

Abstract
Long bones are integral components of the limb skeleton. Recent studies have indicated that embryonic long bone development is altered by mutations in Ext genes and consequent heparan sulfate (HS) deficiency, possibly due to changes in activity and distribution of HS-binding/growth plate-associated signaling proteins. Here we asked whether Ext function is continuously required after birth to sustain growth plate function and long bone growth and organization. Compound transgenic Ext1f/f;Col2CreERT mice were injected with tamoxifen at postnatal day 5 (P5) to ablate Ext1 in cartilage and monitored over time. The Ext1-deficient mice exhibited growth retardation already by 2 weeks post-injection, as did their long bones. Mutant growth plates displayed a severe disorganization of chondrocyte columnar organization, a shortened hypertrophic zone with low expression of collagen X and MMP-13, and reduced primary spongiosa accompanied, however, by increased numbers of TRAP-positive osteoclasts at the chondro-osseous border. The mutant epiphyses were abnormal as well. Formation of a secondary ossification center was significantly delayed but interestingly, hypertrophic-like chondrocytes emerged within articular cartilage, similar to those often seen in osteoarthritic joints. Indeed, the cells displayed a large size and round shape, expressed collagen X and MMP-13 and were surrounded by an abundant Perlecan-rich pericellular matrix not seen in control articular chondrocytes. In addition, ectopic cartilaginous by EXT mutations and HS deficiency. In sum, the data do show that Ext1 is continuously required for postnatal growth and organization of long bones as well as their adjacent joints. Ext1 deficiency elicits defects that can occur in human skeletal conditions including trabecular bone loss, osteoarthritis and HME.
Introduction
Long bones are integral components of the limb skeleton where they provide anatomical definition and structural support and permit movement via their epiphyseal diarthrodial synovial joints. Their development and growth initiate during embryogenesis with the formation of mesenchymal cell condensations that undergo chondrogenesis and assemble the initial cartilaginous anatomical blueprint of each limb element, be it a femur or a metacarpal. Growth and elongation of the long bones continue postnatally and end at puberty when the growth plates located at each epiphyseal end close [1]. In addition to elongation, the growing long bones need to be sculpted into anatomically complex and functional structures in which various portions and regions acquire distinct shapes and organization, an example of which being the proximal femoral head and the distal knee condyles. The elongated and relatively narrow diaphysis is the first to undergo endochondral ossification with ingression of blood vessels and formation of a primary ossification center, while the wider epiphyses ossify starting late in embryogenesis and then postnatally with the formation of the secondary ossification center [2]. Spared the endochondral ossification process are the most epiphyseal chondrocytes that become permanent articular chondrocytes and sustain movement of the joints though life via their abundant and resilient extracellular matrix and production of synovial fluid components including hyaluronate and lubricin [3, 4]. Given the multiplicity of processes and steps required for the genesis, growth and morphogenesis of long bones and joints, it is not at all surprising that the regulation of their development and growth is equally complex [5]. Much has been uncovered in these areas over the last several years, particularly with regard to the roles of members of the hedgehog, bone morphogenetic protein (BMP), fibroblast growth factor (FGF) and Wnt signaling protein families and members of the Sox, Runx and Ets transcription factor families [6]. However, relatively little continues to be understood about several other pertinent processes including how the epiphyses and diaphysis undergo differential morphogenetic processes, how articular chondrocytes acquire their permanent phenotype, and how the hypertrophic zone of growth plate interacts and communicates with the underlying marrow and vascular progenitor cell population to sustain trabecular bone and primary spongiosa formation.
EXT1 is a member of the exostosin protein family that includes the EXT2, EXT3 and EXTL genes [7]. EXT1 and EXT2 form heteromeric glycosyltransferase protein complexes in the Golgi complex and are responsible for the synthesis of heparan sulfate (HS) [7]. This glycosaminoglycan is an important component of cell surface and matrix-associated proteoglycans that include Syndecans, Glypicans and Perlecan. The heparan sulfate proteoglycans (HSPGs) are expressed in unique patterns in different tissues and organs [8] and can influence a variety of developmental and physiologic processes that include cell determination and differentiation, cell-matrix interactions, receptor recycling and organogenesis [9, 10]. Importantly, their HS chains serve as binding partners for key signaling proteins such as hedgehogs, restrict and limit their distribution, activity and targets selection, and can thus influence their roles in development and growth [11-13]. For instance, mouse embryos carrying a hypomorphic gene trap mutation in Ext1 –that results in a severe reduction in Ext1 protein levels and HS production- survive until E14.5-E16.5, but display skeletal growth retardation and deformities, delays in chondrocyte hypertrophy, and a much broader and abnormal distribution of Indian hedgehog (Ihh) within the long bone growth plate [11]. Ablation of Ext1 in early embryonic limb buds causes severe impairment of mesenchymal prechondrogenic cell condensation and cartilaginous primordium formation [12], and Ext1- and HS-deficiency in synovial joint-forming cells leads to defects in joint formation and even joint fusion [13, 14]. Furthermore, heterozygous Ext1-null mouse embryos exhibit changes in growth plate chondrocyte proliferation and hypertrophic differentiation [15]. There is also evidence that Ext deficiency occurring postnatally can cause skeletal abnormalities as well [16, 17]. In a recent study Jones and collaborators created an innovative floxed Ext1 mouse line with head-to-head loxP sites that elicits stochastic generation of Ext1-null or wild type chondrocytes in cartilage after mating with Col2rtTACre mice. The authors found that the resulting mutant mice displayed postnatal skeletal aberrations and in particular, developed several large cartilaginous tumor-like outgrowths near the epiphyses of their long bones [18]. The outgrowths resembled the cartilaginous exostoses that form near the growth plates of children with Hereditary Multiple Exostoses (HME), a syndrome caused by dominant loss-of-function mutations in EXT1 or EXT2 and consequent HS deficiency [19, 20].
The present study was carried out to further test and clarify the roles of Ext genes in postnatal long bone growth, organization and structure. To do so, we created conditional compound mouse mutants bearing a standard floxed Ext1 gene (Ext1f/f) [21] and the Col2CreERT transgene that directs CreERT expression in chondrocytes [22]. We induced Ext1f/f ablation by tamoxifen injection at early postnatal time points and examined the developmental and structural consequences on long bones over time. We found that conditional loss of Ext1 expression postnatally is incompatible with normal skeletal growth and results in a number of structural and functional deficiencies in both the long bones and their joints that also resemble human skeletal pathologies.
Materials and Methods
Transgenic mice
All studies were conducted with approval by the IACUC. Creation of the loxP-modified Ext1 allele and establishment of the Ext1f/f mouse line were described previously [21]. Col2CreERT transgenic mice that encode a Cre recombinase linked to a modified estrogen ligand binding domain under the control of collagen 2a1 promoter sequences were generated and provided by Dr. S. Mackem, NCI [22]. Ext1f/f mice were mated with Col2CreERT mice, and the resulting Ext1f/f;Col2CreERT mice and appropriate controls (Ext1f/f or Ext1f/+) received a single intraperitoneal injection of tamoxifen (100 μg/10 μl/mouse) at postnatal day 5 (P5). Genotyping was performed using tail DNA and amplified by PCR. Mice were sacrificed 2-, 4-, 8-weeks from the time of tamoxifen injection. The total numbers of mice studied in Ext1f/f and Ext1f/f;Col2CreERT groups were: 4 each at 2 weeks; 2 each at 4 weeks; and 3 each at 8 weeks time points. We also created Ext1f/f and Ext1f/f;Col2CreERT in RosaR26R background to monitor CreERT expression and activity by LacZ reporter staining after tamoxifen injection.
Skeletal analyses
Whole skeletons were analyzed by soft x-ray imaging using Bioptics piXarray100 (Kenmore, WA) in utomatic exposure control mode. Total mouse body lengths were measured from nose to sacral tip, excluding the tail length. Lengths of long bones were determined from x-ray images using Image J software.
