) for 5 days (n=5–6/group). Efflux data are representative of 3 independent experiments. Total phospholipids in FPLC purified HDL from Floxed and LivKO mice fed a standard chow diet (C) or 0.2% cholesterol diet (D) and treated for 5 days with vehicle or T0901317 (10 mpk). HDL phospholipid levels were normalized by HDL APOA1 protein levels as determined by Western blottting. Data are mean ± SEM. *Statistically significant difference between vehicle and T0901317-treated animals of the same genotype (p ≤ 0.05%). †Statistically significant difference between Floxed and LivKO mice with the same treatment and diet (p ≤ 0.05%).
Our results indicate that LXR activation can improve the cholesterol acceptor activity of HDL and this effect is influenced by liver LXR activity in a diet-dependent fashion. As an initial characterization of HDL particle composition we measured phospholipid levels in the FPLC-purified HDL fractions. Phospholipids are the major components by mass of HDL and a number of studies suggest that HDL phospholipid levels are a better predictor of cholesterol efflux than other HDL parameters48, 49. As shown in Figure 4C and 4D, T0901317 treatment increases the amount of total phospholipids associated with purified HDL particles (normalized by APOA1 levels) from standard chow fed floxed and LivKO mice (Figure 4C). The increase in HDL-phospholipid levels is consistent with studies demonstrating that LXR agonist treatment increased HDL particle size34, 50. The effect of agonist treatment on HDL-phospholipid levels, however, is lost in 0.2% cholesterol diet challenged LivKO animals (Figure 4D). Phospholipid transfer protein is a HDL-bound protein that plays a major role in regulating HDL size and phospholipid composition through its phospholipid transfer activity51. Phospholipid transfer protein mRNA levels have been shown to be regulated by LXR52 however we did not detect significant differences in plasma phospholipid transfer protein activity between floxed and LivKO mice on either dietary condition (Supplemental Table I).
CETP decreases macrophage-derived cholesterol in plasma
To test the hypothesis that LXR-dependent regulation of HDL levels and activity plays a major role in driving the accumulation of macrophage-derived cholesterol in plasma, we took advantage of the observation that LXR agonist-dependent increases in HDL cholesterol are lost in CETP transgenic mice53. CETP facilitates the transfer of cholesterol esters from HDL to apolipoprotein B containing particles thereby decreasing HDL cholesterol levels54. Importantly, the transgene is under control of the human CETP promoter which has been shown to be directly regulated by LXR in human cells and in transgenic mice55, 56 (Supplemental Figure VIIA–B). Indeed, treatment of CETP transgenic mice with T0901317 decreases HDL cholesterol by approximately 25% and raises the amount of cholesterol associated with apolipoprotein B containing lipoprotein particles (Figure 5A and B and Table 1). To determine the effect of CETP expression on RCT in vivo, CETP transgenic mice and littermate controls were treated with vehicle or T0901317 and injected with 3H-cholesterol loaded C57BL/6J (LXR+) BMM as described in previous experiments. Consistent with a critical role for HDL in promoting the accumulation of macrophage-derived cholesterol in plasma, the amount of 3H-cholesterol in this compartment at 24 and 48 hours is significantly reduced in CETP transgenic mice and the ability of T0901317 to increase plasma cholesterol accumulation is lost (Figure 5C). Similarly, unfractionated plasma and FPLC purified HDL particles from T0901317 treated CETP transgenic mice do not exhibit increased efflux activity as is observed in non-transgenic controls (Figure 5D–E). The ability of LXR agonists to increase HDL phospholipids, however, is not impaired in CETP transgenics (Supplemental Figure VIIC). Taken together, the RCT and in vitro efflux experiments indicate that LXR-dependent up-regulation of CETP expression counters the ability of agonists to enhance the appearance of macrophage-derived cholesterol in the plasma. In contrast to the inhibitory effect of CETP expression on the accumulation of macrophage-derived cholesterol in plasma, LXR agonist treatment increases fecal 3H-sterol levels in both CETP transgenic and littermate controls (Figure 5F). Interestingly, CETP expression also results in a significant increase in fecal bile acids in vehicle treated animals (Supplemental Figure VIID). Increased bile acid synthesis has previously been reported in CETP transgenic mice57, 58. Little or no difference was observed in hepatic 3H-cholesterol levels among the groups (data not shown). Thus as observed with the LXRα liver-specific knockout mice (LivKO), it is possible to functionally sever the transfer of macrophage-derived cholesterol to the plasma from subsequent fecal excretion.
Figure 5. CETP inhibits LXR agonist-dependent RCT.
