

Abstract
Due to the complexity of biological systems, cutting-edge machine-learning methods will be critical for future drug development. In particular, machine-vision methods to extract detailed information from imaging assays and active-learning methods to guide experimentation will be required to overcome the dimensionality problem in drug development.
High-throughput and high-content screening have been widely adopted by pharmaceutical and biotechnology companies as well as many academic labs over the past twenty years, with the goal of rapidly identifying potential drugs that affect specific molecular targets.1-3 These technologies dramatically enhance the rate and amount of information that can be collected about the effects of chemical compounds, and publicly-funded efforts such as the Molecular Libraries Screening Centers of the United States National Institutes of Health have enabled the creation of extensive databases such as PubChem (these databases typically contain the results of many screens in the form of scores for many compounds on a given assay, and also contain information on the structures of compounds and the target of particular assays). However, the premise that effective drugs can be found by screening primarily in single target assays has run aground on the complex network of interactions that occur within cells and tissues; drugs often have unfavorable side effects that are not discovered until late in the drug development process.4 Discovering these effects earlier in the development process by screening simultaneously for a specific desired effect and against many (potentially thousands of) undesired ones is infeasible. One proposed approach is to create panels of assays to capture many aspects of cell or tissue behavior.5 This may reduce the dimensionality of the problem, but relies on knowing in advance which aspects to assay. In any case, the size and complexity of the problem make reliance on human evaluation of results problematic. This type of challenge is being encountered throughout Big Science projects, and is one that machine learning, in which statistical and computational techniques are applied to learn complex relationships and build models, is well suited to address. Thus, machine learning will play increasingly important roles in the drug discovery and development process in the future. Here, I focus on two areas where machine learning can have a profound impact: the use of machine-vision methods to improve information extraction from high-content assays, and the use of active machine learning to drive experimentation.
Seeing more in an assay
High-throughput microscopy and high-content screening are widely used to determine the effects of small-molecule compounds, inhibitory RNAs, or other treatments (collectively referred to as perturbagens) on both specific molecular targets and cell behaviors. Analysis for high-content screens is typically done by calculating features that describe aspects of the images, and training a classifier (Fig. 1a, path i) to recognize the expected patterns (e.g., of positive and negative controls).6, 7 An alternative is to use clustering methods (which do not need to know the patterns ahead of time) to identify compounds that have similar biological effects.8 While these approaches can provide important information, they have two major limitations. The first is that they do not work well when changes occur along a continuum, in which the assumption of discrete populations made by classification and clustering methods does not hold. An example of a continuum is the relocation of a protein from one organelle to another. Classifiers may reveal whether or not the relocation occurs in an endpoint assay, but do not accurately represent the kinetics of the process or whether two compounds differ in the extent of induced relocation. The second limitation is that image features are usually sensitive to differences in cell size and shape such that the same classifier cannot be used for more than one cell type. This not only requires retraining for each cell type, but, more importantly, it does not readily allow comparison of patterns between cell types.
Figure 1.

Machine-vision methods for identifying and resolving drug and disease effects on protein distributions. a) Methods for extracting information about subcellular location from microscope images. (i) Given a collection of images for a cell type with unknown patterns (such as tagged proteins with unknown subcellular distributions) and images for proteins with known (fundamental) location patterns in that cell type, a classifier can be trained to recognize those subcellular patterns and used to assign a label to each unknown image. If the unknown images contain a mixture of patterns, results will be unpredictable. (ii) As an alternative, the fundamental location images can be used to train a supervised pattern-unmixing system so that the fraction of each unknown protein present in each fundamental pattern can be determined. This provides a better representation for the protein's distribution. (iii) If images of the fundamental patterns are not available (or if all patterns are not known), unsupervised pattern unmixing can be used to simultaneously estimate the fundamental patterns and the fraction in each. (iv) Lastly, the fundamental patterns can each be represented by learning generative models, so that each unknown protein can be compactly represented as a set of pattern models plus the fraction of protein in each. b) Methods for comparing location patterns between cell types and conditions. Current methods use features to train classifiers to recognize patterns in each cell type, and then apply them to determine if the pattern has been changed by a compound. This typically detects only major changes. Alternatively, the parameters of generative models provide a more interpretable and consistent means of comparing patterns between cell types, and a more sensitive approach for analysis of compound effects. In this hypothetical example, the first cell type (x) undergoes a change in the morphology of the Golgi that was missed by classification but was reflected in the generative model. In a third cell type (z), a protein is relocalized in response to a drug from Golgi to a population of endosomes that are statistically larger than in untreated cells.