Chondrocyte cultures, DNA isolation and PCR
Cartilage was isolated from epiphyses of tibia, femur and humerus and radius of the P12 Ext1f/f and Ext1f/f;Col2CreERT mice that had received a single tamoxifen injection (100 μg/10 μl/mouse) at P5 as above. Epiphyseal cartilage pieces isolated from individual animals were digested with 0.05% trypsin in HBSS for 1 h at 37°C, and the liberated cells consisting mostly of contaminating fibroblasts were discarded. The remaining fragments were further incubated with 86 U/ml collagenase Type I (Worthington Biochemical Corporation, Lakewood, NJ) in serum-free DMEM overnight. Dissociated cells were filtered and plated on 60 mm agarose coated petri dishes (BD Biosciences, Franklin Lakes, NJ) and cultured in DMEM containing 10% FBS (Gemini Bio-Products, West Sacramento, CA) in suspension for 1 day to eliminate the few additional non-chondrogenic cells. The surviving cells –mostly chondrocytes- were finally collected and used to prepare DNA samples.
DNA was isolated using the Qiagen Mini Kit (Qiagen), according to the manufacturer instructions, quantified with BioMate 3 Spectrophotometer, Thermo Spectronic (Thermo Fisher Scientific Inc. Waltham, MA), and PCR amplified with specific primer pairs for detection of intact or recombined floxed allele as described [16]. Bands were visualized on 2% agarose gels, and GAPDH was used as internal control.
To further confirm the efficacy of Cre recombinase activity, qPCR was performed on genomic DNAs to specifically detect intact Ext1 allele. DNA samples above were combined with SYBR Green PCR Master Mix (Applied Biosystem, Foster City, CA) and primers for non-recombined intact allele or GAPDH as internal control. Reaction was performed using an ABI 7900 HT machine (Applied Biosystem, Foster City, CA) according to the manufacturer's protocols.
Histological, histochemical and immunohistochemical analyses
Knee and wrist joints were dissected and fixed with 4% (v/v) paraformaldehyde, decalcified with EDTA for 7-10 days and embedded in paraffin. Serial longitudinal 5 μm-thick sections of tibiae, femurs and the distal portions of ulnae and radii were processed for staining with hematoxylin/eosin and Safranin O/fast green. LacZ (β-galactosidase) activity was detected using X-Gal Stock and Stain Base Solution (EMD Millipore Corporation, Billerica, MA) according to the manufacturer's protocol, and sections were counter-stained with eosin. For immunohistochemical staining, sections were pretreated with 0.1% pepsin in 0.02N HCl for 10 min at 37°C for demasking, incubated in 10% goat serum in PBS for 1h at room temperature for blocking, and then reacted for 14 hours at 4°C with one of the following primary antibodies: collagen X rabbit polyclonal antibodies (1:1000 dilution, Cosmo Bio Co LTD, Tokyo, Japan); MMP13 rabbit polyclonal antibodies (1:250 dilution, Abcam Ab75606, Cambridge, UK); or rabbit antibodies against Perlecan domains I or V (1:1000 dilution) [23]. For Perlecan staining, antigen demasking was performed with hyalorunidase (1000U/ml in PBS, 30 min at room temperature) instead of pepsin. Following rinsing, sections were incubated with biotinylated anti-rabbit secondary antibodies in HRP/DAB detection IHC kit mixture according to the manufacturer's instructions (Abcam), suitable for colorimetric visualization of signal; counterstaining was with methyl green. Under these conditions, the antibodies specifically recognize Perlecan as determined by staining of Perlecan-deficient mouse sections [23] and staining with non-immune IgGs. Staining data were examined with a Nikon Eclipse TE400 microscope equipped with a SPOT 5.0 advanced software (Diagnostic Instruments Inc., Sterling Heights, MI) to capture and analyze images. Non-specific staining was determined on parallel sections in which the primary antibody was replaced with non-immune rabbit IgGs (5 μg/ml, Vector Laboratories, Burlingame, CA).
TRAP staining was performed on knee joint sections using the Acid Phosphatase Leukocyte Kit (Sigma-Aldrich, St. Louis, MO) according to manufacturer's instructions.
Histomorphometry
Longitudinal tibia and femur sections from Ext1f/f and Ext1f/f;Col2CreERT mice injected with tamoxifen 2 weeks earlier were processed for hematoxylin/eosin or TRAP staining. Images of primary spongiosa were taken and analyzed for quantification of trabecular bone volume and TRAP-positive cells. Four animals for the control and mutant groups were examined, and two images from the central and lateral planes of the femur and tibia were used for quantification. Trabecular bone area in each section was calculated and normalized over total volume, according to the Bioquant Osteo 2011 (Image Analysis Corporation, Nashville, TN) Protocol. TRAP positive cells along the chondro-osseous junction were counted and normalized over total length using Image J.
Statistical methods
Results were analyzed using InStat 3 version 3.1a (GraphPad Software, Inc., La Jolla, CA). One-way analysis of Variance (ANOVA) with a Tukey-Kramer Multiple Comparison Test or Student's t-test was used to establish statistically significance. Threshold for significance for all tests was set as p<0.05.
Results
Postnatal growth deficiency and abnormalities in Ext1-deficient long bones
To examine the roles of Ext1 in postnatal limb skeletal growth and growth plate organization and function, we used Ext1f/f mice that bear standard loxP sites flanking exon 1 and elicit a null allele following Cre-mediated recombination [21] and Col2CreERT transgenic mice that direct CreERT expression to chondrocytes throughout the developing and growing embryonic and early postnatal skeleton, but in which transgene expression dwindles during early postnatal age [22]. To verify CreERT expression patterns and recombination efficiency, we first created triple compound Ext1f/f;Col2CreERT in RosaR26R background and injected them with tamoxifen at postnatal day 5 (P5) (hereafter identified as conditional knock-outs –CKO- in figures and figure legends). Two weeks after injection, we prepared serial longitudinal sections of femurs and tibia at the knee level and processed them for histochemical staining of LacZ (β-galactosidase). We found that LacZ staining was restricted to growth plate chondrocytes and articular chondrocytes (Fig. 1C-1D). No staining was detectable in comparable sections prepared from companion control tamoxifen-injected Ext1f/f mice lacking the Col2CreERT transgene (Fig. 1A-1B) (hereafter identified as controls –Control- in figures and figure legends). To quantify CreERT efficiency, we isolated chondrocytes from epiphyseal cartilage dissected from P12 Ext1f/f and Ext1f/f;Col2CreERT mice that had been injected with tamoxifen at P5 as above. The dissociated chondrocytes were first expanded in suspension to increase their number and eliminate contaminating adhering connective tissue cells, and their DNAs were then subjected to conventional PCR and qPCR to distinguish and quantify the relative amounts of intact un-cleaved floxed Ext1f/f allele and Cre-cleaved allele. DNA samples from Ext1f/f;Col2CreERT cells did direct the amplification of considerable amounts of cleaved allele product (Fig. 1E, lanes 3 and 4) and also a relatively small amount of residual un-cleaved allele product (Fig. 1E, lanes 3 and 4). As to be expected, DNA from control Ext1f/f chondrocytes elicited only un-cleaved allele RT-PCR product (Fig. 1E, lanes 1 and 2). Based on q-PCR analysis, the recombination efficiency was over 80% (Fig. 1F).
Fig. 1.