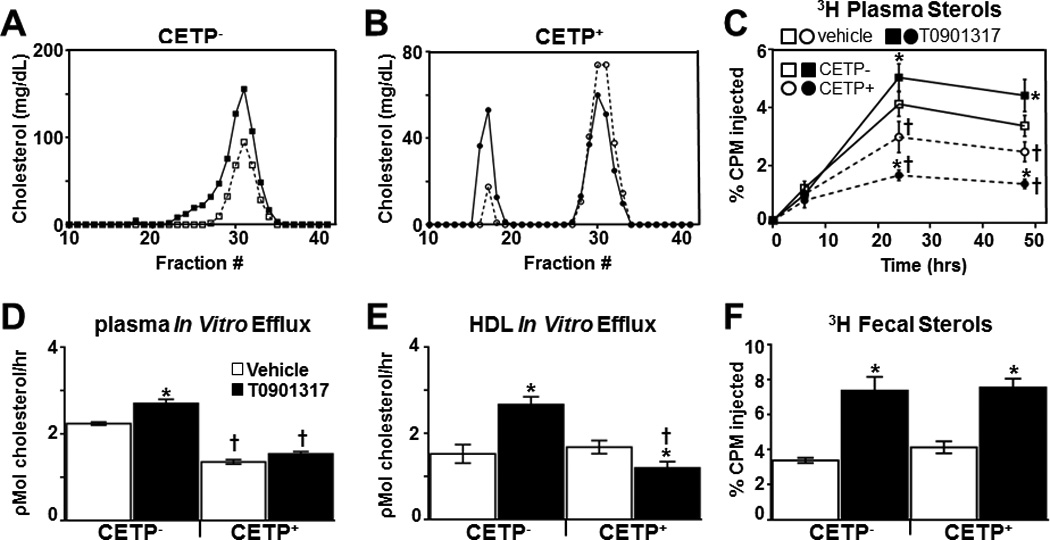
CETP− (A) and CETP+ (B) mice (n=6/group) were treated with vehicle or T0901317 (10 mpk) for 5 days, plasma was pooled, subjected to FPLC and the cholesterol content of each fraction was measured as described in Materials and Methods. 3H-cholesterol and acetylated LDL-loaded C67BL/6 BMDMs were injected into CETP− and CETP+ mice (n=6/group) treated with vehicle or T0901317 and the amount of 3H sterol in plasma (C) and feces (F) was determined as described in Materials and Methods. D) Raw 264.7 cells were incubated with 0.03% pooled plasma (D) or FPLC purified HDL (E) from vehicle or T0901317 treated CETP− and CETP+ mice (n=5/group) and cholesterol efflux was measured as described in Materials and Methods. Efflux data is representative of 3 independent experiments. Data are mean ± SEM. *Statistically significant difference between vehicle and T0901317-treated animals of the same genotype (p ≤ 0.05%). †Statistically significant difference between CETP− and CETP+ with the same treatment (p ≤ 0.05%).
DISCUSSION
The discovery that LXR agonists can promote macrophage cholesterol efflux in vitro via direct regulation of the genes encoding ABCA1, ABCG1 and APOE22, 59 suggested a simple hypothesis for the cardio-protective effect of LXR activation based on promoting cholesterol transfer from macrophage foam cells to HDL; the first step in the RCT pathway. This hypothesis is supported by the finding that macrophage LXR activity is required for the anti-atherogenic activity of LXR agonists38. Combining in vitro cholesterol efflux measurements, in vivo RCT assays and tissue-specific LXR knockouts we now demonstrate that the ability of LXR agonists to stimulate RCT in vivo defined as the transfer of macrophage-derived cholesterol to the feces is largely independent of macrophage LXR activity (Figure 6). Thus macrophage LXRs are neither necessary nor sufficient for LXR agonists to increase RCT, at least when measured in an acute assay over a 48 hour time course. Additionally, our studies suggest that it is the ability of LXR agonists to increase HDL biogenesis and to improve HDL functional activity that is largely responsible for stimulating the appearance of macrophage-derived cholesterol in plasma (Figure 6). The LXR agonist used in these studies, T0901317, has been reported to modulate other nuclear receptors, at least in vitro60–62. Therefore the possibility that another nuclear receptor, such as the pregnane X receptor, contributes to the activity of this molecule in vivo cannot be ruled out. All the activities of T0901317 measured in this work, however, are lost in cells and animals that are deficient in LXRs.
Figure 6. Effect of LXR agonist treatment on in vivo RCT.
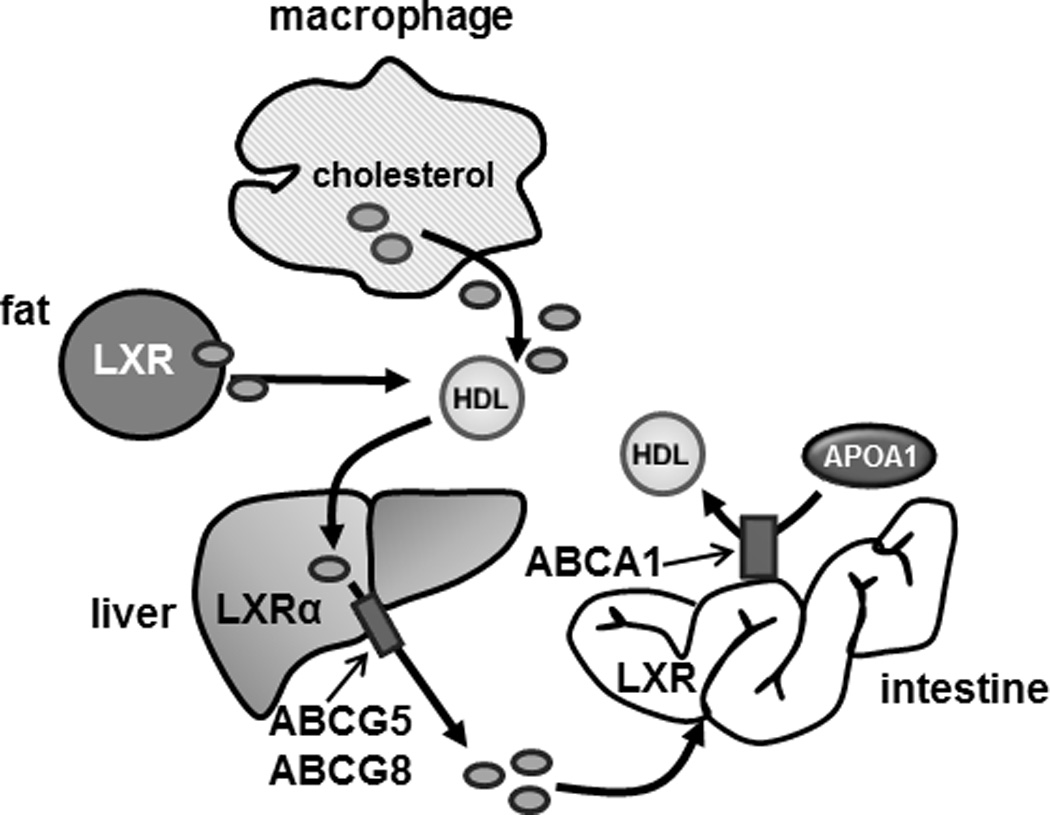
LXR agonist activity in the intestine and adipose tissue contributes to the accumulation of macrophage-derived cholesterol in the plasma during in vivo RCT assays. Agonist activity in the liver promotes fecal cholesterol excretion (see text for details).
