

Abstract
A graft copolymer with pendant drug segment can fold into nanostructures in a protein folding-like manner. The graft copolymer is constructed by directly polymerizing γ-camptothecin-glutamate N-carboxyanhydride (Glu(CPT)-NCA) on multiple sites of poly(ethylene glycol) (PEG)-based main chain via the ring open polymerization (ROP). The “purely” conjugated anticancer agent camptothecin (CPT) is hydrophobic and serves as the principal driving force during the folding process. When exposed to water, the obtained copolymer, together with doxorubicin (Dox), another anticancer agent, can fold into monodispersed nanocarriers (with a diameter of around 50 nm) for dual-drug delivery. Equipped with a PEG shell, the nanocarriers displayed good stability and can be internalized by a variety of cancer cell lines via the lipid raft and clathrin-mediated endocytotic pathway without premature leakage, which showed a high synergetic activity of CPT and Dox toward various cancer cells. In vivo study validated that the nanocarriers exhibited strong accumulation in tumor sites and showed a prominent anticancer activity against the lung cancer xenograft mice model compared with free drugs.
Keywords: graft copolymer, micelle, combination therapy, folding, ring open polymerization
1. Introduction
Evolution of drug resistance in cancer cells has been attributed as a major factor in the failure of many forms of monotherapy [1–2]. To prevent evolutionary development, it often requires a high drug dose to kill the whole cancer cell population; whereas it always induces severe side effects [3–4]. The limitations of monotherapy can be circumvented by synergistic combination of multiple anticancer agents which allows for reduction of the drug dose and offers a potential benefit to simultaneously act on several cancer targets, therefore preventing or delaying the emergence of drug resistance [5]. However, the traditional combination strategy, namely the drug cocktail, shows limited success in clinics due to the non-coordinated distributions of drugs after administration [6]. Pharmacokinetic interactions are also observed in these combination therapies, raising concern about the synergistic toxicity of these cocktails [7–8]. For example, a combination of rapamycin (RAPA) and cyclosporine (CsA) produces renal dysfunction, since the pharmacokinetic interaction between RAPA and CsA highly increases the concentration of CsA in kidney [7]. In addition, the difference in solubility, potency, pharmacokinetics and bioavailability between drugs makes the dose schedule extremely challenging in the cocktail therapy [9–11].
Nanocarriers have been developed as an important strategy for drug delivery due to their capabilities of enhancing drug solubility, improving pharmacokinetics and preferentially accumulating in tumor by the enhanced permeability and retention (EPR) effect [12–17]. Therefore, the nanocarrier-based combination therapy not only has the loading potency of the multiple agents, but also accommodates their biodistribution and plasma elimination, realizing an extremely simple dose optimization [18–19]. Polymeric micelles are one of the most common nanoparticle systems for co-delivery of multiple drugs due to their physicochemical stability and low toxicity [12, 20–21].
Topoisomerase (Top) represents one of the most popular targets in cancer chemotherapy. Its inhibitors can block the DNA ligation step during cell cycling, and induce irreversible single and double stranded DNA breaks during transcription and replication, thereby leading to apoptosis and cell death [22]. Both camptothecin (CPT) and doxorubicin (Dox) are potent Top inhibitors, but they interfere with the action of Top I and II, respectively. CPT as the Top I inhibitor has shown potent anti-tumor efficacy in a broad spectrum of cancers in clinic [23–24]. However, certain clinical limitations such as resistance of cancer cells impair its clinical application [25–26]. It has been postulated that the reduced Top I activity following the CPT treatment could be compensated by the Top of the other class (Top II), because the actions of both enzymes are partially overlapping [27]. It has been proven that the increase of Top II activity occurred in cancer cells resistant to CPT [28], and Top II inhibitor Dox showed a collateral cytotoxicity on the CPT adapted cancer cells [29]. Therefore, the concomitant use of both Top I and Top II inhibitors might elicit synergistic effects and prevent the emergence of drug resistance [30]. Polymeric nanocarriers have provided a novel combination strategy for simultaneously delivering multiple agents to tumor sites via a single vehicle. Coordination of the solubility, pharmacokinetics and biodistribution of multiple drugs by nanocarriers allows this strategy to overcome the problems commonly associated with traditional drug cocktail methodology.
We designed a new graft copolymer, which can fold into a polymeric nanomicelle for co-delivery of two small-molecule anticancer drugs: CPT and Dox. Unlike the traditional micelle assembled from AB type of amphiphilic diblock copolymer, the current nanocarrier was folded by a structurally well-defined graft copolymer in which the side chain associated together as the hydrophobic micelle core and poly(ethylene glycol) (PEG) main chain entangled around it. As depicted in Figure 1A, the graft copolymer was constructed from three monomers by a two-step polymerization. The “tee joint”-like monomer I and monomer II were first condensed into a linear multiblock copolymer, to which a CPT-linked monomer III γ-camptothecin-glutamate N-carboxyanhydride (Glu(CPT)-NCA) was subsequently grafted by an amine-initiated ring open polymerization (ROP) [31–32]. The resulting graft copolymer displayed a well-defined structure where the main chain was composed of the hydrophilic PEG and the side chains were polypeptide chains “purely” conjugated with hydrophobic drugs (Figure 1B). Once reconstituted with water, this graft copolymer (“denatured” state in organic solvents, designated “Sd”) folded into a uniform nanocarrier (“native” state, designated “Sn”), in which the side chains were assembled into a hydrophobic core wrapped around by the main PEG chain as a shell. Unlike the traditional micelles assembled by a linear unit, this nanocarrier was folded from a grafted unit, making the final structure more compact and stable [33]. In addition, the hydrophobic core also provided accommodation for co-encapsulating other hydrophobic anticancer drugs through physical interactions such as π-π stacking [9, 34–35]. In our co-delivery system, Dox was physically encapsulated in the hydrophobic core, while the CPT was covalently linked on side chains which can be released via the degradation of ester linkage by the intracellular esterase [36–37].
Figure 1.

Schematic design of graft copolymer synthesis and stealth nanocarrier folding. (A) Graft copolymer with pendant drug segment is synthesized from three monomers by a two-step polymerization, and then folds into nanostructure with free doxorubicin encapsulated. (B) The chemical structure of graft copolymer. The graft copolymer is comprised of a PEG-containing multiblock copolymer main chain and the polyglutamic acid-based side chains with “purely” conjugated camptothecin.
2. Materials and methods
2.1. Material
All chemicals were obtained from commercial sources and used without further purification. Tris(2-aminoethyl)amine, di-t-butyl dicarbonate (Boc2O), trifluoroacetic acid (TFA), N,O-bis(trimethylsilyl) acetamide (BSA) and triphosgene were purchased from Sigma Aldrich. N,N′-diisopropylcarbodiimide (DIPC), triethylamine (Et3N) and 4-dimethylaminopyridine (DMAP) were obtained from Acros Organics. N-hydroxylsuccinimide (NHS) bifunctionalized PEG5000 (NHS-PEG5000-NHS) was purchased from Nanocs Inc. CPT and Dox were purchased from Alfa Aesar. Boc-L-glutamic acid 5-tert-butyl ester (Boc-Glu-OtBu) was ordered from Chem-Impex International Inc. All the organic solvents for synthesis and analysis were ordered from Fisher Scientific Inc. and used as received.
2.2. Synthesis of graft copolymer 5
2.2.1. Synthesis of monomer I (2)
The monomer I, N,N-bis(2-aminoethyl)-N-[2-(tert-butylcarbamoyl)ethyl-amine (2), was synthesized as reported [38–39]. Briefly, a chloroform solution of Boc2O (1 g, 4.6 mmol, 0.1 equiv) was added dropwise into a stirred solution of tris(2-aminoethyl)amine (1) (6.7 g, 46 mmol) in 300 mL of chloroform at 0 °C. The reaction was allowed to warm to room temperature and stirred overnight. The reaction was stopped by 15 mL of distilled deionized (DD) water. After stirred for 5 min, the organic phase was separated. The aqueous phase was re-extracted with 30 mL of chloroform. The combined organic phase was dried with Na2SO4, then concentrated under vacuum to give crude compound 2 which was purified by silica gel chromatography using CHCl3/MeOH/concentrated aqueous NH4OH (v/v/v, 10/5/1) as eluent. The purified monomer I was given as viscous oil (1.05 g, 92%). 1H NMR (300 MHz, DMSO-d6): δ 6.81 (t, 1H), 2.95 (q, 2H), 2.81 (brs, 4H), 2.52 (t, 4H), 2.38 (q, 6H), 1.39 (s, 9H). ESI-MS calcd for C11H26N4O2 246.21, found 247.21 [M + H]+.