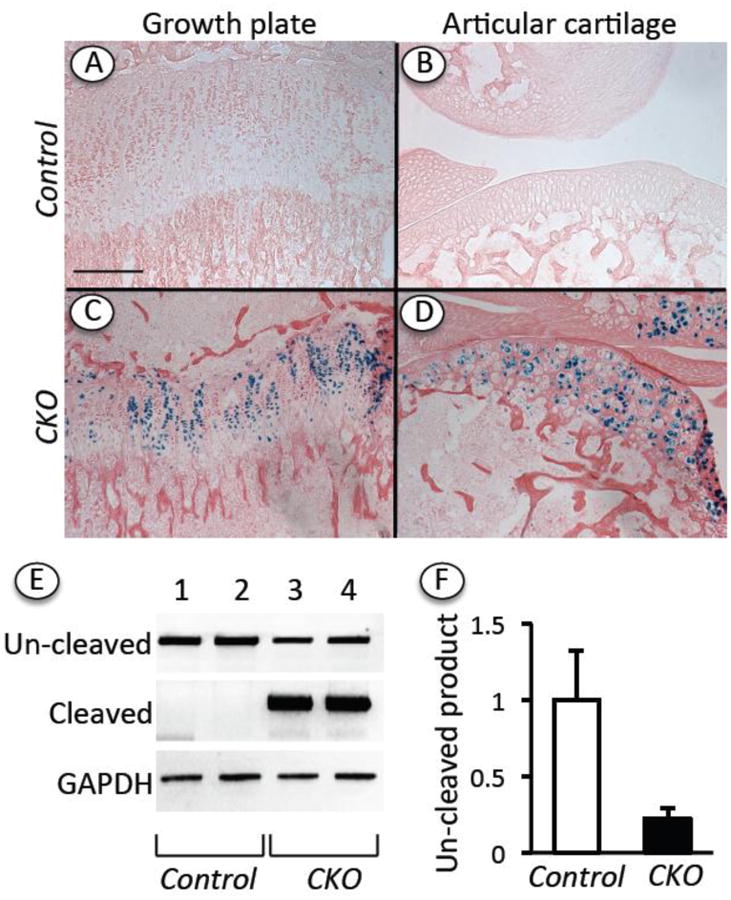
Topographical and genetic analysis of Cre activity and efficiency. Ext1f/f (Control) and Ext1f/f;Col2CreERT (CKO) mice in RosaR26R-LacZ background were injected at P5 with tamoxifen (100 μg/10 μl/mouse). (A-D) Longitudinal sections of knee joints processed for LacZ (β-galactosidase) activity show that strong staining was present in growth plate (C) and articular cartlilage (D) in Ext1f/f;Col2CreERT specimens but not control Ext1f/f specimens. (E) PCR product analysis on agarose gel shows that DNA from mutant CKO chondrocytes mostly directed amplification of cleaved Ext1 gene product, while DNA from control cells (Control) directed amplification of un-cleaved product only. (F) qPCR shows un-cleaved DNA product levels in control and mutant cells after normalization to GAPDH and indicates that Cre efficiency averaged about 80%. Bar for A-D = 500 μm.
Next, we monitored the overall growth of Ext1f/f mice (hereafter called controls) versus Ext1f/f;Col2CreERT mice (hereafter called mutants) over postnatal time following a single tamoxifen injection at P5. Interestingly, we found that the mutant mice exhibited an appreciable reduction in overall body length already by about 2 weeks after injection compared to control tamoxifen-injected mice (Fig. 2A) (and also wild type mice injected with tamoxifen; not shown). The reduction of body length from nose tip to sacral area amounted to about 20% and was statistically significant. A similar trend was observed when we measured the longitudinal lengths of individual long bones, with reductions of about 20% and 15% in mutant femurs and tibias compared to respective controls (Fig. 2B). Tamoxifen-injected heterozygous Ext1f/+;Col2CreERT mice were essentially normal signifying that deficiency of both alleles was needed to elicit a phenotype and that tamoxifen treatment did not cause significant skeletal abnormality in control Col2CreERT mice.
Fig. 2.
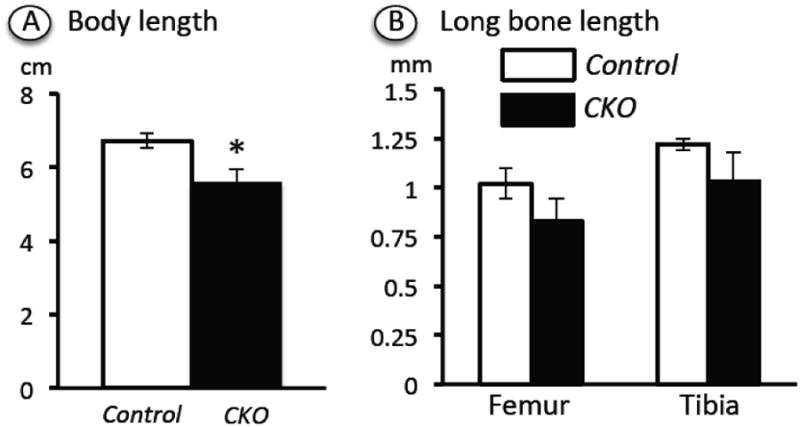
Determination of total body and long bone length. Ext1f/f and Ext1f/f;Col2CreERT mice injected at P5 with tamoxifen were analyzed at the 2 week time point. (A) Body length was measured from nose to sacral tip by visual inspection and a ruler. (B) Femur and tibia lengths were determined from soft x-ray images quantified with Image J software. A total of 5 CKO mutants and 4 controls were examined. Note that body length was significantly reduced in CKO versus controls. A similar and consistent trend was observed in long bone average length. *, p<0.05.
Growth plate disorganization and primary spongiosa deficiency
To account for the growth retardation, we carried out histological, histochemical and immunohistochemical analyses of control and mutant long bone growth plates. We harvested hindlimbs and forelimbs at 2 and 8 weeks after tamoxifen injection at P5 and processed the resulting longitudinal serial sections for H&E staining first. As to be expected, the control growth plates exhibited a normal and stereotypic organization and structure (Fig. 3A and 3C). The chondrocytes were aligned in well-defined longitudinal columns and were arranged in readily recognizable proliferative (PZ), prehypertrophic (PHZ) and hypertrophic (HZ) zones based on cell size and architecture (Fig. 3C). The border between growth plate and perichondrium was continuous and well defined (Fig. 3A, arrow). The growth plates were followed by typical primary spongiosa (trabecular bone) embedded in bone marrow (Fig. 4A). In sharp contrast, the growth plates in mutant mice were obviously aberrant (Fig. 3B and 3D). The mutant chondrocytes had failed to align in columns (Fig. 3D). The individual growth plate zones were not easily discernable and recognizable, and the hypertrophic zone was consistently reduced in height (Fig. 3D). The reduction of hypertrophic zone height was verified by immunostaining for collagen X; the staining region for this hypertrophic chondrocyte marker [24] was in fact reduced considerably in mutant (Fig. 3H and 3J, bracket) versus control growth plates (Fig. 3G and 3I, bracket). In addition, the mutant growth plates occasionally displayed a cartilaginous outgrowth seemingly emanating from their lateral bottom side (Fig. 3B, double arrow), thus altering and distorting the alignment of the growth plate/perichondrium border. We also noted the presence of small clusters of tightly-packed cells near/within the prospective growth plate reserve zone just below the secondary ossification center (Fig. 3F, arrows) that were not seen in controls (Fig. 3E). The development of the ossification center itself was also clearly delayed in mutants versus controls (Fig. 3A and 3B; see below for further details).
Fig. 3.