On a standard mouse chow diet the ability of LXR agonists to stimulate the accumulation of macrophage-derived cholesterol in plasma is independent of LXR activity in both macrophages and the liver. Previous studies have determined that LXR agonists increase HDL cholesterol by inducing ABCA1 expression in the intestine34, 40, 63. Consistent with an important role for intestinal LXR activity in regulating RCT is the finding that selective activation of LXRs in the intestine using either a poorly absorbed “intestine-specific” LXR agonist41 or intestine-specific transgenic over expression of a hyperactive LXR (VP16-LXRα)64 increases RCT when measured using assays similar to those described in this work. Furthermore, our studies indicate that intestinal LXR activation can increase the cholesterol acceptor activity of HDL particles (Figure 6) most likely by increasing the production of immature nascent particles that have been shown to be preferred cholesterol acceptors65–67. Interestingly, this work also describes a potential role for LXR activity in white adipose in regulating cholesterol trafficking.
To test the hypothesis that agonist dependent increases in HDL mass and function drive the accumulation of macrophage-derived cholesterol in plasma during RCT assays we took advantage of the observation that the ability of LXR agonists to raise HDL cholesterol is lost in CETP transgenic mice53, 56. CETP, an enzyme that transfers cholesterol esters from HDL to apolipoprotein B containing lipoprotein particles in exchange for triglycerides, is not expressed in rodents but the human gene used in this study is regulated by LXRs55, 56, 68. Importantly CETP activity in the plasma is increased following LXR agonist treatment, HDL levels are lowered and plasma cholesterol accumulation measured during RCT assays is decreased. The cholesterol acceptor activity of unfractionated plasma and FPLC-purified HDL from T0901317 treated CETP transgenic mice is also reduced relative to non-transgenic controls. Finally, the conclusion that increasing CETP activity impairs HDL particle function is consistent with reports that inhibition of CETP activity improves the cholesterol acceptor activity of human HDL particles69. Taken together, the data supports the hypothesis that the ability of LXR agonists to increase the accumulation of macrophage-derived cholesterol in plasma is primarily determined by the quantity and quality of the HDL particles. Nevertheless, in CETP transgenic animals LXR agonist treatment still increases fecal excretion of macrophage-derived cholesterol. Therefore we cannot rule out the possibility that CETP expression decreases the levels of macrophage-derived cholesterol in plasma by increasing hepatic clearance via receptors for apolipoprotein B containing particles. Similar to CETP expression, Bi et al. found that liver-specific deletion of ABCA1 reduces plasma HDL levels and decreases plasma accumulation of 3H-cholesterol in RCT assays without altering fecal sterol excretion63. Bi et al. suggest the small plasma HDL pool that remains in the liver ABCA1 knockout mice may be quantitatively sufficient to mediate the transport of macrophage-derived cholesterol to the liver for excretion63. Our study with CETP transgenic mice together with the work of Bi et al. raises the possibility, at least under these experimental conditions, that the appearance of macrophage-derived cholesterol in the plasma is a not a rate limiting step for fecal cholesterol excretion.
In contrast to CETP transgenic expression, liver-specific deletion of LXRα (LivKO) has little or no effect on the accumulation of macrophage-derived cholesterol in plasma (on a standard chow diet) but strongly inhibits LXR agonist-stimulated fecal cholesterol excretion (Figure 6). Thus our analysis of CETP transgenic and LXRα LivKO mice indicate that it is possible to functionally separate plasma cholesterol accumulation from fecal excretion. Plasma cholesterol accumulation is primarily controlled by the ability of LXRs to regulate the quantity and quality of HDL while fecal excretion is controlled by LXR-dependent regulation of hepatic ABCG5 and ABCG8 levels allowing a single transcription factor pair (LXRα and LXRβ) to coordinate cholesterol movement throughout the body. These results raise the question regarding the potential therapeutic benefit of regulating either macrophage cholesterol efflux or fecal excretion independently. Current therapeutic approaches for atherosclerotic cardiovascular disease all involve reducing low density lipoprotein (LDL) cholesterol in the blood. Therefore if increasing fecal cholesterol excretion ultimately reduces plasma LDL levels one might predict a therapeutic benefit. On the other hand, APOA Milano and other APOA1-derived peptides have been shown to increase macrophage cholesterol efflux and to improve cardiovascular endpoints, although it not clear that the beneficial effects of these agents are dependent on promoting cholesterol efflux70, 71. Future studies that for instance combine macrophage selective over expression of ABCA1 with LXR liver-specific knockouts may be a way to address the therapeutic benefits of increased macrophage efflux in the absence of fecal cholesterol excretion.