Machine-vision methods have the potential to extract more detailed information from high-content assays than methods currently in use. Pattern-unmixing methods seek to address the continuous nature of relocation events by estimating the fraction of a target that is in each of the subcellular locations. This can be done by both supervised methods (Fig. 1a, path ii) in which the locations are specified, and unsupervised methods (Fig. 1a, path iii) in which the patterns are discovered at the same time that the fractions are estimated. Once discovered, the patterns can be represented by learning generative models (Fig. 1a, path iv), as discussed below.
In addition to their role in drug development, perturbagen studies also improve our understanding of cellular mechanisms, which in turn accelerates future drug development. Constructing models of cellular organization and how it changes through the cell cycle, through development, and in response to disease or drugs is critical for understanding these mechanisms. However, current classification methods for comparing different cell types and conditions do not capture changes in sufficient detail (Fig. 1b, upper). The alternative is to build generative models directly from images to capture not just average behavior but how that behavior varies from cell to cell (Fig. 1b, lower). We can view this problem as trying to infer a generative model of cell behavior from example images under specified conditions.
Unlike descriptive, feature-based approaches, generative models are able to synthesize new images that in a statistical sense are drawn from the same population as the images they were trained on. In this way, they are analogous to Hidden Markov Models for sequence motifs, which describe the input training motifs but can also generate new sequences. A critical question in designing approaches for learning generative models is how to decompose different aspects of cell organization. One approach is to first build a model of nuclear size and shape, and then use it as a basis for a model of cell size and shape. The distribution of other components can then be learned relative to the nuclear and plasma membrane. Such models have a conditional structure, that is, the model of a given organelle (e.g., an endosome) depends upon the models for the plasma membrane and nucleus, and the model for the plasma membrane depends on the model for the nucleus. This approach has been used to learn models of HeLa cells, first in two dimensions9 and more recently in three dimensions10, 11. Many aspects of these models were simplistic, so additional work will be needed to improve them, including more realistic models of organelle shape and the distribution of proteins within organelles. In addition, it will be critical to capture which distributions are conditional upon which (e.g., whether the distributions of mitochondria are determined by the distribution of microtubules, and to what extent).
A major advantage of learning generative models is that the parameters of those models provide a method for comparing distributions between cell types or conditions. For example, we can ask whether the distribution of mitochondria within one cell type follows the same model as another cell type once differences in cell size and shape are factored out. Similarly, we can ask whether the effects of a perturbagen that appears to change both cell shape and microtubule distribution does so independently or whether the change in the latter can be accounted for by the change in the former.
Improving how we use machine learning to extract and represent information from cell images is an area of ongoing research and much work remains to be done. Of particular importance will be methods for learning and modeling cell organization not just in space but in time. Since continuous imaging of the same cell at subsecond resolution for many days is not currently feasible, adding temporal information involves not only determining the time scales with which changes occur, but also combining information from different time series. The ultimate goal is a machine model of the spatiotemporal behavior of each cell type. This brings us to the question of how those behaviors change in response to disease or drugs.
Active learning to the rescue
At a fundamental level, the central problem of screening for potential drugs is the dimensionality of the experimental space within which screening takes place. As highlighted in Fig. 2, the number of experiments required to directly screen for compounds that affect one target while not affecting others can quickly become intractable. The only practical solution is to carry out a subset of the possible experiments. Current approaches in drug development require scientists to choose a path through experimental space guided by existing knowledge (e.g., signaling pathways), investigator insight and intuition. This process is often hindered by incomplete or incorrect pathway information and the difficulty of making predictions about complex pathway interactions. An alternative described here involves the use of active machine-learning methods to build statistical models of the entire space and iteratively choose experiments that are expected to best improve the model. The major strength of this approach is that experiment choice is guided on a purely empirical basis and in full consideration of the potential complexity of the system. Active learning is well established in some domains, and it has been applied in a few cases to biological problems (Box 1).