2.2.2. Synthesis of multiblock copolymer 3 by condensation polymerization
Multiblock copolymer 3 was synthesized as the method we reported before [40]. Briefly, Monomer I (2) and NHS-PEG5000-NHS (monomer II) was dried in CaCl2 desiccator for two days before synthesis. One gram of NHS-PEG5000-NHS (0.2 mmol, 1 equiv) was dissolved in 4 mL of dry dimethyl formamide (DMF), followed by adding 0.5 mL of monomer I (52.1 mg, 0.2 mmol, 1 equiv) solution in DMF at N2 atmosphere. The reaction was initiated by adding 140 μL of dry Et3N. Two days later, the reaction mixture was poured into 50 mL of diethyl ether. The precipitate was washed twice with diethyl ether, dried in vacuum and yield as white powder (860 mg, 81%). 1H NMR (300 MHz, CDCl3): δ 4.21 (t, 2H), 3.63 (s, 448H), 3.17 (m, 4H), 2.55 (t, 6H), 2.37 (brs, 4H), 1.43 (s, 9H).
2.2.3. TMS activated polymer 4
To remove the protecting group Boc, polymer 3 was dissolved in 10 mL of dichloromethane (DCM) and TFA mixture (v/v, 1/1). The scavenger, phenol (140 mg), was also added into the mixture to minimize the side reaction. The reaction mixture was stirred at room temperature for 1 h. The organic solvent was evaporated in vacuum and the residue was washed with diethyl ether. After removing the remaining organic solvent in vacuum, the residue was dissolved in water and dialyzed against DD water with cellulose tubing (molecular weight cut-off (MWCO): 12 kDa). The resulting solution was lyophilized and dried in CaCl2 desiccator.
The amine groups on the multiblock copolymer were activated with N-trimethylsilyl (TMS) as initiator for graft polymerization. The dry polymer (200 mg) was mixed with 5 mL of anhydrous benzene. After polymer was completely dissolved, 2 mL of N,O-bis(trimethylsilyl) acetamide (BSA) was added. The reaction was stirred at room temperature for 24 h. Anhydrous hexane (20 mL) was added into the reaction mixture to precipitate the product. The white precipitate was washed three times with anhydrous hexane and dried under vacuum to give polymer 4 in quantitative yield. 1H NMR (300 MHz, CDCl3): δ 4.21 (t, 2H), 3.64 (s, 448H), 3.20 (m, 4H), 2.60 (m, 6H), 2.12 (m, 4H), 0.01 (s, 9H).
2.2.4. Graft copolymer 5
The ROP of the side chain was initiated by TMS activated amines [31]. Briefly, monomer III (121.1 mg) and polymer 4 (62.1 mg) was suspended in 5 mL of dry chloroform. The yellow suspension gradually turned clear after adding 75 μL of dry Et3N. The reaction gradually became turbid over time, indicating the successful polymerization. After 48 h, the reaction mixture was poured into 30 mL of diethyl ether to precipitate the product. After washing three times with diethyl ether, the precipitate was dried under vacuum to give graft copolymer 5. Note: extensively washing of polymer 4 is essential for the graft polymerization. The trace amount of BSA in polymer 4 can induce considerable side reaction, resulting in failure of graft polymerization and large aggregation during folding. Directly initiating graft polymerization with amine (the intermediate of polymer 4) is also applicable, but with less ROP efficiency [31]. 1H NMR (300 MHz, DMSO-d6): δ 8.68 (s, 10H), 8.14 (m, 20H), 7.82 (m, 10H), 7.69 (m, 10H), 7.05 (s, 10H), 5.47 (m, 20H), 5.25 (br, 20H), 4.01 (s, 10H), 3.51 (s, 448H), 2.12 (br, 50H), 0.86 (br, 30H).
2.3. Synthesis of monomer III
2.3.1. Synthesis of Boc-Glu(CPT)-OtBu
To the suspension of CPT (0.8 g) in chloroform (50 mL), Boc-Glu-OtBu (2.4 g), DIPC (2.4 mL) and DMAP (0.154 g) were separately added. The suspension was stirred at room temperature and the reaction gradually turned clear in around 1 h. The reaction was stopped by adding 30 mL of saturated aqueous NH4Cl solution. The organic phase was separated, washed with brine and dried by Na2SO4. The organic solvent was removed by evaporation under vacuum and the crude product was purified by silica gel chromatography eluted with hexane/EtOAc (v/v = 2/1, then 1/1 and then EtOAc only). The pure product was given as white powder with yield of 80%. 1H NMR (300 MHz, DMSO-d6): δ 8.68 (s, 1H), 8.14 (t, 2H), 7.86 (t, 1H), 7.71 (t, 1H), 7.22 (d, 1H), 7.07 (s, 1H), 5.50 (s, 1H), 5.28 (s, 1H), 3.88 (m, 1H), 2.62 (m, 2H), 2.17 (t, 2H), 1.89 (q, 2H), 1.37 (s, 18H), 0.92 (t, 3H). ESI-MS calcd for C34H39N3O9 633.28, found 634.27 [M + H]+.
2.3.2. Glu(CPT)-OH
The compound Boc-Glu(CPT)-OtBu was dissolved in 10 mL of DCM at 0 °C. TFA (10 mL) was then added into the reaction. The mixture was allowed to warm to room temperature and stirred for 1.5 h. The solvent was removed under vacuum and the product was then precipitated with diethyl ether. After washing three times with ether, the precipitate was dried under vacuum for next step reaction (yield, 90%). 1H NMR (300 MHz, DMSO-d6): δ 8.68 (s, 1H), 8.15 (t, 2H), 7.86 (t, 1H), 7.71 (t, 1H), 7.09 (s, 1H), 5.51 (s, 2H), 5.28 (s, 2H), 3.96 (t, 1H), 3.46 (brs, 2H), 2.76 (m, 2H), 2.16 (m, 2H), 2.05 (m, 2H), 0.93 (t, 3H). ESI-MS calcd for C25H23N3O7 477.16, found 478.16 [M + H]+.
2.3.3. Glu(CPT)-NCA (monomer III)
Glu(CPT)-OH (280 mg) was added into 300 mL of anhydrous tetrahydrofuran (THF). The mixture was sonicated in ultrasonic water tank for couple minutes to disperse Glu(CPT)-OH in THF. After adding triphosgene (170 mg), the suspension was stirred at 48 °C for 2 h. The suspensions gradually become clear, indicating the completion of the reaction. Note: sufficient solvent THF in reaction is important; otherwise it will not turn clear in 2 h. The reaction was concentrated under vacuum and the yellow residue was re-dissolved in 15 mL of anhydrous THF. The crude product was precipitated with 30 mL of anhydrous hexane. The precipitate was collected by centrifuge, washed twice with anhydrous THF/hexane mixture (v/v, 1/2), dried in the vacuum desiccators (yield, 70%). 1H NMR (300 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.69 (s, 1H), 8.15 (t, 2H), 7.87 (t, 1H), 7.71 (t, 1H), 7.13 (s, 1H), 5.51 (s, 2H), 5.27 (s, 2H), 4.46 (m, 0.5H), 3.59 (m, 0.5H), 2.74 (t, 2H), 2.15 (q, 2H), 1.98 (m, 2H), 0.93 (t, 3H).
2.4. Folding graft copolymer into nanocarriers
Graft copolymer 5 was first dissolved in N-methylpyrrolidone (NMP) to obtain a clear solution with concentration of 10 mg/mL. To obtain the MB-20 nanocarriers, 1 mL of the graft copolymer solution was quickly added into 6 mL of DD water with vigorous shaking. The micelle solution was then sonicated for 20 cycles (1 s each with a duty cycle of 20%). The homogenous micelle solution was dialyzed against DD water for 24 h at room temperature to completely remove the organic solvent NMP. The DD water was frequently changed during the dialysis process. The nanocarrier solution was passed through 0.45 μm SFCA syringe filter (Corning Inc, Corning, NY). The folded micelle was stored at 4 °C for further characterization. For long-term storage, 10 mL of micelle solution was mixed with 100 mg of cryoprotectant hydroxypropyl-β-cyclodextrin (HPβCD), frozen in liquid nitrogen and lyophilized [41]. The lyophilization cake was stored at 4 °C for reconstitution with DD water. To encapsulate Dox, Et3N-treated Dox (1 mg) was added into 1 mL of graft copolymer solution in NMP and added slowly into DD water as described above. The loading efficiency of Dox under this condition was 10 wt% of the total polymer weight. Higher loading can be obtained by increasing the Dox amount during nanocarrier folding.