Interestingly, the contribution of liver LXR activity to RCT can be influenced by the cholesterol content of the diet. As described above, on a standard mouse chow diet knocking out LXRα in the liver has little or no effect on the accumulation of macrophage-derived cholesterol in plasma while completely eliminating agonist-stimulated fecal excretion (Figure 6). When cholesterol (0.2%) is added to the diet, however, LXR agonist-dependent plasma cholesterol accumulation is significantly decreased in LivKO mice. The absence of agonist-dependent accumulation of macrophage-derived cholesterol in plasma when cholesterol is included in the diet correlates with the inability of agonist treatment to increase HDL cholesterol and to improve the acceptor capacity of purified HDL in LivKO mice under these conditions. LXR agonist treatment still increases ABCA1 expression in the intestines of LivKO on the 0.2% cholesterol diet and the reason(s) why HDL cholesterol levels are not increased in these mice remains to be determined. Compared to littermate floxed controls on the 0.2% cholesterol diet, LivKO mice have increased hepatic cholesterol levels although we did not detect any evidence for increased hepatic inflammation, endoplasmic reticulum stress or liver damage in these mice. We and others have shown that the ability of LXR agonists to increase HDL levels in LXR positive animals is lost under severe hyperlipidemic conditions such as Ldlr−/− or Apoe−/− mice on Western diets34, 36, 37, 39, 72. Thus the ability of LXR agonists to regulate HDL metabolism can be influenced by dietary cholesterol levels. Interestingly, Kalaany et al. demonstrated that Lxrα−/−/Lxrβ−/− mice are resistant to high fat diet-induced obesity, however, this resistance is only observed when the high fat diet also contains cholesterol73. These observations raise the possibility that hepatic cholesterol accumulation leads to the generation of a paracrine signal that can influence lipid metabolism in other tissues.
Bone marrow transplantation experiments and over expression studies indicate that macrophages are the site of LXR agonist-dependent anti-atherogenic activity38, 42, 43. The studies described in this work, however, indicate that macrophage LXR activity does not make a significant contribution to RCT. Similarly using LivKO mice in a severe hyperlipidemic environment (Ldlr−/− + Western diet) we demonstrated that LXR agonists can reduce atherosclerosis without increasing RCT34. Kappus et al. also reached an analogous conclusion in a recent study using mice with myeloid-specific double knockout of Abca1 and Abcg174. Together, these observations suggests that while hematopoietic LXR expression is required for the beneficial effects of LXR agonists an increase in RCT or macrophage efflux is not. LXR activation inhibits NFκβ signaling suggesting decreased inflammation as an obvious mechanism for LXR-dependent anti-atherogenic activity75, 76. A dominant role for anti-inflammatory activity as the beneficial effect of LXR activation on atherosclerosis has important implications for the potential therapeutic use of LXR agonists. In particular, in vitro experiments have suggested that LXR agonists can have pro-inflammatory activities in human macrophages77 in contrast to the anti-inflammatory effects measured in rodents. Additionally, as described above, pre-clinical studies examining the anti-atherogenic activity of LXR ligands generally have been carried out under severe hyperlipidemic conditions where the ability of LXR agonists to increase HDL mass is lost34, 37, 78. Since human cardiovascular disease patients do not usually present with the supra-physiological plasma cholesterol levels observed in genetic mouse models, the ability of LXR agonists to stimulate RCT may be maintained in humans and could be therapeutic. As we observe in CETP transgenic mice, however, the ability of LXR agonists to increase HDL cholesterol appears to be lost in non-human primates that express CETP79, 80.
Recent clinical trials with niacin7 and CETP inhibitors6 have called into question the hypothesis that raising HDL cholesterol has beneficial effects on human cardiovascular disease. The clinical trials together with experiments suggesting that the cholesterol acceptor activity of HDL isolated from patients can be a more accurate measurement of cardiovascular disease risk has led to the proposal that assessing HDL function may be more relevant than measurements of HDL cholesterol mass9, 15, 20. Along with increasing the levels of HDL cholesterol, LXR agonist treatment also increases the cholesterol acceptor activity of HDL particles that were normalized by the quantity of APOA1. HDL particles are heterogeneous in size and composition making it difficult to discern the LXR-dependent modifications that improve cholesterol acceptor activity. Nevertheless, our initial analysis of HDL particle composition found increased levels of phospholipids (normalized to APOA1) in the HDL particles purified from agonist treated animals. The phospholipid:APOA1 ratio in HDL has been shown to be an important determining factor in predicting macrophage efflux. Studies using mice and rats expressing human APOA1 indicate that the prime component of HDL that modulates cholesterol efflux is HDL phospholipid81, 82. Furthermore, the correlation between macrophage cholesterol efflux and HDL phospholipid in human sera is stronger than with any other measured lipoprotein parameter, including HDL cholesterol, APOA1 and triglycerides48. CETP expression, however, appears to impact HDL function without modulating phospholipid levels suggesting that multiple components of HDL can influence particle function. LXRs likely regulate multiple pathways that modulate HDL activity and future studies employing detailed lipidomic and proteomic approaches can be used to further define the LXR-dependent changes in HDL composition that regulate HDL particle function. These studies that define particle function may open the door to new therapeutic approaches for targeting HDL.
Supplementary Material
SIGNIFICANCE.