Figure 2.
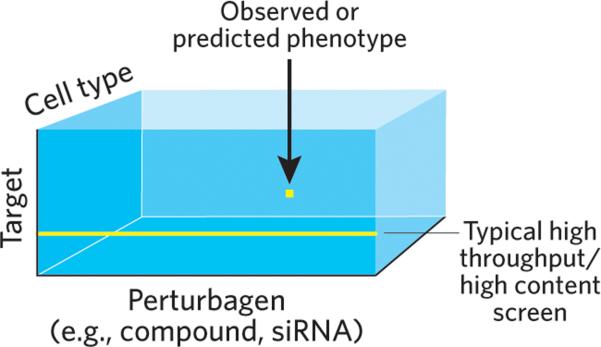
The perturbagen effect hyper-rectangle. Each element of the 3D matrix contains the phenotype observed or predicted for a given combination of perturbagen, target and cell type. For 104 protein targets, 102 cell types and 106 compounds, filling the matrix would require 1012 assays. With triplicate wells for each assay and 384 wells per plate, this corresponds to approximately 10 billion plates. At a rate of 1 plate per minute, this would take 15,000 years. Varying the concentration of compounds or testing combinations of compounds makes the problem exponentially worse.
The two main components of an active-learning system are a method for constructing a predictive model from currently available data and a method for using the model to determine future data collection. Passive machine-learning applications consist of just the first component. The system is “active” because the learner is able to iteratively choose one or more data points to be collected and added to the existing data. The construction of models and their application to guide experimentation are already central to the field of systems biology. The critical difference between active learning and systems biology is that the latter typically seeks to test or validate a model. Systems biology therefore describes only one round of model construction and testing, and chooses a high (often the highest) confidence prediction to test. However, except in cases where the model construction was seriously flawed, the prediction is typically correct and therefore provides little or no information for improving the model. By contrast, the essence of active-learning methods is to choose experiments for which predictions are likely to be wrong, since (as discussed below) these experiments are expected to most directly lead to model improvement.
Constructing predictive models
The choice of a model construction method should be specific to the problem being studied. In the case considered here, we wish to determine the effects of perturbagens upon many targets (typically proteins) in many cell types. As shown in Fig. 2, we can view the problem as a three-dimensional matrix in which the contents of each element contain a label specifying the phenotype that was, or is predicted to be, observed for that particular combination of variables. Given the size of this matrix and the explicit goal of filling it without doing exhaustive experimentation, the modeling challenge is straightforward: learn a set of rules much smaller in number than the size of the matrix that enable the value of all elements to be imputed. We must assume that such a set exists, since the alternative is that nothing but exhaustive (or nearly exhaustive) experimentation will do. Fortunately, the accuracy of model predictions for future experiments provides a measure of the correctness of this assumption.
For a screen, information is available not only on the results of assays (which we refer to below as “internal” data) but also on the properties of the targets and perturbagens (“external” data). We can therefore consider two variations on the modeling problem: one in which we ignore external information and one in which we take it into account. Ignoring external data may provide a less biased exploration of the assay results. This is possible because not only can external data be wrong (e.g., a protein-protein interaction listed in a database may not occur under the conditions of the experiment) but they might not be distributed randomly over the sets of targets and perturbagens (much more information is available on certain classes of targets than others, sets of motifs are biased towards systems that have been more heavily studied, and chemical descriptors may be missing essential moieties not previously observed to be important).
Once a model is constructed, the challenge is choosing which data to collect next. A number of active learning methods have been described to perform this task that typically balance between choosing points for which the current model is uncertain and points in regions that have not been explored. The goal is not to test the current model (given that all models are wrong to some extent) but rather to improve it as much and as quickly as possible. My group is currently developing systems for both the “internal only” and “internal plus external” cases.