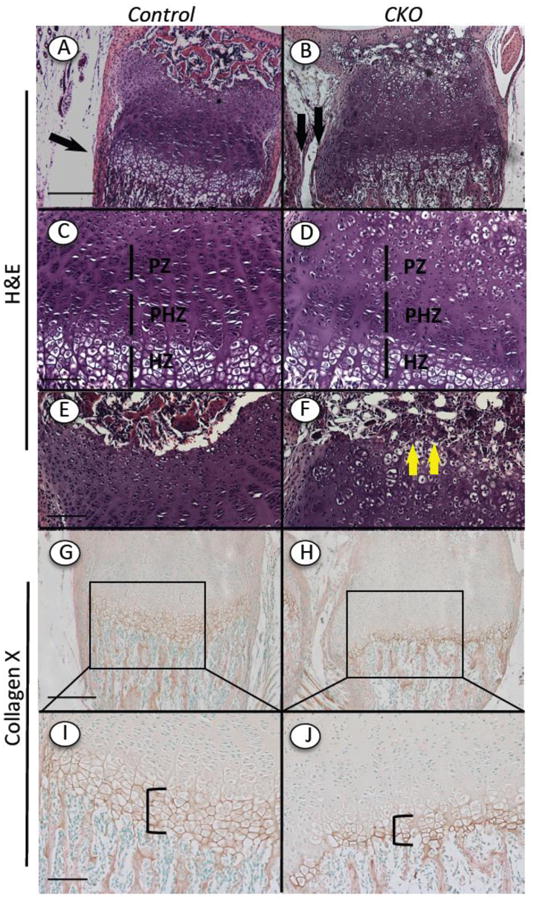
Histological and immunohistochemical analyses of growth plate organization. Continuous longitudinal sections of tibias from Ext1f/f and Ext1f/f;Col2CreERT mice injected at P5 with tamoxifen and harvested 2 weeks later were processed for routine histology or immunostaining. (A-F) H&E staining shows that control growth plate displayed normal proliferative (PZ), prehypertrophic (PHZ) and hypertrophic (HZ) zones and typical chondrocyte columns (A, C), while mutant CKO growth plates were disorganized (B, D). Note the presence of clusters of tightly associated cells near/within the reserve zone in mutants (F, arrows) that are not appreciable in controls (E). Development of secondary ossification center itself appeared to be delayed in mutants. (G-J) Immunostaining shows that collagen X displayed its typical abundance in the hypertrophic zone (G, I, bracket), but was appreciably reduced in mutant hypertrophic zone (H, J, bracket). Bars= 500 μm (A, B, G, H); 200 μm (C, D, E, F,I, J).
Fig. 4.
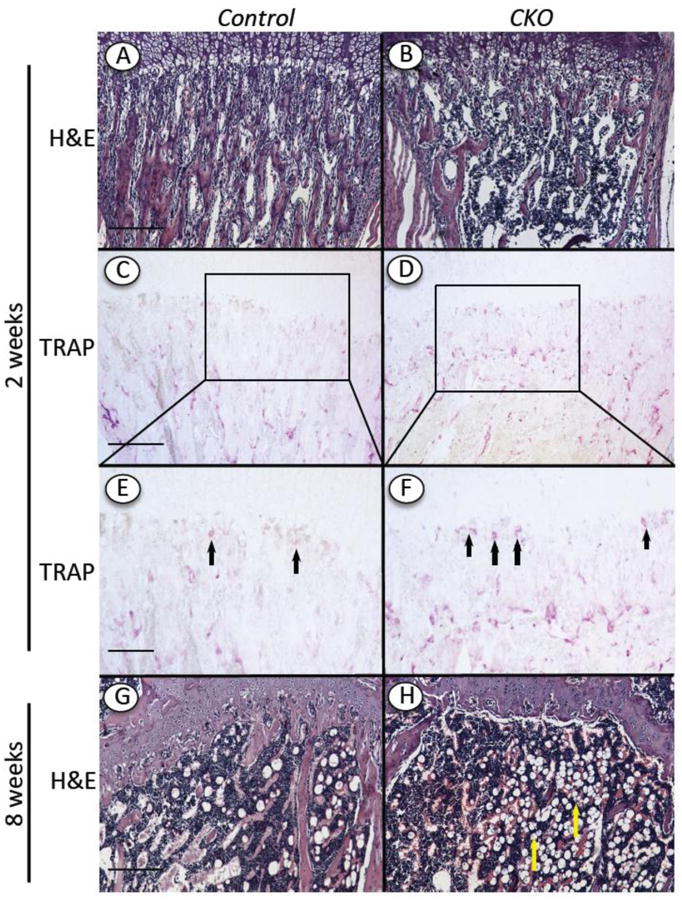
Changes in trabecular bone and TRAP-positive cells. Tibia longitudinal sections from Ext1f/f and Ext1f/f;Col2CreERT mice tamoxifen-injected at P5 and harvested 2 and 8 weeks later were processed for H&E (A, B, G and H) or TRAP staining (C-F). Note that control specimens displayed typical organization of trabecular bone and marrow at the 2 and 8 weeks (A, G) and several TRAP-positive osteoclastic cells at the chondro-osseous border (C, E, arrows), whereas mutant specimens contained appreciably lower levels of trabecular bone (B, H), a higher number of TRAP-positive cells (D, F, arrows) and an increased frequency of adipocyte-like cells in marrow at 8 weeks (H, arrows). Boxed areas in C and D are shown at higher magnification in E and F. Bars= 500 μm (A, B, C, D, G, H); 200 μm (E, F).
Defects in the hypertrophic zone are often accompanied by, and may directly cause, defects in formation of primary spongiosa [6]. Indeed, the amount of trabecular bone present in the mutants was appreciably decreased by 2 weeks post-injection (Fig. 4B) and obviously so by 4 (not shown) and 8 weeks (Fig. 4H) compared to controls (Fig. 4A and 4G). We used Image J-assisted histomorphometry to quantify the differences in trabecular bone, and found that the decrease in mutants varied somewhat from specimen to specimen but averaged about 45% at 2 weeks (Fig. 5A). Scanning by μCT confirmed the overall drop in trabecular bone (Suppl. Fig. 1). Several mechanisms could have led to the decrease in primary spongiosa in addition to a defective hypertrophic chondrocyte zone [25]. We did find that there were more than twice as many TRAP-positive osteoclasts along the mutant chondro-osseous border (Fig. 4D and 4F, arrows) compared to controls (Fig. 4C, 4E and 5B). The defects in growth plate and primary spongiosa organization persisted over time and were still clear at 8 weeks when the marrow had in fact become richer in adipose cells compared to controls (Fig. 4H cfr 4G, arrows).
Fig. 5.
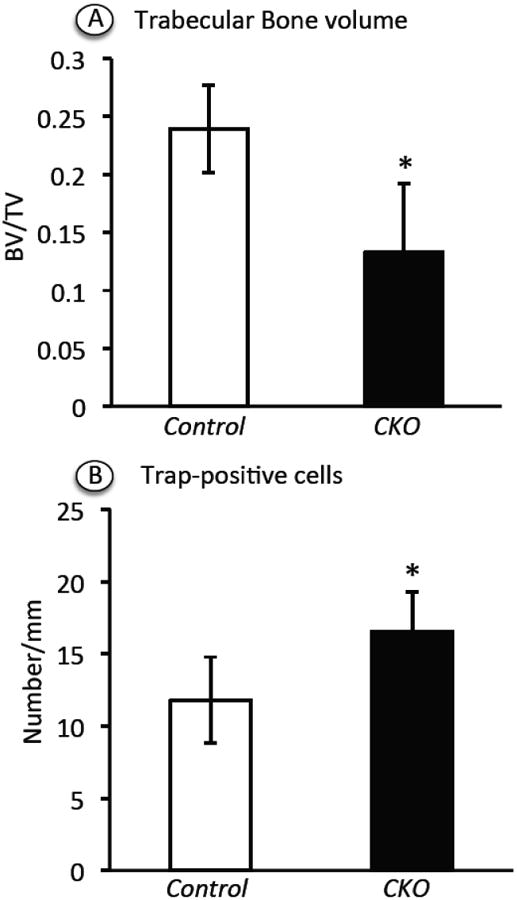
Histomophometric determination of trabecular bone volume and TRAP-positive cell number. Longitudinal distal femoral sections from Ext1f/f and Ext1f/f;Col2CreERT mice tamoxifen-injected at P5 and harvested at 2 weeks were stained with H&E or TRAP (4 controls and 4 mutants) and subjected to histomorphometry. (A) Trabecular bone volume in primary spongiosa was measured and normalized over total volume using BioQuant OSTEO 2011 Protocol. (B) TRAP staining positive cells along the chondro-osseous junction were counted and normalized to overall length, using Image J. Note that both parameters changes in mutants versus controls in a statistically significant manner (*p<0.05).