The liver X receptors, LXRα and LXRβ, are important regulators of cholesterol transport. Treatment with LXR agonists promotes the efflux of cholesterol from macrophages and the excretion of cholesterol from the liver resulting in a net movement of cholesterol from the periphery out of the body. Utilizing tissue-specific LXR deletions we demonstrate that macrophage LXR activity is neither necessary nor sufficient for LXR agonist stimulated RCT. In contrast the ability of LXR agonists to increase HDL mass and HDL function primarily acting in the intestine appears to underlie the ability of LXR agonists to stimulate RCT in vivo.
Our studies suggest that the ability of macrophages to efflux cholesterol to HDL in vivo is not regulated in a cell autonomous fashion but is primarily determined by the quantity and functional activity of HDL.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Norbert Leitinger and Dr. Irena Ignatova (U. of Virginia) for comments on the manuscript and Dr.s Yuan Zhang, Steven Kliewer and David Mangelsdorf (UT Southwestern) for providing the LXR liver knockout mice.
SOURCES OF FUNDING
Work in the author’s laboratory is supported by Grants to I.G.S. from the NIH (1R01HL096864-01A1) and the AHA (13GRNT1650022).
Nonstandard Abbreviations and Acronyms
- ABCA1
ATB binding cassette transporter A1
- ABCG1
ATB binding cassette transporter G1
- ABCG5
ATB binding cassette transporter G5
- ABCG8
ATB binding cassette transporter G8
- APOA1
apolipoprotein A1
- CETP
cholesteryl ester transfer protein
- CVD
cardiovascular disease
- FPLC
fast liquid protein chromatography
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- LXR
liver X receptor
- RCT
reverse cholesterol transport
Footnotes
DISCLOSURES
The authors have nothing to disclose.
REFERENCES
- 1.Chyu KY, Shah PK. Emerging therapies for atherosclerosis prevention and management. Cardiol Clin. 2011;29:123–135. doi: 10.1016/j.ccl.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward S, Lloyd Jones M, Pandor A, Holmes M, Ara R, Ryan A, Yeo W, Payne N. A systematic review and economic evaluation of statins for the prevention of coronary events. Health technology assessment (Winchester, England) 2007;11:1–160. iii–iv. doi: 10.3310/hta11140. [DOI] [PubMed] [Google Scholar]
- 4.Fruchart JC, Sacks FM, Hermans MP, Assmann G, Brown WV, Ceska R, Chapman MJ, Dodson PM, Fioretto P, Ginsberg HN, Kadowaki T, Lablanche JM, Marx N, Plutzky J, Reiner Z, Rosenson RS, Staels B, Stock JK, Sy R, Wanner C, Zambon A, Zimmet P. The residual risk reduction initiative: A call to action to reduce residual vascular risk in dyslipidaemic patient. Diabetes & vascular disease research : official journal of the International Society of Diabetes and Vascular Disease. 2008;5:319–335. doi: 10.3132/dvdr.2008.046. [DOI] [PubMed] [Google Scholar]
- 5.Gordon DJ, Rifkind BM. High-density lipoprotein--the clinical implications of recent studies. The New England journal of medicine. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 6.Mohammadpour AH, Akhlaghi F. Future of cholesteryl ester transfer protein (cetp) inhibitors: A pharmacological perspective. Clinical pharmacokinetics. 2013;52:615–626. doi: 10.1007/s40262-013-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low hdl cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 8.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma hdl cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Triolo M, Annema W, Dullaart RP, Tietge UJ. Assessing the functional properties of high-density lipoproteins: An emerging concept in cardiovascular research. Biomarkers in medicine. 2013;7:457–472. doi: 10.2217/bmm.13.35. [DOI] [PubMed] [Google Scholar]
- 10.von Eckardstein A, Nofer JR, Assmann G. High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arteriosclerosis, thrombosis, and vascular biology. 2001;21:13–27. doi: 10.1161/01.atv.21.1.13. [DOI] [PubMed] [Google Scholar]
- 11.Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. Journal of lipid research. 1995;36:211–228. [PubMed] [Google Scholar]
- 12.Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: Key to the regression of atherosclerosis? Circulation. 2006;113:2548–2555. doi: 10.1161/CIRCULATIONAHA.104.475715. [DOI] [PubMed] [Google Scholar]
- 13.van Eck M, Bos IS, Kaminski WE, Orso E, Rothe G, Twisk J, Bottcher A, Van Amersfoort ES, Christiansen-Weber TA, Fung-Leung WP, Van Berkel TJ, Schmitz G. Leukocyte abca1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:6298–6303. doi: 10.1073/pnas.092327399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore RE, Navab M, Millar JS, Zimetti F, Hama S, Rothblat GH, Rader DJ. Increased atherosclerosis in mice lacking apolipoprotein a–i attributable to both impaired reverse cholesterol transport and increased inflammation. Circulation research. 2005;97:763–771. doi: 10.1161/01.RES.0000185320.82962.F7. [DOI] [PubMed] [Google Scholar]
- 15.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Rader DJ. Molecular regulation of macrophage reverse cholesterol transport. Curr Opin Cardiol. 2007;22:368–372. doi: 10.1097/HCO.0b013e3281ec5113. [DOI] [PubMed] [Google Scholar]
- 17.Lewis GF, Rader DJ. New insights into the regulation of hdl metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 18.van der Velde AE, Brufau G, Groen AK. Transintestinal cholesterol efflux. Current opinion in lipidology. 2010;21:167–171. doi: 10.1097/MOL.0b013e3283395e45. [DOI] [PubMed] [Google Scholar]