2.5. Characterization of graft copolymer and nanocarrier
Gel permeation chromatography (GPC) was performed on Waters 2695 Alliance® separation model equipped with RI 2414 (410) detector. The separation of polymer was achieved on Styrogel® HR4E column (Waters, 5 μm, 7.8 mm × 300 mm) at 50°C using DMF containing 0.1 M LiBr as mobile phase. The molecular weight of polymer was calibrated against standard PEG ranging from 112k–0.4k Dalton. For transmission electron microscope (TEM) characterization, the copper TEM grid (Ted Pella Inc.) was plasma glow-discharged for 20 s to create a hydrophilic surface on the carbon surface. Nanocarrier solution (20 μL) was absorbed onto the freshly plasma-discharged carbon membrane for 30 s and then blotted with a filter paper to remove excess solution. The grid was examined with JEOL 2000FX at 100 kV. The particle size of nanocarrier was measured on the Malvern Zetasizer NanoZS by dynamic light scattering (DLS).
2.6. In vitro drug release
The release of CPT from nanocarriers was monitored in 100% mouse serum by high performance liquid chromatography (HPLC). Briefly, MB-20 solution (or Glu(CPT)-OH solution) was mixed with 200% mouse serum to achieve a final concentration of 40 μM (CPT equivalent). The mixtures were incubated at 37 °C. At indicated time points, 20 μL of aliquot was sampled and precipitated with 80 μL of acetonitrile containing 0.5% TFA. After 10 min of incubation, the precipitate was spin down at 20,000g for 10 min and the supernatant was analyzed by HPLC for quantification of the released CPT. The HPLC system was equipped with Agilent pump, controller, autosampler and detector. The sample was analyzed on Waters C-18 reverse phase column (4.6 × 250 mm, 5 μm) with a mobile phase of acetonitrile/water (v/v, 35/65) plus 0.1% formic acid. The flow rate was set at 1 mL/min and absorbance of CPT was monitored at 370 nm.
The release of Dox was conducted using the dialysis method in the phosphate buffer (50 mM, pH 5.0 and 7.4) as reported [42]. MB-20/Dox in different pH phosphate buffers (400 μL, final concentration: 5 μM) was sealed in the dialysis tubing with MWCO 12 kDa and was dialyzed against 25 mL of the corresponding pH phosphate buffer with stirring at 37 °C. At indicated time points, 100 μL of release media was sampled for fluorescence quantification of Dox (Ex/Em = 480/596 nm), and then returned to the release media to maintain a constant volume. The release experiments were conducted in triplicate.
2.7. Dye micellization and fluorescence dequenching assay
To monitor the nanocarrier formation, dye merocyanine 540 was added into a serial concentration of micelle solutions (from 0 to 1 mg/mL) to reach a final dye concentration of 40 μM [43]. After equilibrating at room temperature overnight, the absorbance spectra of dye/micelle mixtures were scanned by the microplate reader (Infinite M200 PRO, Tecan).
The fluorescence dequenching assay was determined by monitoring the fluorescence spectra of CPT and Dox after disrupting the nanocarrier with surfactant sodium dodecyl sulfate (SDS). Briefly, MB-20/Dox nanocarrier was incubated with 1% SDS solution at room temperature. At the indicated time points, the fluorescence spectra of CPT and Dox were scanned, respectively. Note: Dox fluorescence was scanned from the top of well, while CPT signal was detected from the bottom. The spectra were merged by the Graphpad Prism 5 software (Graphpad software Inc).
2.8. In vitro cytotoxicity assay
In vitro cytotoxicity of CPT, Dox, MB-20 and MB-20/Dox was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, the cancer cells was seeded in 96-well plate at a density of 5,000 cells per well. After 12 h incubation, a series of drug dilution (1 nM to 300 μM) were added into wells. After 48 h of drug treatment, the cytotoxicity was measured using the MTT assay. To enhance the assay sensitivity, Sorensen’s glycine buffer (0.1 M glycine, 0.1 M NaCl adjust pH to 10.5 with 0.1 N NaOH) was added into wells before absorbance assay [44–46]. The absorbance at wavelength of 570 nm was measured using a microplate reader (Infinite M200 PRO, Tecan). The half-maximal inhibitory concentration (IC50) was calculated by fitting a concentration – absorbance curve using the Graphpad Prism 5 software (Graphpad software Inc.).
2.9. Cellular uptake study
For in vitro cellular uptake study, A549 cells were seeded into 6-well plate one day before uptake study. When it was confluent, the monolayer was washed twice with FBS-free DMEM medium and subsequently incubated with MB-20/Dox solution in DMEM (100 μM, CPT equivalent) at 37°C. At the indicated time points, the drug was removed and the monolayer was washed four times with Dulbecco’s phosphate-buffered saline (PBS). The cells were lysed with 0.5 mL of 1% SDS solution for 10 min at room temperature. The viscous solution was transferred into 1.5 mL centrifuge tube and sonicated for 2 cycles (1 s each with a duty cycle of 20%). The drug concentration in the cell lysate was quantified by fluorescence plate reader (CPT: Ex/Em = 370/450 nm; Dox: Ex/Em = 480/596 nm). The total protein concentration of cell lysate was measured by the bicinchoninic acid assay (BCA assay) kit (Sigma), and the cellular uptake was normalized to the total amount of cell protein for each sample.
To study the endocytotic pathway, the cells were pretreated with sucrose (450 mM), chlorpromazine (30 μM), nystatin (25 μg/mL), methyl-β-cyclodextrin (3 mM), sodium azide/2-deoxyglucose (0.1%/50 mM) and wortmannin (0.8 mM) in serum-free DMEM media for 1 h at 37 °C, respectively [47]. Nanocarriers were then added into each well at CPT equivalent concentration of 6 μM and incubated with cells for another 2 h in the presence of inhibitors. The cell monolayer was then washed and lysed. The drug concentrations in the lysate were quantified as described above.
To track the intracellular location of nanocarrier, the fluorescence of CPT and Dox in cancer cells was monitored by the confocal laser scanning microscope (CLSM). Briefly, A549 cells (40,000 cells) were seeded in a confocal microscope dish (MatTek) one day before experiments. Cell monolayer was then washed with serum-free DMEM and incubated with MB-20/Dox in DMEM (4 μM CPT equivalent) at 37 °C. At the indicated time points, drugs were removed and the late endosomes and lysosomes were stained with LysoTracker® Green DND-26 (50 nM) for 30 min (Life Tech, Grand Island, NY). The intracellular distribution of nanocarrier was observed by CLSM (LSM 710, Zeiss).
2.10. Near-Infrared optical imaging of tumor-bearing mouse
The graft copolymer was labeled with Cy5.5-NHS ester as reported [48–49]. Briefly, 20 μL of Cy5.5-NHS solution in chloroform (2.5 mg/mL) was added into 1 mL of graft copolymer solution in NMP (10 mg/mL). The reaction was stirred in dark at room temperature overnight, and then 500 μL of PBS was added. The reaction continued to stir for another 5 h and then the Cy5.5 labeled graft copolymer was folded into nanocarrier as described above. The nanocarrier solution was dialyzed against DD water for 48 h to remove the organic solvent and free Cy5.5. After passing through 0.45 μm syringe filter, the micelle solution was lyophilized with cryoprotectant HPβCD (1%).
Female naive athymic nude mouse (Strain name, J:NU) was purchased from Jackson Lab, USA. The animal study protocol was approved by the Institutional Animal Care and Use Committee of North Carolina State University. Tumor xenograft model was established in the right dorsum of the nude mice by injecting 200 μL of A549 cells/Matrigel mixture (5 × 106 cells per mouse; v/v = 1/1) (BD Biosciences). When tumors reached 300–400 mm3, mice were intravenously injected by the MB-20/Cy5.5 nanocarrier at the Cy5.5 dose of 26 nmol/kg. The near-infrared (near-IR) optical images were taken on IVIS Lumina imaging system (Caliper, USA) at 4, 24, 48 hand 72 h post injection. At 72 h post injection, the mice were euthanized, and tumors as well as other tissues were harvested for ex vivo imaging. Region-of-interest (ROI) was circled around the organs, and the fluorescence intensities were analyzed by Living Image Software.
2.11. In vivo tumor ablation study
Tumor xenograft model was established in the right dorsum of the nude mice as described above. Seventeen days after implantation when tumors reached an average volume of 80–100 mm3, treatment was initiated by giving multiple doses (once every four days for a total of six injections, q4d × 6) of PBS, free CPT (5 mg/kg), free Dox (5 mg/kg), MB-20 (10 mg/kg, CPT equivalent) and MB-20/Dox (10 mg/kg, CPT equivalent). All the groups were intravenously injected via the tail vein with an exception of CPT (intraperitoneal injection) due to its low solubility. The tumor sizes and body weights were closely monitored every other day since the ninth day after implantation. The tumor volume for each time point was calculated using the following formulation: length× width2/2.