Lateral outgrowths
The lateral cartilaginous outgrowths in mutant growth plates described above were reminiscent of outgrowths seen in previous studies with Ext1-deficient mouse embryos [14] and of exostoses seen in HME patients [26]. To delineate the nature of these outgrowths more clearly and uncover their implications, we carried out a natural history analysis of their genesis and evolution. We harvested limb skeletal elements from control and mutant mice at 1, 2, 4 and 8 weeks after tamoxifen injection at P5 and used the wrist area as a representative test region. Already by 1 week, we did observe small but distinct outgrowths on the lower lateral side of growth plate in the mutants (Fig. 6A and 6E, arrows). By 2 weeks, the outgrowths had become very conspicuous and elongated and were present on both radius and ulna (Fig. 6B, arrows). Medially, they faced each other and were separated by connective tissue (Fig. 6F, asterisk); internally, they faced diaphyseal bone and marrow. Each outgrowth was largely cartilaginous, and the bulk of the chondrocytes were not organized in typical growth plate-like zones (Fig. 6B and 6F). By 4 weeks, the outgrowths became quite large and still faced each other, and the chondrocytes had become more organized into a growth plate-like structure that was oriented at about 90° with respect of the long bone's longitudinal axis and contained a hypertrophic zone facing bone and marrow (Fig. 6C and 6G, arrow). In specimens harvested at 8 weeks, the medial connective tissue separating the outgrowths was no longer clear and the outgrowths often fused along their mid-line into amorphous and heterogeneous cartilaginous tissue masses (Fig. 6D and 6H, arrow) intermingled with trabecular bone and marrow.
Fig. 6.
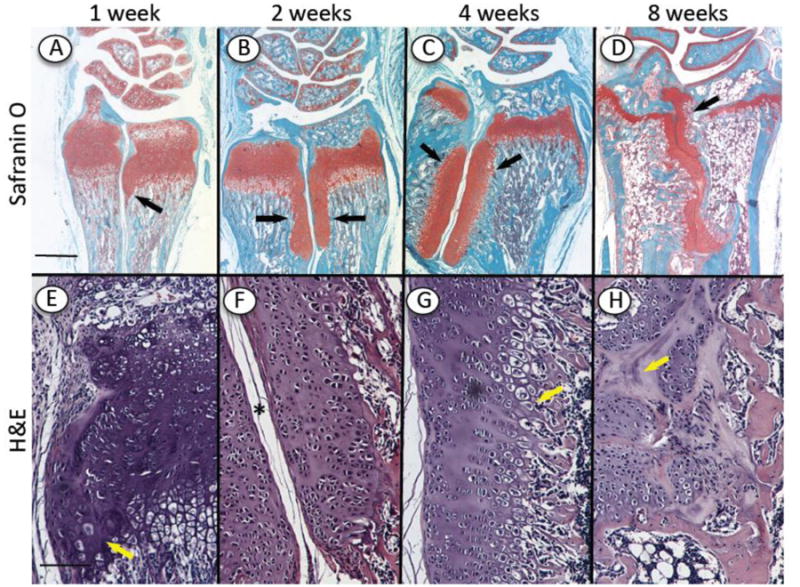
Natural history of outgrowth formation over time. Longitudinal serial sections were prepared of the wrist region of Ext1f/f;Col2CreERT mice tamoxifen-injected at P5 and harvested 1, 2, 4 and 8 weeks later. (A-H) Staining with Safranin O (A-D) and H&E (E-H) shows (i) the cartilaginous growth plates and articular cartilage in radius, ulna and wrist elements and (ii) the presence of an incipient outgrowth at 1 week (A, E, arrow) and large and long outgrowths at 2 and 4 weeks (B, C, arrows). The outgrowths are separated by connective tissue medially (F, asterisk) and face bone and marrow internally (G, arrow). By 8 weeks, the connective tissue separation was reduced and the outgrowths merged into each other (D, H). Bars= 1 mm for A-D; 300 μm for E-H.
Epiphyseal defects and articular chondrocyte hypertrophy
The epiphyses of long bones are the site where permanent articular chondrocytes are generated and function through life and where an underlying secondary ossification center forms to provide physical stability and support to the joint [3]. As pointed out above, the development of the secondary ossification center appeared delayed in mutant long bones harvested at 2 weeks after tamoxifen injection (see Fig. 3A and 3B). Closer analysis of multiple mutant specimens confirmed that the formation of the ossification center was consistently delayed in each mutant and regardless of anatomical site, be it wrist or knee (not shown). In addition and quite interestingly, many articular chondrocytes in the mutants had undergone ectopic maturation and hypertrophy and were very large in size and round in shape (Fig. 7B, 7D and 7F, arrows) compared to the typical small-sized articular chondrocytes present in controls (Fig. 7A, 7C and 7E). The hypertrophic-like cells were present in every mutant specimen analyzed, but their frequency varied and was not overtly related to weight-bearing area within the joint. Their hypertrophic phenotype was confirmed by positive immunostaining for collagen X and matrix metalloprotease 13 (MMP-13), two well-known markers of hypertrophic chondrocytes (Fig. 7H and 7J). Joint sections from control mice were negative (Fig. 7G and 7I).
Fig.7.
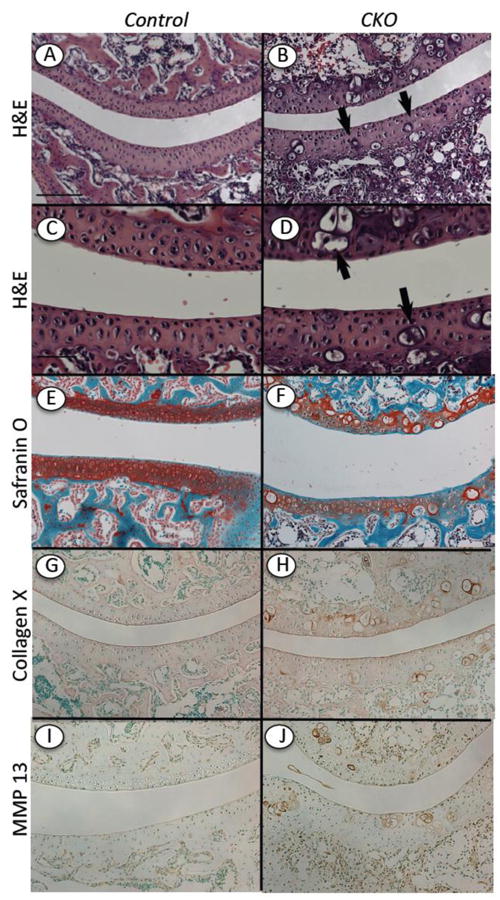
Ectopic hypertrophic-like chondrocytes in mutant articular cartilage. Longitudinal serial sections were prepared of knees from Ext1f/f and Ext1f/f;Col2CreERT mice tamoxifen-injected at P5 and harvested at 2 weeks. (A-F) Staining with H&E (A-D) and Safranin O (E-F) shows that articular cartilage in controls displayed typical cellular organization and strong and uniform matrix staining (A, C, E), whereas articular cartilage in mutants exhibited several hypertrophic-like chondrocytes and uneven matrix staining (B, D, F). (G-J) Immunostaining for collagen X and MMP-13 shows that these hypertrophic markers were essentially undetectable in control articular cartilage (G, I), but were obvious and associated with hypertrophic-like cells in mutant tissue (H, J). Bar= 500 μm (A-B, E-J); 200 μm (C, D).