Conclusion
While the complexity of cellular systems is expected to continue to challenge both cell biologists and drug developers for many years, machine-learning methods hold tremendous promise for determining the critical relationships governing cell behaviors. This promise will be realized through better extraction of information from high content screens and more effective experimentation driven by active learning. The result will be that drug discovery and development will be dramatically improved by being able to assess effects of potential drugs more comprehensively. Clearly much work remains to be done, not the least of which is to convince practitioners of the value of ceding some important decisions to machines.
Box 1. Examples of active learning applications in biology.
Reconstructing gene networks14
Cancer classification15
Finding cancer rescue mutations16, 17
Predicting macromolecular structure18
Learning protein-protein interactions19
Acknowledgments
Much of the work from my group referred to here was supported by NIH grant GM075205.
Footnotes
Competing financial interests
The author declares no competing financial interest.
References
- 1.Bleicher KH, Bohm HJ, Muller K, Alanine AI. Nat Rev Drug Discov. 2003;2:369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 2.Taylor DL. Methods Mol Biol. 2007;356:3–18. doi: 10.1385/1-59745-217-3:3. [DOI] [PubMed] [Google Scholar]
- 3.Macarron R, et al. Nat Rev Drug Discov. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 4.Merino A, Bronowska AK, Jackson DB, Cahill DJ. Drug Discov Today. 2010;15:749–756. doi: 10.1016/j.drudis.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Giuliano KA, Premkumar DR, Strock CJ, Johnston P, Taylor L. Combinatorial chemistry & high throughput screening. 2009;12:838–848. doi: 10.2174/138620709789383286. [DOI] [PubMed] [Google Scholar]
- 6.Carpenter AE, et al. Genome biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shariff A, Kangas J, Coelho LP, Quinn S, Murphy RF. Journal of biomolecular screening : the official journal of the Society for Biomolecular Screening. 2010;15:726–734. doi: 10.1177/1087057110370894. [DOI] [PubMed] [Google Scholar]
- 8.Perlman ZE, et al. Science. 2004;306:1194–1198. doi: 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- 9.Zhao T, Murphy RF. Cytometry Part A. 2007;71A:978–990. doi: 10.1002/cyto.a.20487. [DOI] [PubMed] [Google Scholar]
- 10.Shariff A, Murphy RF, Rohde GK. Cytometry Part A. 2010;77A:457–466. doi: 10.1002/cyto.a.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng T, Murphy RF. Cytometry Part A. 2011;79A doi: 10.1002/cyto.a.21066. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warmuth MK, et al. Journal of Chemical Information and Computer Sciences. 2003;43:667–673. doi: 10.1021/ci025620t. [DOI] [PubMed] [Google Scholar]
- 13.Fujiwara Y, et al. Journal of Chemical Information and Modeling. 2008;48:930–940. doi: 10.1021/ci700085q. [DOI] [PubMed] [Google Scholar]
- 14.Pournara I, Wernisch L. Bioinformatics. 2004;20:2934–2942. doi: 10.1093/bioinformatics/bth337. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y. Journal of Chemical Information and Computer Sciences. 2004;44:1936–1941. doi: 10.1021/ci049810a. [DOI] [PubMed] [Google Scholar]
- 16.Danziger SA, et al. PLoS Comput Biol. 2009;5:e1000498. doi: 10.1371/journal.pcbi.1000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danziger SA, Zeng J, Wang Y, Brachmann RK, Lathrop RH. Bioinformatics. 2007;23:i104–114. doi: 10.1093/bioinformatics/btm166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stegle O, Payet L, Mergny JL, MacKay DJ, Leon JH. Bioinformatics. 2009;25:i374–382. doi: 10.1093/bioinformatics/btp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohamed TP, Carbonell JG, Ganapathiraju MK. BMC Bioinformatics. 2010;11(Suppl 1):S57. doi: 10.1186/1471-2105-11-S1-S57. [DOI] [PMC free article] [PubMed] [Google Scholar]