2.12. Statistical analysis
Student’s t-test or ANOVA were utilized to determine statistical significance between different groups. A p value < 0.05 was considered to be statistically significant.
3. Results and discussion
3.1. Synthesis of graft copolymer
The graft copolymer was prepared via a facile two-step polymerization as illustrated in Figure 2A and 2B. As a “tee joint”, partially protected tris(2-aminoethyl)amine 2 (monomer I) condensed with bifunctional NHS-PEG5000-NHS (monomer II) to form an alternating multiblock copolymer. After removing the Boc group, the third amino group on the “tee joint” was liberated and then served as the initiator for side-chain polymerization. The ROP of amino acid N-carboxyanhydrides, facilitated by TMS, gradually proceeded on the main chain, leading to the chemically well-defined graft copolymer. It is notable that, for the first time, CPT was directly polymerized into the side chains through ROP of Glu(CPT)-NCA, different from the traditional polyglutamic acid CPT conjugation method [50–51]. Direct polymerization of Glu(CPT)-NCA can guarantee 100% CPT conjugation to the residues of polyglutamic acid, which maximizes the drug loading efficiency and reduces the polymer heterogeneity. The traditional polyglutamic acid CPT conjugation method, however, can only achieve 20 – 30% conjugation efficiency (calculated from the loading capacity) [37, 52]. Moreover, 100% CPT conjugation in the graft copolymer completely masked the carboxylic acid residues of polyglutamic acid, making the graft copolymer, as well as the nanocarrier, electrically neutral. The partial conjugation in traditional methods, however, always maintained some free carboxylic acid groups and makes the polymer negatively charged, which might adversely affects the cellular uptake and in vivo biodistribution [53–55]. The resulting graft copolymer presented a well-controlled structure and relatively narrow distributed molecular weight (Figure 1B and Table 1). The content of CPT in the graft copolymer can be readily tuned by the side-chain length which was controlled by the feed ratios during the second-step polymerization. As shown in Table 1, the graft copolymer MB-10 (“MB” designates multiblock, 10 indicates the second-step polymerization feed ratio of monomer III is 10/1) showed a CPT loading capacity of 15.2wt%. By increasing the feed ratio to 20/1 (designated as MB-20), the CPT loading capacity was tuned as 23.9 wt%. However, when further increasing feed ratio to 30/1 (designated as MB-30), only slightly enhanced the drug loading capacity (25.1 wt%), which might be caused by the declined efficiency of ROP as the side chains elongated [56].
Figure 2.

The synthetic schemes of graft copolymer 5 and monomer III. Monomer I and Monomer II were firstly condensed into a linear alternating multiblock copolymer by NHS mediated amidation. After liberating the third amino group of monomer I, the side chains were gradually elongated on the main chain via a TMS controlled ROP of monomer III. To maximize the drug loading capacity, CPT was conjugated with glutamic acid and directly polymerized into the side chain, presenting a new method for drug loading.
Table 1.
Characterization of polymers and nanocarriers.
| Sample | Mw (g/mol) | Mn (g/mol) | Mw/Mn | CPT loading (wt%)a | Size (nm) | PDI | CMCb (mg/mL) |
|---|---|---|---|---|---|---|---|
| Polymer 3 | 30,798 | 21,638 | 1.42 | −/− | −/− | −/− | −/− |
| MB-10 | 34,003 | 23,041 | 1.47 | 15.2±0.4 | 30.3±8.2 | 0.166 | 0.0025 |
| MB-20 | 38,843 | 24,248 | 1.60 | 23.9±0.5 | 62.8±14.4 | 0.158 | 0.0023 |
| MB-30 | 39,899 | 23,301 | 1.71 | 25.1±0.4 | 65.1±15.5 | 0.137 | 0.0023 |
wt% is expressed as weight percentage of CPT in polymer.
The CMC was determined by eosin Y mediated dye micellization method (Ab 520 nm) as reported [43].
3.2. Graft copolymer folding and nanocarrier characterization
To fold the graft copolymer and encapsulate Dox, graft copolymer and Dox were dissolved into organic solvent NMP and then simply mixed with large quantities of water. Together with Dox encapsulation, the folding was processed instantly and efficiently. All the three graft copolymers showed strong encapsulation capability toward Dox. Within MB-20’s folding, as much as 30.0 wt% of Dox (percentage of Dox weight compared to polymer) can be encapsulated into the nanocarriers. The structure of the nanocarriers was characterized by the TEM imaging. The nanocarriers folded by MB-20 (Dox: 10 wt%) appeared as monodispersed particles with a diameter of around 50 nm, which was in good agreement with the results determined by the DLS assay (Figure 3A). In addition, nanocarriers with either CPT only or two drugs exhibited good stability at 37 °C up to 5 days (Figure 4). The folding process of graft copolymers can be monitored by the dye micellization (Figure 3B) [43]. Dye merocyanine 540 is known to show a shift of the maximum wavelength (λmax) during the formation of micelle [57–58]. As shown in Figure 3B, the absorbance intensity of the micelled dye peak at 575 nm, associated with a micellar environment, steadily increased as the concentration of the graft copolymer increased, clearly indicating the formation of a micelle-like folded nanostructure. Furthermore, the core-shell corona structure of the nanocarriers was characterized by 1H NMR spectra and fluorescence dequenching assay. As shown in Figure 3C, the proton of CPT, polypeptide and PEG segments were clearly detectable under “Sd” in deuterated dimethyl sulfoxide (DMSO-d6). In contrast, only signals associated with PEG was detected under “Sn” in deuterated water (D2O), further indicating a core-shell structure of the nanocarrier in water [42]. The folded structure was also confirmed by fluorescence dequenching measurement, as shown in Figure 3D and E. When the graft copolymer folded into nanocarriers, the hydrophobic side chains as well as the Dox molecules were expected to stack together to form a hydrophobic core. The drug aggregation in the core caused the “static quenching effect” [59–60], which dramatically decreased the fluorescence intensities of both CPT and Dox, as displayed in Figure 3D and E. However, when the corona structure was disrupted by adding the surfactant SDS, the fluorescence of CPT and Dox was substantially recovered (from “Sn” to “Sd”).
Figure 3.
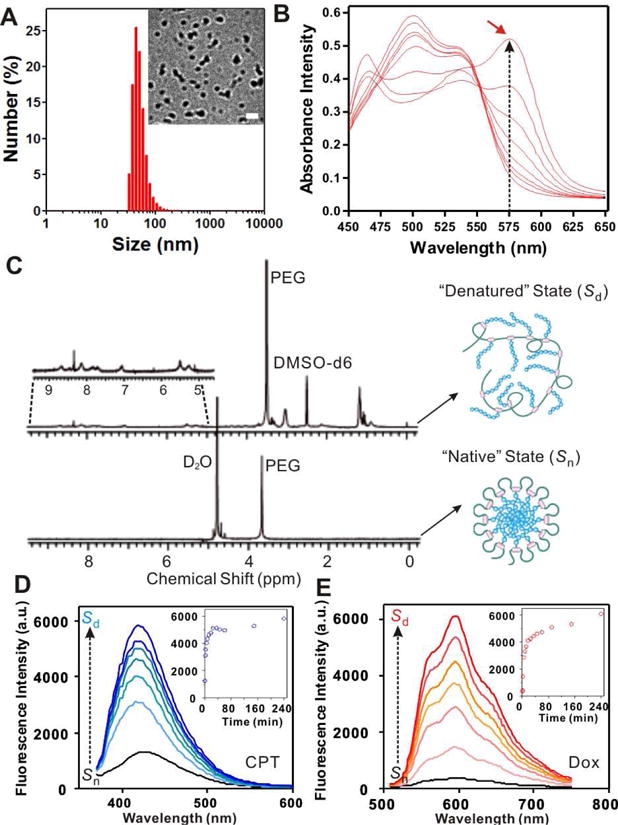
Characterization of graft copolymer and its folded structure. (A) The hydrodynamic size of MB-20/Dox measured by DLS. Inset: the TEM image of the folded nanocarriers encapsulated with Dox (MB-20/Dox). Scale bar: 100 nm. (B) Absorbance spectra of merocyanine 540 in aqueous solution of graft copolymer MB-20. The maximum wavelength (λmax) shifted toward 575 nm as the polymer concentration increased (from bottom to top: 0 to 1 mg/mL). (C) 1H NMR spectra of graft copolymer (MB-20) in DMSO-d6 (“denatured” state, Sd) and its folded nanocarrier in D2O (“native” state, Sn). Insert shows the NMR signals of CPT in the side chains. (D)–(E) The fluorescence spectra of CPT (D) and Dox (E) in nanocarriers after disrupting the structure of nanocarriers by SDS. Insets: plots of recovery fluorescence intensities over time (CPT: monitored at Ex/Em=370/418 nm; Dox: monitored at Ex/Em=480/596 nm).