- 19.Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50(Suppl):S189–S194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sankaranarayanan S, de la Llera-Moya M, Drazul-Schrader D, Phillips MC, Kellner-Weibel G, Rothblat GH. Serum albumin acts as a shuttle to enhance cholesterol efflux from cells. Journal of lipid research. 2013;54:671–676. doi: 10.1194/jlr.M031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calkin AC, Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1513–1518. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Role of lxrs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Regulation of mouse sterol regulatory element-binding protein-1c gene (srebp-1c) by oxysterol receptors, lxralpha and lxrbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW. Enzymatic reduction of oxysterols impairs lxr signaling in cultured cells and the livers of mice. Cell Metab. 2007;5:73–79. doi: 10.1016/j.cmet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor lxr alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 27.Edmondson AC, Braund PS, Stylianou IM, Khera AV, Nelson CP, Wolfe ML, Derohannessian SL, Keating BJ, Qu L, He J, Tobin MD, Tomaszewski M, Baumert J, Klopp N, Doring A, Thorand B, Li M, Reilly MP, Koenig W, Samani NJ, Rader DJ. Dense genotyping of candidate gene loci identifies variants associated with high-density lipoprotein cholesterol. Circulation. Cardiovascular genetics. 2011;4:145–155. doi: 10.1161/CIRCGENETICS.110.957563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oram JF, Lawn RM. Abca1. The gatekeeper for eliminating excess tissue cholesterol. J Lipid Res. 2001;42:1173–1179. [PubMed] [Google Scholar]
- 30.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. Hdl, abc transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Yu L, Gupta S, Xu F, Liverman AD, Moschetta A, Mangelsdorf DJ, Repa JJ, Hobbs HH, Cohen JC. Expression of abcg5 and abcg8 is required for regulation of biliary cholesterol secretion. The Journal of biological chemistry. 2005;280:8742–8747. doi: 10.1074/jbc.M411080200. [DOI] [PubMed] [Google Scholar]
- 32.Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ. Regulation of atp-binding cassette sterol transporters abcg5 and abcg8 by the liver x receptors alpha and beta. J Biol Chem. 2002;277:18793–18800. doi: 10.1074/jbc.M109927200. [DOI] [PubMed] [Google Scholar]
- 33.Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent abc transporters. Science (New York, NY) 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Breevoort SR, Angdisen J, Fu M, Schmidt DR, Holmstrom SR, Kliewer SA, Mangelsdorf DJ, Schulman IG. Liver lxralpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J Clin Invest. 2012;122:1688–1699. doi: 10.1172/JCI59817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu L, York J, von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH. Stimulation of cholesterol excretion by the liver x receptor agonist requires atp-binding cassette transporters g5 and g8. J Biol Chem. 2003;278:15565–15570. doi: 10.1074/jbc.M301311200. [DOI] [PubMed] [Google Scholar]
- 36.Bischoff ED, Daige CL, Petrowski M, Dedman H, Pattison J, Juliano J, Li AC, Schulman IG. Non-redundant roles for lxralpha and lxrbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J Lipid Res. 2010;51:900–906. doi: 10.1194/jlr.M900096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P. Synthetic lxr ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A. 2002;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levin N, Bischoff ED, Daige CL, Thomas D, Vu CT, Heyman RA, Tangirala RK, Schulman IG. Macrophage liver x receptor is required for antiatherogenic activity of lxr agonists. Arterioscler Thromb Vasc Biol. 2005;25:135–142. doi: 10.1161/01.ATV.0000150044.84012.68. [DOI] [PubMed] [Google Scholar]
- 39.Lund EG, Peterson LB, Adams AD, Lam MH, Burton CA, Chin J, Guo Q, Huang S, Latham M, Lopez JC, Menke JG, Milot DP, Mitnaul LJ, Rex-Rabe SE, Rosa RL, Tian JY, Wright SD, Sparrow CP. Different roles of liver x receptor alpha and beta in lipid metabolism: Effects of an alpha-selective and a dual agonist in mice deficient in each subtype. Biochem Pharmacol. 2006;71:453–463. doi: 10.1016/j.bcp.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Brunham LR, Kruit JK, Pape TD, Parks JS, Kuipers F, Hayden MR. Tissue-specific induction of intestinal abca1 expression with a liver x receptor agonist raises plasma hdl cholesterol levels. Circ Res. 2006;99:672–674. doi: 10.1161/01.RES.0000244014.19589.8e. [DOI] [PubMed] [Google Scholar]
- 41.Yasuda T, Grillot D, Billheimer JT, Briand F, Delerive P, Huet S, Rader DJ. Tissue-specific liver x receptor activation promotes macrophage reverse cholesterol transport in vivo. Arterioscler Thromb Vasc Biol. 2010;30:781–786. doi: 10.1161/ATVBAHA.109.195693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tangirala RK, Bischoff ED, Joseph SB, Wagner BL, Walczak R, Laffitte BA, Daige CL, Thomas D, Heyman RA, Mangelsdorf DJ, Wang X, Lusis AJ, Tontonoz P, Schulman IG. Identification of macrophage liver x receptors as inhibitors of atherosclerosis. Proc Natl Acad Sci U S A. 2002;99:11896–11901. doi: 10.1073/pnas.182199799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teupser D, Kretzschmar D, Tennert C, Burkhardt R, Wilfert W, Fengler D, Naumann R, Sippel AE, Thiery J. Effect of macrophage overexpression of murine liver x receptor-alpha (lxr-alpha) on atherosclerosis in ldl-receptor deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:2009–2015. doi: 10.1161/ATVBAHA.108.175257. [DOI] [PubMed] [Google Scholar]