Perlecan is the most abundant HS-rich proteoglycan present in articular and growth plate cartilage and is located in interterritorial, territorial and pericellular matrices where it likely plays distinct roles [27, 28]. Thus it became of interest to study whether Ext1 deficiency in our mutants would result in changes in presence and/or distribution of Perlecan. Sections from control and mutant limb specimens were processed for immunostaining with antibodies to Perlecan domain 1 or domain 5, thus spanning the N- and C-terminal ends of the core protein that can be subjected to differential cleavage in certain physiological and pathological circumstances [23]. In joint sections from control mice, Perlecan staining was intense in the superficial zone of articular cartilage and was much lower in middle and deep zones (Fig. 8A and 8C). The superficial zone in mutant specimens also exhibited positive immunostaining but in addition, the ectopic hypertrophic chondrocytes displayed very strong and distinct staining pericellularly (Fig. 8B and 8D, arrows). The underlying growth plates in mutants and controls displayed typical and similar patterns of staining, confirming that Perlecan is usually distributed throughout the matrix (Fig. 8E-8H). On the other hand, the small cell clusters present near/within the top reserve zone in mutant growth plates displayed extremely positive staining (Fig. 8H, arrows). Given that this zone is considered to be a source of progenitor cells for growth plate [29], Perlecan might be involved in stem/progenitor cell function and niche regulation. We examined also the cartilaginous outgrowths present in mutants and found that they exhibited positive staining throughout their structure, a clear reflection of their growth plate-like character (Supplement Fig. 2A, 2C and 2E). Interestingly, the staining pattern became inhomogeneous over time (Supplement Fig. 2B, 2D and 2F). Because the staining patterns above were obtained with antibodies to domain 1 or domain 5, the data suggest that Perlecan did not undergo major changes in cleavage or processing in control versus mutant tissues.
Fig. 8.
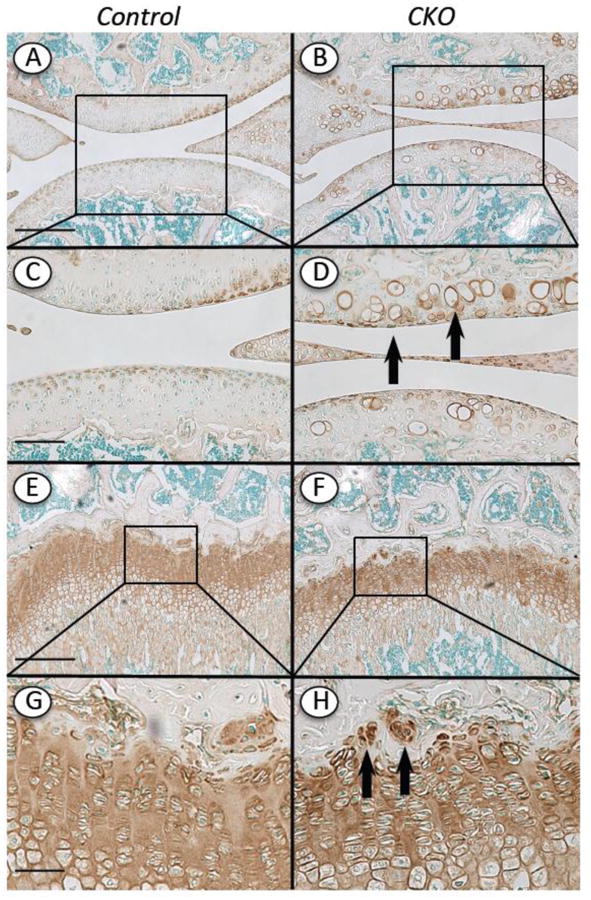
Distribution of Perlecan in articular cartilage and growth plate. Knee sections from Ext1f/f and Ext1f/f;Col2CreERT mice tamoxifen-injected at P5 and harvested at 4 weeks were processed for Perlecan immunostaining using domain 5 antibodies. (A, C, E and G) In controls positive Perlecan staining was evident in top articular cartilage zone and throughout the growth plate. (B, D, F and H) In CKO mutants, positive Perlecan staining was also very strong in the pericellular matrix of the hypertrophic-like chondrocytes in articular cartilage (D, arrows) and the cell clusters present near/within the growth plate reserve zone (H, arrows). Boxed areas in A-B and E-F are shown at higher magnification in C-D and G-H, respectively. Bars= 500 μm (A, B, E, F); 200 μm (C, D, G, H)
Discussion
The data clearly indicate that Ext1 deficiency is not compatible with normal elongation, organization and morphogenesis of long bones in postnatal mice. The Ext1-deficient long bones acquire and display an increasingly abnormal phenotype over time, and the changes include a distortion of growth plate chondrocyte columnar organization, deficiencies in both primary spongiosa and secondary ossification center development, and ectopic formation of hypertrophic-like chondrocytes within articular cartilage. These are encompassing and pleiotropic changes that affect diverse cell populations at diverse sites and with diverse function, reflecting the multiple essential roles that HS and HSPGs play in cell and tissue development, function and physiology both in the skeleton and several other organs [30]. Thus, the levels of Ext1 expression, HS production and HSPG function must be kept under strict control for skeletogenesis and skeletal growth and functioning to proceed and be maintained during postnatal life [10].
The multiple roles that HS and HSPGs exert in development and growth are a reflection of their complex structural composition, genetic makeup and expression patterns [9]. The HS chains have distinct sulfation patterns and levels that are thought to constitute a “sulfation code” capable of regulating interactions and function of hedgehogs, BMPs, Wnts and other HS-binding signaling proteins [8]. Thus, HS and HSPGs can act at multiple levels to regulate physiologic and pathologic processes. The ability of HS chains to affect activity and distribution of signaling proteins is of particular relevance here. As originally shown by Vortkamp and coworkers, the distribution and signaling activity of Indian hedgehog were abnormally broad and ectopic in the growth plates of Ext1-hypomorphic mouse mutants. We showed recently that BMP signaling became broad and ectopic in conditional Ext1-deficient long bone epiphyses [14]. Thus, it is possible that several if not many of the defects in mutant long bones described here reflect alterations in patterns of activity and distribution of signaling proteins. The misalignment of chondrocytes in mutant growth plates may reflect defects in hedgehog signaling topography and targeting given that a similar chondrocyte misalignment was observed in Ihh-null cartilaginous long bones [31]. The misalignment may have resulted also from defects in the planar cell polarity pathway, a form of non-canonical Wnt pathway mediated by primary cilia essential for cell orientation in many organs and thought to operate in chondrocyte stacking in the growth plate as well [32]. Notably, we observed growth plate chondrocyte misalignment in mice lacking the essential primary cilia protein Kif3a accompanied by excess hedgehog signaling in perichondrium and formation of ectopic cartilage [33]. Indeed, defects in primary cilia have also been observed in human exostosis chondrocytes in HME patients [34].
The reduction in trabecular bone in primary spongiosa and the delay in secondary ossification center formation could have multiple origins. As pointed out above, they may reflect the fact that the growth plate was disorganized in the mutants and that the hypertrophic zone was markedly reduced in height and cell number. The mutant hypertrophic chondrocytes –be it at the bottom of the growth plate or at the center of the incipient secondary ossification center- may have been unable to facilitate the transition to endochondral ossification [6]. Hypertrophic chondrocytes normally produce a number of proteins needed for a seamless transition from cartilage to bone, including vascular endothelial growth factors (VEGFs), MMPs and transferrin, and one or more of these proteins may have been present at suboptimal levels in the mutants. In addition, bone marrow- and perichondrial-associated osteoprogenitor cells require signaling by hypertrophic chondrocyte-derived Indian hedgehog (Ihh) for their osteogenic differentiation [1, 5, 6, 25], and recruitment of osteoclasts is induced by VEGFs produced and released by hypertrophic chondrocytes [35]. Hypertrophic chondrocytes are also known to normally produce RANKL, a powerful inducer of osteoclast differentiation [36]. The significantly larger number of osteoclasts we observe at the chondro-osseous border in mutant growth plates suggests that the mutant hypertrophic chondrocytes may have expressed higher RANKL or M-CSF levels or there may have been a local decrease in Osteoprotegerin production [37]. The ultimate overall result of all these multiple changes would have been an abnormal environment in which the hypertrophic zone was defective, bone formation was deficient, development of osteoclasts was excessive, and trabecular bone and bone marrow were deranged in both extent and cell composition.