Figure 4.
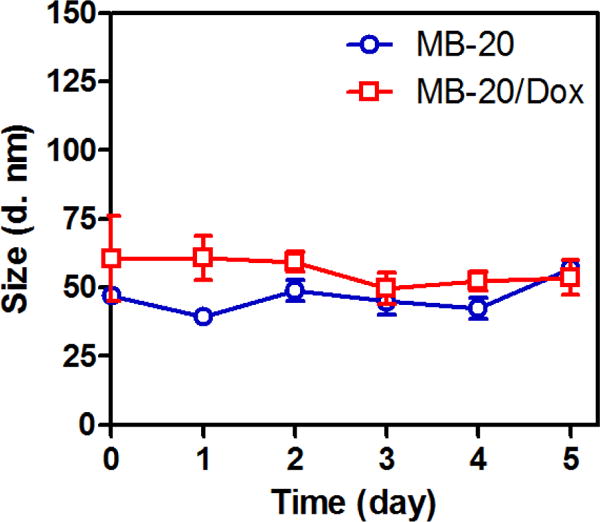
The hydrodynamic size of MB20 and MB-20/Dox measured by DLS over time. The data were expressed as mean ± SD (n=3).
3.3. In vitro drug release
The in vitro CPT release kinetics of MB-20 was quantified by HPLC. The data in Figure 5A indicated that less than 10% of CPT was released from nanocarriers in the presence of the mouse serum within 6 days, indicating its high stability in the serum. In sharp contrast, Glu(CPT)-OH, the monomer III derivative, showed a burst release profile of CPT, although they had the same drug-ester linker, which provided a substantial evidence that the presence of PEG shell around MB-20 was able to effectively shield off esterase attack and prevent premature drug release. In addition, the encapsulated Dox displayed a preferential release under a weak-acid condition, as depicted in Figure 5B. This accelerated release under an acidic condition was attributed to the re-protonation of the amino group of Dox and its subsequently higher aqueous solubility at a lower pH value [35, 61]. Providing the route that the nanocarriers were trapped in endosomes (pH 5–6) and lysosomes (pH 4–5) after cellular uptake, the acid-facilitated release can be a benefit to the enhanced cytotoxicity.
Figure 5.
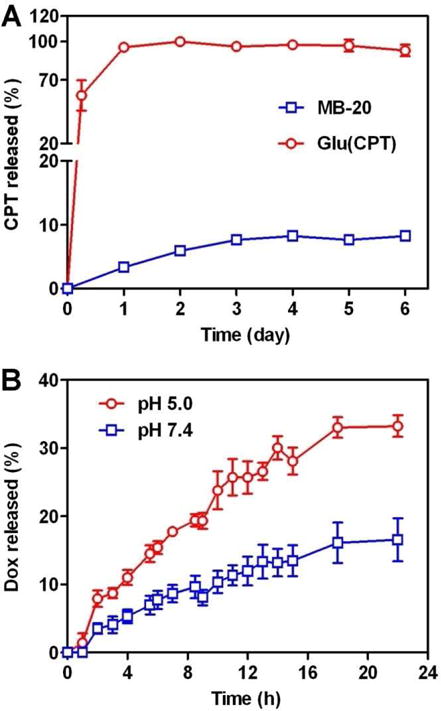
(A) The CPT release from Glu(CPT) and MB-20 nanocarrier in 100% mouse serum at 37 °C. MB-20 nanocarrier has the same drug ester linkage with Glu(CPT). However, its ester linkage is protected by PEG shell and prevented from the esterase attack, resulting in better stability in the serum. (B) The accumulative release of Dox from MB-20/Dox nanocarrier over time at different pH buffers at 37 °C.
3.4. In vitro cytotoxicity
As shown in Table 2, MB-20 and MB-20/Dox exhibited potent in vitro cytotoxicity against a panel of 9 cancer cell lines. CPT showed potent cytotoxicity in most of the cancer cell lines, with IC50 ranging from 0.02 to 3.30 μM. Interestingly, MB-20 exhibited comparable or only slightly weaker potency than CPT, with the exception of cell line MDA-MB-231. Since the ester linker cleavage is the prerequisite for MB-20 to elicit its activity, the results demonstrated that CPT of nanocarriers is releasable in cancer cells. After co-delivery with Dox, nanocarrier MB-20/Dox presented 1.5- to 60-fold more potent than MB-20, confirming the enhanced anticancer efficacy of the co-delivery system.
Table 2.
In vitro cytotoxicity (IC50, μM). The cancer cells were treated with the agents for 48 h before MTT assay. Values shown are mean ± SD (n =3).
| Cancer Type | Cell Line | CPT | Dox | MB-20a | MB-20/Doxa |
|---|---|---|---|---|---|
| Breast | MCF-7 | 1.2±1.0 | 0.3±1.0 | 6.7±1.2 | 2.1±1.3 |
| MDA-MB-231 | 0.6±0.5 | 0.1±0.6 | 17.5±0.7 | 0.3±0.6 | |
| MDA-MB-468 | 0.09±0.09 | 0.1±1.1 | 0.3±1.5 | 0.2±1.6 | |
| Lung | A549 | 0.3±1.0 | 0.5±0.8 | 1.2±1.0 | 0.8±1.1 |
| NCI-H441 | 0.1±0.2 | 0.2±0.4 | 0.3±0.4 | 0.2±0.4 | |
| Liver | HepG2 | 0.6±1.4 | 0.5±1.4 | 2.0±1.5 | 1.0±1.1 |
| Prostate | PC-3 | 0.02±0.3 | 0.2±0.4 | 0.3±0.5 | 0.1±0.3 |
| Cervix | HeLa | 3.3±1.5 | 1.0±1.7 | 6.2±1.7 | 2.3±1.8 |
| Leukemia | HL-60b | 0.4±1.0 | 0.3±1.0 | 1.0±0.2 | 0.3±0.2 |
The IC50 values are expressed in term of CPT equivalent.
The IC50 values of this suspension cell line were determined by CellTiter-Glo® Luminescent Cell Viability Assay.
3.5. Cellular uptake and intracellular nanocarrier tracking
The drug accumulation and subcellular localization of nanocarriers were determined by the in vitro uptake study and CLSM observation. As shown in Figure 6A, the accumulated uptake of CPT and Dox in A549 cells was gradually increased over the uptake time. Moreover, the molar ratio of the cellular accumulated CPT and Dox at most time points in Figure 6A was around 2.1/1, which well agreed with the drugs’ molar ratios in nanocarriers, indicating that the nanocarrier was internalized by the cancer cells without significant premature leakage of Dox. To elucidate the uptake pathway of the nanocarrier, several specific endocytic inhibitors were used. As shown in Figure 6B, sucrose and chlorpromazine (clathrin-mediated endocytosis inhibitors) [62–63] reduced the uptake of nanocarrier by about 70% and 50%, respectively. In contrast, insignificant inhibition on the cellular uptake of nanocarrier was found in the cells pretreated with nystatin (an inhibitor of caveolin-mediated endocytosis) [64] or sodium azide/2-deoxyglucose and wortmannin (inhibitors of macropinocytosis) [65–66]. Additionally, the uptake of nanocarrier was strongly inhibited by the lipid raft inhibitor, methyl-β-cyclodextrin [67]. The similar phenomenon was also observed in the cellular uptake of other polymeric micelles [68]. These results indicated that lipid raft and clathrin-mediated endocytosis might play a major role in the internalization of the nanocarrier. Similar to the observation of in vitro uptake study in Figure 6A, the fluorescence of CPT and Dox in cells gradually increased until reaching the plateau at 4 h. The CLSM images at 1 h pointed out that the fluorescence of CPT (blue) and Dox (red) was well merged with that of LysoTracker (green), revealing that nanocarriers were mainly localized in the acidic organelles (endosomes and lysosomes) at the first hour of incubation. After 4 h of incubation, parts of drugs migrated outside from endosomes/lysosomes as evidenced by the presence of CPT and Dox fluorescence beyond acidic organelles (Figure 6C). Further incubation lead to more escape of drugs from lysosomes at 8 h. A considerable amount of nanocarriers/drugs are still trapped in the acidic organelles, which might be attributed by the lack of endosome/lysosome disrupting feature in the nanocarrier. However, lysosome might play important roles in the drug release. The free drugs might be released through degradation of polymers by lysosomal enzymes, followed by diffusion of the active agents into the cytoplasm [69].