- 44.Schulman IG. Nuclear receptors as drug targets for metabolic disease. Advanced drug delivery reviews. 2010;62:1307–1315. doi: 10.1016/j.addr.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP, Billheimer JT, Rothblat GH, Rader DJ. Pharmacological activation of liver x receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113:90–97. doi: 10.1161/CIRCULATIONAHA.105.560177. [DOI] [PubMed] [Google Scholar]
- 46.Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, Schulman IG, Glass CK. Promoter-specific roles for liver x receptor/corepressor complexes in the regulation of abca1 and srebp1 gene expression. Mol Cell Biol. 2003;23:5780–5789. doi: 10.1128/MCB.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor lxr alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 48.Fournier N, Paul JL, Atger V, Cogny A, Soni T, de la Llera-Moya M, Rothblat G, Moatti N. Hdl phospholipid content and composition as a major factor determining cholesterol efflux capacity from fu5ah cells to human serum. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:2685–2691. doi: 10.1161/01.atv.17.11.2685. [DOI] [PubMed] [Google Scholar]
- 49.Yancey PG, de la Llera-Moya M, Swarnakar S, Monzo P, Klein SM, Connelly MA, Johnson WJ, Williams DL, Rothblat GH. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor bi. The Journal of biological chemistry. 2000;275:36596–36604. doi: 10.1074/jbc.M006924200. [DOI] [PubMed] [Google Scholar]
- 50.Okazaki H, Goldstein JL, Brown MS, Liang G. Lxr-srebp-1c-phospholipid transfer protein axis controls very low density lipoprotein (vldl) particle size. J Biol Chem. 2010;285:6801–6810. doi: 10.1074/jbc.M109.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yazdanyar A, Yeang C, Jiang XC. Role of phospholipid transfer protein in high-density lipoprotein- mediated reverse cholesterol transport. Curr Atheroscler Rep. 2011;13:242–248. doi: 10.1007/s11883-011-0172-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laffitte BA, Joseph SB, Chen M, Castrillo A, Repa J, Wilpitz D, Mangelsdorf D, Tontonoz P. The phospholipid transfer protein gene is a liver x receptor target expressed by macrophages in atherosclerotic lesions. Mol Cell Biol. 2003;23:2182–2191. doi: 10.1128/MCB.23.6.2182-2191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Honzumi S, Shima A, Hiroshima A, Koieyama T, Ubukata N, Terasaka N. Lxralpha regulates human cetp expression in vitro and in transgenic mice. Atherosclerosis. 2010;212:139–145. doi: 10.1016/j.atherosclerosis.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 54.Masson D, Jiang XC, Lagrost L, Tall AR. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J Lipid Res. 2009;50(Suppl):S201–S206. doi: 10.1194/jlr.R800061-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luo Y, Tall AR. Sterol upregulation of human cetp expression in vitro and in transgenic mice by an lxr element. J Clin Invest. 2000;105:513–520. doi: 10.1172/JCI8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masson D, Staels B, Gautier T, Desrumaux C, Athias A, Le Guern N, Schneider M, Zak Z, Dumont L, Deckert V, Tall A, Jiang XC, Lagrost L. Cholesteryl ester transfer protein modulates the effect of liver x receptor agonists on cholesterol transport and excretion in the mouse. J Lipid Res. 2004;45:543–550. doi: 10.1194/jlr.M300432-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Agarwal-Mawal A, Murray CM, Belkhode S, Cheema SK. Differential regulation of cholesterol homeostasis in transgenic mice expressing human cholesterol ester transfer protein. Canadian journal of physiology and pharmacology. 2007;85:430–438. doi: 10.1139/y07-019. [DOI] [PubMed] [Google Scholar]
- 58.Cappel DA, Palmisano BT, Emfinger CH, Martinez MN, McGuinness OP, Stafford JM. Cholesteryl ester transfer protein protects against insulin resistance in obese female mice. Molecular metabolism. 2013;2:457–467. doi: 10.1016/j.molmet.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zelcer N, Tontonoz P. Liver x receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Houck KA, Borchert KM, Hepler CD, Thomas JS, Bramlett KS, Michael LF, Burris TP. T0901317 is a dual lxr/fxr agonist. Molecular genetics and metabolism. 2004;83:184–187. doi: 10.1016/j.ymgme.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Kumar N, Solt LA, Conkright JJ, Wang Y, Istrate MA, Busby SA, Garcia-Ordonez RD, Burris TP, Griffin PR. The benzenesulfoamide t0901317 [n-(2,2,2-trifluoroethyl)-n-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluorometh yl)ethyl]phenyl]-benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-alpha/gamma inverse agonist. Molecular pharmacology. 2010;77:228–236. doi: 10.1124/mol.109.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mitro N, Vargas L, Romeo R, Koder A, Saez E. T0901317 is a potent pxr ligand: Implications for the biology ascribed to lxr. FEBS letters. 2007;581:1721–1726. doi: 10.1016/j.febslet.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 63.Bi X, Zhu X, Duong M, Boudyguina EY, Wilson MD, Gebre AK, Parks JS. Liver abca1 deletion in ldlrko mice does not impair macrophage reverse cholesterol transport or exacerbate atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:2288–2296. doi: 10.1161/ATVBAHA.112.301110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lo Sasso G, Murzilli S, Salvatore L, D'Errico I, Petruzzelli M, Conca P, Jiang ZY, Calabresi L, Parini P, Moschetta A. Intestinal specific lxr activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab. 2010;12:187–193. doi: 10.1016/j.cmet.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 65.Favari E, Calabresi L, Adorni MP, Jessup W, Simonelli S, Franceschini G, Bernini F. Small discoidal pre-beta1 hdl particles are efficient acceptors of cell cholesterol via abca1 and abcg1. Biochemistry. 2009;48:11067–11074. doi: 10.1021/bi901564g. [DOI] [PubMed] [Google Scholar]