Given that the number and function of hypertrophic chondrocytes were decreased in the mutant growth plate, it may appear paradoxical that simultaneously, ectopic hypertrophic-like chondrocytes actually formed in the synovial joints of the mutants. By their large cell size and round shape and expression of both collagen X and MMP-13, the cells did closely resemble growth plate hypertrophic chondrocytes. Interestingly, cell hypertrophy and expression of hypertrophic traits are often seen in osteoarthritic joints in both experimental animals and patients [38]. What could then account for the decrease in hypertrophic cells in mutant growth plate and excess hypertrophy in articular cartilage? One interesting possibility is that articular and growth plate chondrocytes responded differently to Ext1 deficiency. It is known that immature and hypertrophic chondrocytes react in opposite manners to factors including retinoic acid and FGFs [39-41]. Thus, the Ext1 deficiency could have elicited different responses in articular and growth plate chondrocytes because the cells are intrinsically different. It was shown recently that ectopic expression and activity of hedgehog factors induce chondrocyte hypertrophy and an osteoarthritic phenotype in postnatal transgenic mouse limb joints [42]. Thus, excess signaling and/or distribution of hedgehog proteins or other HS-dependent factors may have occurred in the joints in our Ext1 mutants as well, contributing to joint's phenotypic de-stabilization, ectopic chondrocyte hypertrophy and inception of an osteoarthritic-like phenotype. Ext mutations and/or decreases in HS or HSPGs might thus be additional culprits in osteoarthritis.
Amongst the several members of the HSPG family, we have specifically analyzed Perlecan here because it is abundant in growth plate and articular cartilage and has multiple important roles and functions [43]. The Perlecan hypomorphic mice exhibit disease phenotypes comparable to those of Ext1 mutants shown here, including ∼15% skeletal growth reduction, delayed endochondral ossification and secondary ossification center formation, disorganized chondrocyte polarity and column formation within growth plates, altered arrangement and reduction of trabecular bone, and increased osteoclast activity. Moreover, the mice exhibit postnatal joint abnormalities, including degeneration of articular cartilage and chondro-osteonecrosis of the femoral and humeral heads, but not ectopic articular chondrocyte hypertrophy [23]. Thus, deficiencies in Perlecan or Ext1 can cause similar disease-like outcomes such as growth plate chondrocyte disorientation and aberrant transition to trabecular bone, consistent with the interesting possibility that the prevalent chondrodyslasia phenotypes seen in Perlecan hypomorphic mice and Schwartz-Jampel patients may ensue from reduced HS levels.
At this regard, it is important to discuss the conspicuous cartilaginous outgrowths forming on the lateral side of the Ext1-deficient growth plates starting as early as 1-week post-tamoxifen injection (Figs. 3 and 6), not observed in Perlecan mutants [23]. The outgrowths display a growth plate-like structure, and do resemble the exostoses forming in HME patients in terms of anatomical location, orthogonal orientation with respect to long bone main axis, and persistent cartilaginous character [26]. They also resemble the outgrowths described previously in the long bones of double Ext1+/-;Ext2+/- heterozygous [17] and Ext1f/f;Col2CreERT [16] mice. Interestingly, they are also reminiscent of the ectopic cartilaginous outgrowths forming after conditional postnatal ablation in chondrocytes of the canonical Wnt signaling mediator β-catenin [44]. Given our previous observation that b-catenin expression was quite low in Ext1-deficient growth plates including the hypertrophic zone where it is normally strong [44, 45], the data suggest that Wnt signaling was decreased in the outgrowths as well and may have contributed to both their genesis and persistent cartilaginous character. What do the data here and in previous studies tell us about HME pathogenesis and clinical implications? The data certainly reaffirm the notion that the growth plate is very vulnerable to destabilization and aberrant behavior if Ext-, HS- and HSPG-dependent mechanisms do not work appropriately. They also tell us that a simple heterozygous mutation in EXT1 or EXT2 usually seen in HME patients may not be sufficient to trigger exostosis formation and cause a full blown clinical phenotype. Using mouse models of stochastic Ext1 ablation, recent studies have in fact revealed that exostosis formation can be triggered by a relatively small number of Ext1-null chondrocytes [16, 18]. One of those studies was carried out by one of our labs [16] and relied on basal leakiness of the Col2CreERT mouse line, eliciting 5 to 15% Ext1f/f ablation levels without tamoxifen injection and indicating that the floxed Ext1 gene is very sensitive to CreERT leakage activity. The resulting mutant embryos displayed exostosis-like outgrowths in long bones as well as changes in growth plate and articular cartilage [16]. We have observed similar but less apparent and consistent skeletal changes in uninjected Ext1f/f;Col2CreERT mice, though heterozygous Ext1f/+;Col2CreER uninjected mice were normal (not shown). In HME patients then, exostosis formation would require mechanisms by which EXT expression and/or HS production are markedly reduced in some cells well beyond the partial reduction (about 50%) that is caused by a simple heterozygous EXT mutation [46, 47]. Loss-of-heterozygosity, increased heparanase expression or other genetic alterations have been invoked to occur in HME patients that, singly or in concert, would decrease EXT expression and HS levels substantially in some cells and would lead to exostosis formation [48-51].
In conclusion, we have shown here that postnatal conditional loss of Ext1 expression in cartilage is incompatible with normal skeletal growth and results in a number of structural and functional deficiencies in both the long bones and joints. We have shown also that Ext1 deficiency elicits defects that can occur in human skeletal conditions including trabecular bone loss, osteoarthritis and HME.
Supplementary Material
Highlights.
Postnatal Ext1-deficiency in cartilage causes skeletal growth retardation in mice
Ext1-deficient long bone growth plates are disorganized and display a shallow hypertrophic zone
Mutant long bones exhibit reduced primary spongiosa and increased TRAP-positive cells
Hypertrophic-like chondrocytes develop within Ext1-deficient articular cartilage
The mutant cells express hypertrophic genes and abundant Perlecan-rich pericellular matrix
Acknowledgments
We thank Dr. S. Mackem (NCI) for providing Col2CreERT mice, the microCT Imaging Core in McKay Orthopaedic Research Laboratory, University of Philadelphia for microCT scans, and Ms. C. Saunders and Ms. L. Cantley for their technical assistance.
*This study was supported by Grants AR058382, AR061758, AR062908, and a Foerderer Award (FY2012) from the Children's Hospital of Philadelphia.