Figure 6.
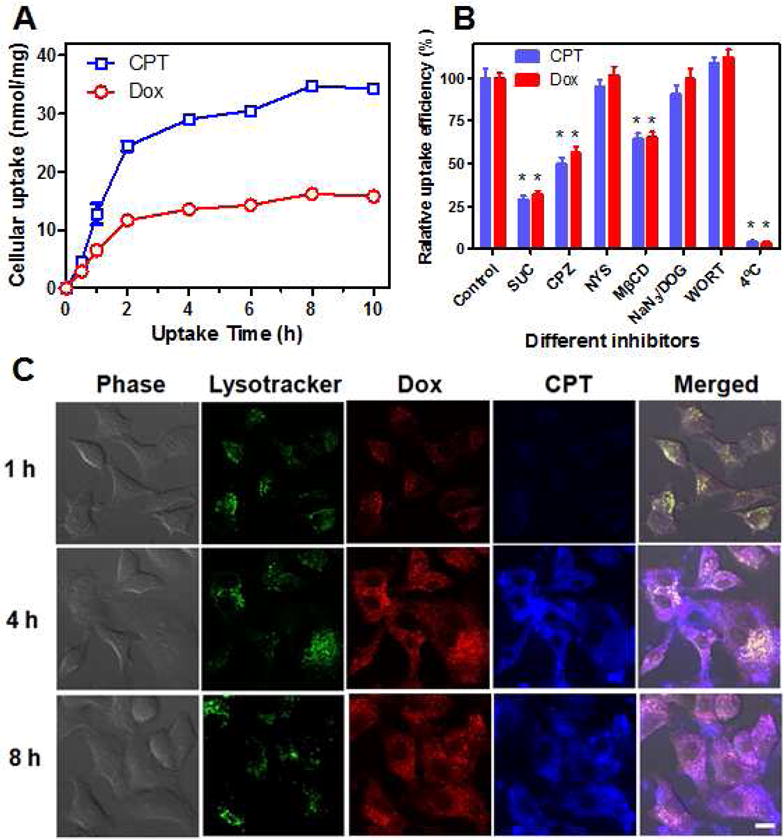
The cellular uptake behavior of nanocarriers (MB-20/Dox) toward cancer cells. (A) The accumulated uptake of MB-20/Dox in A549 cells was steadily increased over the uptake time. (B) The relative uptake efficiency of nanocarriertowardA549 cells in the presence of various endocytosis inhibitors. SUC: sucrose; CPZ: chlorpromazine; NYS: nystatin; MβCD: methyl-β-cyclodextrin; NaN3/DOG: sodium azide/2-deoxyglucose; WORT: wormannin. *p < 0.01 with respect to control groups. (C) Cellular internalization of MB-20/Dox toward A549 cells at different time observed by CLSM. The late endosomes and lysosomes were stained by LysoTracker Green. Scale bar: 10 μm. The data were expressed as mean ± SD (n=3).
3.6. In vivo evaluation in lung tumor
The in vivo accumulation of nanocarrier in tumor was evaluated by the near-IR optical imaging. The N-terminals of the side chains in the graft copolymer MB-20 were labeled with Cy5.5, and the resulting graft copolymer was folded into nanocarriers which were then intravenously administered into the mice bearing the A549 tumor model. The mice receiving MB-20/Cy5.5 nanocarriers displayed strong fluorescence at the tumor sites after 4 h (Figure 7A). To assess biodistribution, tumors and other tissues were harvested for ex vivo imaging at 72 h post injection (Figure 7B). The quantitative analysis in Figure 7C showed that the tumor exhibited significantly higher accumulation of nanocarrier than heart, spleen and kidney. Furthermore, the anticancer efficacy of nanocarriers was assessed toward A549 tumor-bearing nude mice via tail vain injection of different formulations. Compared with the groups treated with PBS, CPT and Dox, the single-drug nanocarrier MB-20 and dual-drug nanocarrier MB-20/Dox possessed significantly higher anti-tumor activities (Figure 7D), regardless of their comparable IC50 values on cancer cells in vitro (Table 2). Noticeably, the dual-drug nanocarrier exhibited a significantly higher tumor inhibition than single-drug loaded MB-20. Moreover, the current platform can dramatically increase the administration dose of CPT to more than 10 mg/kg, which is usually intolerable by mice if injected as a free drug [70]. These results revealed that graft copolymer folded nanocarrier produced measurable inhibitory effect on tumor growth, and their therapeutic efficacy might be attributed to the superior accumulation at tumor sites and insignificant toxicity in vivo (Figure 7E).
Figure 7.
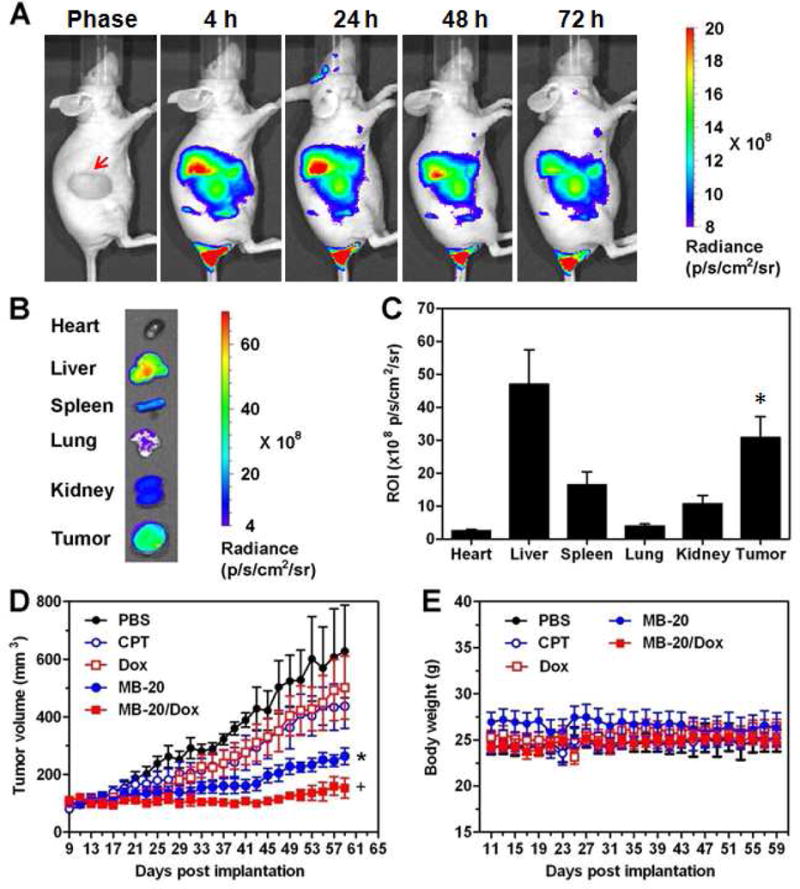
In vivo biodistribution and anti-tumor efficacy of nanocarriers in tumor-bearing mice. (A) Near-IR optical imaging of A549 tumor-bearing nude mice at 4, 24, 48 and 72 h after intravenous injection of MB-20/Cy5.5 at Cy5.5 dose of 26 nmol/kg. The red arrow in the light phase indicated the tumor site. (B) Ex vivo imaging of tumors and other tissues of A549 tumor-bearing nude mice at 72 h post injection. (C) The quantitative analysis of fluorescence signals from tumors and other tissues. Values are means ± SD (n=3). *p < 0.05 with respect to heart, spleen and kidney groups. (D) The A549 tumor growth curves after treating with either PBS, CPT, Dox, MB-20 nanocarrier or MB-20/Dox nanocarrier. Values are means ± SEM (n=5). *p< 0.05 compared to PBS, CPT and Dox groups; +p < 0.05 compared to MB-20. (E) The body weight variation of mice after treated with different formulations.
4. Conclusion
In summary, we have designed and synthesized a new nanocarrier “folded” from a graft copolymer. CPT was “purely” polymerized on the side chain of the graft polymer, while Dox was non-covalently encapsulated in the hydrophobic core. This nanocarrier demonstrated a series of attractive properties as an anticancer drug delivery vehicle, including ease of preparation, uniform formulation, high loading capacity of drugs, good stability, PEG-based stealth character and tumor accumulation ability [71].