- 66.Castro GR, Fielding CJ. Early incorporation of cell-derived cholesterol into pre-beta-migrating high-density lipoprotein. Biochemistry. 1988;27:25–29. doi: 10.1021/bi00401a005. [DOI] [PubMed] [Google Scholar]
- 67.Asztalos B, Zhang W, Roheim PS, Wong L. Role of free apolipoprotein a–i in cholesterol efflux. Formation of pre-alpha-migrating high-density lipoprotein particles. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:1630–1636. doi: 10.1161/01.atv.17.9.1630. [DOI] [PubMed] [Google Scholar]
- 68.Barter PJ, Brewer HB, Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: A novel target for raising hdl and inhibiting atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 69.Tall AR. The effects of cholesterol ester transfer protein inhibition on cholesterol efflux. The American journal of cardiology. 2009;104:39E–45E. doi: 10.1016/j.amjcard.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 70.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant apoa-i milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 71.Zhu X, Parks JS. New roles of hdl in inflammation and hematopoiesis. Annual review of nutrition. 2012;32:161–182. doi: 10.1146/annurev-nutr-071811-150709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Terasaka N, Hiroshima A, Koieyama T, Ubukata N, Morikawa Y, Nakai D, Inaba T. T-0901317, a synthetic liver x receptor ligand, inhibits development of atherosclerosis in ldl receptor-deficient mice. FEBS Lett. 2003;536:6–11. doi: 10.1016/s0014-5793(02)03578-0. [DOI] [PubMed] [Google Scholar]
- 73.Kalaany NY, Gauthier KC, Zavacki AM, Mammen PP, Kitazume T, Peterson JA, Horton JD, Garry DJ, Bianco AC, Mangelsdorf DJ. Lxrs regulate the balance between fat storage and oxidation. Cell metabolism. 2005;1:231–244. doi: 10.1016/j.cmet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 74.Kappus MS, Murphy AJ, Abramowicz S, Ntonga V, Welch CL, Tall AR, Westerterp M. Activation of liver x receptor decreases atherosclerosis in ldlr−/− mice in the absence of atp-binding cassette transporters a1 and g1 in myeloid cells. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:279–284. doi: 10.1161/ATVBAHA.113.302781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fowler AJ, Sheu MY, Schmuth M, Kao J, Fluhr JW, Rhein L, Collins JL, Willson TM, Mangelsdorf DJ, Elias PM, Feingold KR. Liver x receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: Liver-x-receptor-specific inhibition of inflammation and primary cytokine production. The Journal of investigative dermatology. 2003;120:246–255. doi: 10.1046/j.1523-1747.2003.12033.x. [DOI] [PubMed] [Google Scholar]
- 76.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver x receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 77.Fontaine C, Rigamonti E, Nohara A, Gervois P, Teissier E, Fruchart JC, Staels B, Chinetti-Gbaguidi G. Liver x receptor activation potentiates the lipopolysaccharide response in human macrophages. Circ Res. 2007;101:40–49. doi: 10.1161/CIRCRESAHA.106.135814. [DOI] [PubMed] [Google Scholar]
- 78.Claudel T, Leibowitz MD, Fievet C, Tailleux A, Wagner B, Repa JJ, Torpier G, Lobaccaro JM, Paterniti JR, Mangelsdorf DJ, Heyman RA, Auwerx J. Reduction of atherosclerosis in apolipoprotein e knockout mice by activation of the retinoid x receptor. Proc Natl Acad Sci U S A. 2001;98:2610–2615. doi: 10.1073/pnas.041609298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Groot PH, Pearce NJ, Yates JW, Stocker C, Sauermelch C, Doe CP, Willette RN, Olzinski A, Peters T, d'Epagnier D, Morasco KO, Krawiec JA, Webb CL, Aravindhan K, Jucker B, Burgert M, Ma C, Marino JP, Collins JL, Macphee CH, Thompson SK, Jaye M. Synthetic lxr agonists increase ldl in cetp species. J Lipid Res. 2005;46:2182–2191. doi: 10.1194/jlr.M500116-JLR200. [DOI] [PubMed] [Google Scholar]
- 80.Quinet EM, Basso MD, Halpern AR, Yates DW, Steffan RJ, Clerin V, Resmini C, Keith JC, Berrodin TJ, Feingold I, Zhong W, Hartman HB, Evans MJ, Gardell SJ, DiBlasio-Smith E, Mounts WM, LaVallie ER, Wrobel J, Nambi P, Vlasuk GP. Lxr ligand lowers ldl cholesterol in primates, is lipid neutral in hamster, and reduces atherosclerosis in mouse. J Lipid Res. 2009;50:2358–2370. doi: 10.1194/jlr.M900037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Atger V, de la Llera Moya M, Bamberger M, Francone O, Cosgrove P, Tall A, Walsh A, Moatti N, Rothblat G. Cholesterol efflux potential of sera from mice expressing human cholesteryl ester transfer protein and/or human apolipoprotein ai. The Journal of clinical investigation. 1995;96:2613–2622. doi: 10.1172/JCI118326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fournier N, de la Llera Moya M, Burkey BF, Swaney JB, Paterniti J, Jr, Moatti N, Atger V, Rothblat GH. Role of hdl phospholipid in efflux of cell cholesterol to whole serum: Studies with human apoa-i transgenic rats. Journal of lipid research. 1996;37:1704–1711. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.