Acknowledgments
This work was supported by the start-up grant from the Joint Biomedical Engineering Department at UNC-CH and NC State University, the grant 550KR51307 from NC TraCS, NIH’s Clinical and Translational Science Awards (CTSA) at UNC-CH and the NC State Faculty Research and Professional Development Award to Z.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Persidis A. Cancer multidrug resistance. Nat Biotechnol. 1999;17:94–5. doi: 10.1038/5289. [DOI] [PubMed] [Google Scholar]
- 2.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 3.Scripture CD, Figg WD, Sparreboom A. Peripheral neuropathy induced by paclitaxel: recent insights and future perspectives. Curr Neuropharmacol. 2006;4:165–72. doi: 10.2174/157015906776359568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bovelli D, Plataniotis G, Roila F. Cardiotoxicity of chemotherapeutic agents and radiotherapy-related heart disease: ESMO Clinical Practice Guidelines. Ann Oncol. 2010;21(Suppl 5):v277–82. doi: 10.1093/annonc/mdq200. [DOI] [PubMed] [Google Scholar]
- 5.LoRusso PM, Canetta R, Wagner JA, Balogh EP, Nass SJ, Boerner SA, et al. Accelerating cancer therapy development: the importance of combination strategies and collaboration. Summary of an Institute of Medicine workshop. Clin Cancer Res. 2012;18:6101–9. doi: 10.1158/1078-0432.CCR-12-2455. [DOI] [PubMed] [Google Scholar]
- 6.Joensuu H, Holli K, Heikkinen M, Suonio E, Aro AR, Hietanen P, et al. Combination chemotherapy versus single-agent therapy as first- and second-line treatment in metastatic breast cancer: a prospective randomized trial. J Clin Oncol. 1998;16:3720–30. doi: 10.1200/JCO.1998.16.12.3720. [DOI] [PubMed] [Google Scholar]
- 7.Podder H, Stepkowski SM, Napoli KL, Clark J, Verani RR, Chou TC, et al. Pharmacokinetic interactions augment toxicities of sirolimus/cyclosporine combinations. J Am Soc Nephrol. 2001;12:1059–71. doi: 10.1681/ASN.V1251059. [DOI] [PubMed] [Google Scholar]
- 8.Bisogno G, Cowie F, Boddy A, Thomas HD, Dick G, Pinkerton CR. High-dose cyclosporin with etoposide–toxicity and pharmacokinetic interaction in children with solid tumours. Br J Cancer. 1998;77:2304–9. doi: 10.1038/bjc.1998.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S, et al. Smart pH-Sensitive and Temporal-Controlled Polymeric Micelles for Effective Combination Therapy of Doxorubicin and Disulfiram. ACS Nano. 2013;7:5858–69. doi: 10.1021/nn4010796. [DOI] [PubMed] [Google Scholar]
- 10.Lehar J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659–66. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tai W, Qin B, Cheng K. Inhibition of breast cancer cell growth and invasiveness by dual silencing of HER-2 and VEGF. Mol Pharm. 2010;7:543–56. doi: 10.1021/mp9002514. [DOI] [PubMed] [Google Scholar]
- 12.Hu CM, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Deliv. 2010;1:323–34. doi: 10.4155/tde.10.13. [DOI] [PubMed] [Google Scholar]
- 13.Khdair A, Chen D, Patil Y, Ma L, Dou QP, Shekhar MP, et al. Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance. J Control Release. 2010;141:137–44. doi: 10.1016/j.jconrel.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 15.Shi J, Votruba AR, Farokhzad OC, Langer R. Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett. 2010;10:3223–30. doi: 10.1021/nl102184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chow EK, Ho D. Cancer nanomedicine: from drug delivery to imaging. Sci Transl Med. 2013;5:216rv4. doi: 10.1126/scitranslmed.3005872. [DOI] [PubMed] [Google Scholar]
- 17.Mo R, Jiang T, DiSanto R, Tai W, Gu Z. ATP-triggered anticancer drug delivery. Nat Commun. 2014;5 doi: 10.1038/ncomms4364. [DOI] [PubMed] [Google Scholar]
- 18.Ma L, Kohli M, Smith A. Nanoparticles for combination drug therapy. ACS Nano. 2013;7:9518–25. doi: 10.1021/nn405674m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mieszawska AJ, Kim Y, Gianella A, van Rooy I, Priem B, Labarre MP, et al. Synthesis of polymer-lipid nanoparticles for image-guided delivery of dual modality therapy. Bioconjug Chem. 2013;24:1429–34. doi: 10.1021/bc400166j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Ma S, Xie Z, Zhang H. A synergistic combination therapy with paclitaxel and doxorubicin loaded micellar nanoparticles. Colloids Surf B. 2013;116C:41–8. doi: 10.1016/j.colsurfb.2013.12.051. [DOI] [PubMed] [Google Scholar]
- 21.Jiang T, Mo R, Bellotti A, Zhou J, Gu Z. Gel–liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater. 2013;24:2295–304. [Google Scholar]
- 22.Binaschi M, Zunino F, Capranico G. Mechanism of action of DNA topoisomerase inhibitors. Stem Cells. 1995;13:369–79. doi: 10.1002/stem.5530130408. [DOI] [PubMed] [Google Scholar]
- 23.Wall ME, Wani MC. Camptothecin. Discovery to clinic. Ann N Y Acad Sci. 1996;803:1–12. doi: 10.1111/j.1749-6632.1996.tb26371.x. [DOI] [PubMed] [Google Scholar]
- 24.Wall ME, Wani MC. Camptothecin and taxol: discovery to clinic–thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1995;55:753–60. [PubMed] [Google Scholar]
- 25.Beretta GL, Perego P, Zunino F. Mechanisms of cellular resistance to camptothecins. Curr Med Chem. 2006;13:3291–305. doi: 10.2174/092986706778773121. [DOI] [PubMed] [Google Scholar]
- 26.Saleem A, Edwards TK, Rasheed Z, Rubin EH. Mechanisms of resistance to camptothecins. Ann N Y Acad Sci. 2000;922:46–55. doi: 10.1111/j.1749-6632.2000.tb07024.x. [DOI] [PubMed] [Google Scholar]
- 27.Tan KB, Mattern MR, Eng WK, McCabe FL, Johnson RK. Nonproductive rearrangement of DNA topoisomerase I and II genes: correlation with resistance to topoisomerase inhibitors. J Natl Cancer Inst. 1989;81:1732–5. doi: 10.1093/jnci/81.22.1732. [DOI] [PubMed] [Google Scholar]
- 28.Sugimoto Y, Tsukahara S, Oh-hara T, Liu LF, Tsuruo T. Elevated expression of DNA topoisomerase II in camptothecin-resistant human tumor cell lines. Cancer Res. 1990;50:7962–5. [PubMed] [Google Scholar]
- 29.Oguro M, Seki Y, Okada K, Andoh T. Collateral drug sensitivity induced in CPT-11 (a novel derivative of camptothecin)-resistant cell lines. Biomed Pharmacother. 1990;44:209–16. doi: 10.1016/0753-3322(90)90026-6. [DOI] [PubMed] [Google Scholar]
- 30.Pavillard V, Kherfellah D, Richard S, Robert J, Montaudon D. Effects of the combination of camptothecin and doxorubicin or etoposide on rat glioma cells and camptothecin-resistant variants. Br J Cancer. 2001;85:1077–83. doi: 10.1054/bjoc.2001.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu H, Cheng J. N-Trimethylsilyl amines for controlled ring-opening polymerization of amino acid N-carboxyanhydrides and facile end group functionalization of polypeptides. J Am Chem Soc. 2008;130:12562–3. doi: 10.1021/ja803304x. [DOI] [PubMed] [Google Scholar]
- 32.Lu H, Wang J, Lin Y, Cheng J. One-pot synthesis of brush-like polymers via integrated ring-opening metathesis polymerization and polymerization of amino acid N-carboxyanhydrides. J Am Chem Soc. 2009;131:13582–3. doi: 10.1021/ja903425x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurmond Ii KB, Huang H, Clark CG, Jr, Kowalewski T, Wooley KL. Shell cross-linked polymer micelles: stabilized assemblies with great versatility and potential. Colloids Surf B. 1999;16:45–54. [Google Scholar]
- 34.Shi Y, van Steenbergen MJ, Teunissen EA, Novo L, Gradmann S, Baldus M, et al. Pi-pi stacking increases the stability and loading capacity of thermosensitive polymeric micelles for chemotherapeutic drugs. Biomacromolecules. 2013;14:1826–37. doi: 10.1021/bm400234c. [DOI] [PubMed] [Google Scholar]
- 35.Kataoka K, Matsumoto T, Yokoyama M, Okano T, Sakurai Y, Fukushima S, et al. Doxorubicin-loaded poly(ethylene glycol)-poly(beta-benzyl-L-aspartate) copolymer micelles: their pharmaceutical characteristics and biological significance. J Control Release. 2000;64:143–53. doi: 10.1016/s0168-3659(99)00133-9. [DOI] [PubMed] [Google Scholar]
- 36.Sapra P, Zhao H, Mehlig M, Malaby J, Kraft P, Longley C, et al. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin Cancer Res. 2008;14:1888–96. doi: 10.1158/1078-0432.CCR-07-4456. [DOI] [PubMed] [Google Scholar]
- 37.Chen KJ, Tang L, Garcia MA, Wang H, Lu H, Lin WY, et al. The therapeutic efficacy of camptothecin-encapsulated supramolecular nanoparticles. Biomaterials. 2012;33:1162–9. doi: 10.1016/j.biomaterials.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boon JM, Lambert TN, Smith BD, Beatty AM, Ugrinova V, Brown SN. Structure/activity study of tris(2-aminoethyl)amine-derived translocases for phosphatidylcholine. J Org Chem. 2002;67:2168–74. doi: 10.1021/jo016416s. [DOI] [PubMed] [Google Scholar]
- 39.Benito JM, Gomez-Garcia M, Ortiz Mellet C, Baussanne I, Defaye J, Garcia Fernandez JM. Optimizing saccharide-directed molecular delivery to biological receptors: design, synthesis, and biological evaluation of glycodendrimer-cyclodextrin conjugates. J Am Chem Soc. 2004;126:10355–63. doi: 10.1021/ja047864v. [DOI] [PubMed] [Google Scholar]
- 40.Tai W, Chen Z, Barve A, Peng Z, Cheng K. A novel rapamycin-polymer conjugate based on a new poly(ethylene glycol) multiblock copolymer. Pharm Res. 2013;31:706–19. doi: 10.1007/s11095-013-1192-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moretton MA, Chiappetta DA, Sosnik A. Cryoprotection-lyophilization and physical stabilization of rifampicin-loaded flower-like polymeric micelles. J R Soc Interface. 2012;9:487–502. doi: 10.1098/rsif.2011.0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding M, Song N, He X, Li J, Zhou L, Tan H, et al. Toward the next-generation nanomedicines: design of multifunctional multiblock polyurethanes for effective cancer treatment. ACS Nano. 2013;7:1918–28. doi: 10.1021/nn4002769. [DOI] [PubMed] [Google Scholar]
- 43.Patist A, Bhagwat SS, Penfield KW, Aikens P, Shah DO. On the measurement of critical micelle concentrations of pure and technical-grade nonionic surfactants. J Surfact Deterg. 2000;3:53–8. [Google Scholar]
- 44.Plumb JA, Milroy R, Kaye S. Effects of the pH dependence of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide-formazan absorption on chemosensitivity determined by a novel tetrazolium-based assay. Cancer research. 1989;49:4435–40. [PubMed] [Google Scholar]
- 45.Tai W, Shukla RS, Qin B, Li B, Cheng K. Development of a peptide-drug conjugate for prostate cancer therapy. Mol Pharm. 2011;8:901–12. doi: 10.1021/mp200007b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tai W, Chen Z, Cheng K. Expression profile and functional activity of peptide transporters in prostate cancer cells. Mol Pharm. 2013;10:477–87. doi: 10.1021/mp300364k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao L, Xiong X, Sun X, Zhu Y, Yang H, Chen H, et al. Role of cellular uptake in the reversal of multidrug resistance by PEG-b-PLA polymeric micelles. Biomaterials. 2011;32:5148–57. doi: 10.1016/j.biomaterials.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 48.Tsai H-C, Chang W-H, Lo C-L, Tsai C-H, Chang C-H, Ou T-W, et al. Graft and diblock copolymer multifunctional micelles for cancer chemotherapy and imaging. Biomaterials. 2010;31:2293–301. doi: 10.1016/j.biomaterials.2009.11.059. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y-C, Liao L-C, Lu P-L, Lo C-L, Tsai H-C, Huang C-Y, et al. The accumulation of dual pH and temperature responsive micelles in tumors. Biomaterials. 2012;33:4576–88. doi: 10.1016/j.biomaterials.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 50.Matsumura Y. Poly (amino acid) micelle nanocarriers in preclinical and clinical studies. Adv Drug Deliv Rev. 2008;60:899–914. doi: 10.1016/j.addr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 51.Bhatt R, de Vries P, Tulinsky J, Bellamy G, Baker B, Singer JW, et al. Synthesis and in vivo antitumor activity of poly(l-glutamic acid) conjugates of 20S-camptothecin. J Med Chem. 2003;46:190–3. doi: 10.1021/jm020022r. [DOI] [PubMed] [Google Scholar]
- 52.Singer JW, Bhatt R, Tulinsky J, Buhler KR, Heasley E, Klein P, et al. Water-soluble poly-(L-glutamic acid)-gly-camptothecin conjugates enhance camptothecin stability and efficacy in vivo. J Control Release. 2001;74:243–7. doi: 10.1016/s0168-3659(01)00323-6. [DOI] [PubMed] [Google Scholar]
- 53.He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–66. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 54.Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–91. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krasnici S, Werner A, Eichhorn ME, Schmitt-Sody M, Pahernik SA, Sauer B, et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer. 2003;105:561–7. doi: 10.1002/ijc.11108. [DOI] [PubMed] [Google Scholar]
- 56.Deming TJ, Curtin SA. Chain Initiation efficiency in cobalt- and nickel-mediated polypeptide synthesis. J Am Chem Soc. 2000;122:5710–17. [Google Scholar]
- 57.Basu S, De S, Bhowmik BB. Photophysical studies of merocyanine 540 dye in aqueous micellar dispersions of different surfactants and in different solvents. Spectrochim Acta A Mol Biomol Spectrosc. 2007;66:1255–60. doi: 10.1016/j.saa.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 58.Kaschny P, Goni FM. The components of merocyanine-540 absorption spectra in aqueous, micellar and bilayer environments. Eur J Biochem. 1992;207:1085–91. doi: 10.1111/j.1432-1033.1992.tb17145.x. [DOI] [PubMed] [Google Scholar]
- 59.Sung J, Shin KJ, Lee S. Effects of static quenching and light pulse intensity on the time-dependent fluorescence quenching kinetics. Chem Phys. 1994;179:23–37. [Google Scholar]
- 60.Berlier JE, Rothe A, Buller G, Bradford J, Gray DR, Filanoski BJ, et al. Quantitative comparison of long-wavelength alexa fluor dyes to cy dyes: fluorescence of the dyes and their bioconjugates. J Histochem Cytochem. 2003;51:1699–712. doi: 10.1177/002215540305101214. [DOI] [PubMed] [Google Scholar]
- 61.Shuai X, Ai H, Nasongkla N, Kim S, Gao J. Micellar carriers based on block copolymers of poly(epsilon-caprolactone) and poly(ethylene glycol) for doxorubicin delivery. J Control Release. 2004;98:415–26. doi: 10.1016/j.jconrel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 62.Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123:1107–17. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh RD, Puri V, Valiyaveettil JT, Marks DL, Bittman R, Pagano RE. Selective caveolin-1-dependent endocytosis of glycosphingolipids. Mol Biol Cell. 2003;14:3254–65. doi: 10.1091/mbc.E02-12-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nishimura S, Takahashi S, Kamikatahira H, Kuroki Y, Jaalouk DE, O’Brien S, et al. Combinatorial targeting of the macropinocytotic pathway in leukemia and lymphoma cells. J Biol Chem. 2008;283:11752–62. doi: 10.1074/jbc.M708849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol. 1996;135:1249–60. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kabouridis PS, Janzen J, Magee AL, Ley SC. Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur J Immunol. 2000;30:954–63. doi: 10.1002/1521-4141(200003)30:3<954::AID-IMMU954>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 68.Xiao L, Xiong X, Sun X, Zhu Y, Yang H, Chen H, et al. Role of cellular uptake in the reversal of multidrug resistance by PEG-b-PLA polymeric micelles. Biomaterials. 2011;32:5148–57. doi: 10.1016/j.biomaterials.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 69.Chipman SD, Oldham FB, Pezzoni G, Singer JW. Biological and clinical characterization of paclitaxel poliglumex (PPX, CT-2103), a macromolecular polymer-drug conjugate. Int J Nanomedicine. 2006;1:375–83. doi: 10.2147/nano.2006.1.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schluep T, Hwang J, Cheng J, Heidel JD, Bartlett DW, Hollister B, et al. Preclinical efficacy of the camptothecin-polymer conjugate IT-101 in multiple cancer models. Clin Cancer Res. 2006;12:1606–14. doi: 10.1158/1078-0432.CCR-05-1566. [DOI] [PubMed] [Google Scholar]
- 71.Gabizon AA. Stealth liposomes and tumor targeting: one step further in the quest for the magic bullet. Clin Cancer Res. 2001;7:223–25. [PubMed] [Google Scholar]