Footnotes
Abbreviations: heparan sulfate (HS); bone morphogenetic protein (BMP); fibroblast growth factor (FGF); heparan sulfate proteoglycans (HSPGs); Indian hedgehog (Ihh); Hereditary Multiple Exostoses (HME); matrix metalloprotease 13 (MMP-13); vascular endothelial growth factors (VEGFs); tartrate-resistant acid phosphatase (TRAP).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 2.Engsig MT, Chen QJ, Vu TH, Pedersen AC, Therkidsen B, Lund LR, Henriksen K, Lenhard T, Foged NT, Werb Z, Delaisse JM. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. 2000;151:879–89. doi: 10.1083/jcb.151.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pacifici M, Koyama E, Iwamoto M. Mechanisms of synovial joint and articular cartilage formation: recent advances, but many lingering mysteries. Birth Defects Res C Embryo Today. 2005;75:237–48. doi: 10.1002/bdrc.20050. [DOI] [PubMed] [Google Scholar]
- 4.Pacifici M, Koyama E, Shibukawa Y, Wu C, Tamamura Y, Enomoto-Iwamoto M, Iwamoto M. Cellular and molecular mechanisms of synovial joint and articular cartilage formation. Ann N Y Acad Sci. 2006;1068:74–86. doi: 10.1196/annals.1346.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 6.de Crombrugghe B, Lefebvre V, Nakashima K. Regulatory mechanisms in the pathways of cartilage and bone formation. Curr Opin Cell Biol. 2001;13:721–7. doi: 10.1016/s0955-0674(00)00276-3. [DOI] [PubMed] [Google Scholar]
- 7.Busse M, Feta A, Presto J, Wilen M, Gronning M, Kjellen L, Kusche-Gullberg M. Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J Biol Chem. 2007;282:32802–10. doi: 10.1074/jbc.M703560200. [DOI] [PubMed] [Google Scholar]
- 8.Bulow HE, Hobert O. The molecular diversity of glycosaminoglycans shapes animal development. Annu Rev Cell Dev Biol. 2006;22:375–407. doi: 10.1146/annurev.cellbio.22.010605.093433. [DOI] [PubMed] [Google Scholar]
- 9.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–7. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 10.Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–41. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 11.Koziel L, Kunath M, Kelly OG, Vortkamp A. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev Cell. 2004;6:801–13. doi: 10.1016/j.devcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Matsumoto Y, Matsumoto K, Irie F, Fukushi J, Stallcup WB, Yamaguchi Y. Conditional ablation of the heparan sulfate-synthesizing enzyme Ext1 leads to dysregulation of bone morphogenic protein signaling and severe skeletal defects. J Biol Chem. 2010;285:19227–34. doi: 10.1074/jbc.M110.105338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mundy C, Yasuda T, Kinumatsu T, Yamaguchi Y, Iwamoto M, Enomoto-Iwamoto M, Koyama E, Pacifici M. Synovial joint formation requires local Ext1 expression and heparan sulfate production in developing mouse embryo limbs and spine. Dev Biol. 2011;351:70–81. doi: 10.1016/j.ydbio.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huegel J, Mundy C, Sgariglia F, Nygren P, Billings PC, Yamaguchi Y, Koyama E, Pacifici M. Perichondrium phenotype and border function are regulated by Ext1 and heparan sulfate in developing long bones: A mechanism likely deranged in Hereditary Multiple Exostoses. Dev Biol. 2013 doi: 10.1016/j.ydbio.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilton MJ, Gutierrez L, Martinez DA, Wells DE. EXT1 regulates chondrocyte proliferation and differentiation during endochondral bone development. Bone. 2005;36:379–86. doi: 10.1016/j.bone.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto K, Irie F, Mackem S, Yamaguchi Y. A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of heterozygosity in multiple hereditary exostoses. Proc Natl Acad Sci U S A. 2010;107:10932–7. doi: 10.1073/pnas.0914642107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zak BM, Schuksz M, Koyama E, Mundy C, Wells DE, Yamaguchi Y, Pacifici M, Esko JD. Compound heterozygous loss of Ext1 and Ext2 is sufficient for formation of multiple exostoses in mouse ribs and long bones. Bone. 2011;48:979–87. doi: 10.1016/j.bone.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones KB, Piombo V, Searby C, Kurriger G, Yang B, Grabellus F, Roughley PJ, Morcuende JA, Buckwalter JA, Capecchi MR, Vortkamp A, Sheffield VC. A mouse model of osteochondromagenesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci U S A. 2010;107:2054–9. doi: 10.1073/pnas.0910875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pedrini E, Jennes I, Tremosini M, Milanesi A, Mordenti M, Parra A, Sgariglia F, Zuntini M, Campanacci L, Fabbri N, Pignotti E, Wuyts W, Sangiorgi L. Genotype-phenotype correlation study in 529 patients with multiple hereditary exostoses: identification of “protective” and “risk” factors. J Bone Joint Surg Am. 2011;93:2294–302. doi: 10.2106/JBJS.J.00949. [DOI] [PubMed] [Google Scholar]
- 20.Jennes I, Pedrini E, Zuntini M, Mordenti M, Balkassmi S, Asteggiano CG, Casey B, Bakker B, Sangiorgi L, Wuyts W. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb) Hum Mutat. 2009;30:1620–7. doi: 10.1002/humu.21123. [DOI] [PubMed] [Google Scholar]
- 21.Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–6. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura E, Nguyen MT, Mackem S. Kinetics of tamoxifen-regulated Cre activity in mice using a cartilage-specific CreER(T) to assay temporal activity windows along the proximodistal limb skeleton. Dev Dyn. 2006;235:2603–12. doi: 10.1002/dvdy.20892. [DOI] [PubMed] [Google Scholar]
- 23.Rodgers KD, Sasaki T, Aszodi A, Jacenko O. Reduced perlecan in mice results in chondrodysplasia resembling Schwartz-Jampel syndrome. Hum Mol Genet. 2007;16:515–28. doi: 10.1093/hmg/ddl484. [DOI] [PubMed] [Google Scholar]
- 24.Lefebvre V, Garofalo S, de Crombrugghe B. Type X collagen gene expression in mouse chondrocytes immortalized by a temperature-sensitive simian virus 40 large tumor antigen. J Cell Biol. 1995;128:239–45. doi: 10.1083/jcb.128.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zelzer E, Olsen BR. The genetic basis for skeletal diseases. Nature. 2003;423:343–8. doi: 10.1038/nature01659. [DOI] [PubMed] [Google Scholar]
- 26.Hecht JT, Hayes E, Haynes R, Cole WG, Long RJ, Farach-Carson MC, Carson DD. Differentiation-induced loss of heparan sulfate in human exostosis derived chondrocytes. Differentiation. 2005;73:212–21. doi: 10.1111/j.1432-0436.2005.00025.x. [DOI] [PubMed] [Google Scholar]
- 27.Melrose J, Smith S, Cake M, Read R, Whitelock J. Perlecan displays variable spatial and temporal immunolocalisation patterns in the articular and growth plate cartilages of the ovine stifle joint. Histochem Cell Biol. 2005;123:561–71. doi: 10.1007/s00418-005-0789-y. [DOI] [PubMed] [Google Scholar]
- 28.SundarRaj N, Fite D, Ledbetter S, Chakravarti S, Hassell JR. Perlecan is a component of cartilage matrix and promotes chondrocyte attachment. J Cell Sci. 1995;108(Pt 7):2663–72. doi: 10.1242/jcs.108.7.2663. [DOI] [PubMed] [Google Scholar]
- 29.Ohlsson C, Nilsson A, Isaksson O, Lindahl A. Growth hormone induces multiplication of the slowly cycling germinal cells of the rat tibial growth plate. Proc Natl Acad Sci U S A. 1992;89:9826–30. doi: 10.1073/pnas.89.20.9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–77. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 31.Razzaque MS, Soegiarto DW, Chang D, Long F, Lanske B. Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J Pathol. 2005;207:453–61. doi: 10.1002/path.1870. [DOI] [PubMed] [Google Scholar]
- 32.Dabdoub A, Kelley MW. Planar cell polarity and a potential role for a Wnt morphogen gradient in stereociliary bundle orientation in the mammalian inner ear. J Neurobiol. 2005;64:446–57. doi: 10.1002/neu.20171. [DOI] [PubMed] [Google Scholar]
- 33.Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, Maeda Y, Lanske B, Song B, Serra R, Pacifici M. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development. 2007;134:2159–69. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Andrea CE, Wiweger M, Prins F, Bovee JV, Romeo S, Hogendoorn PC. Primary cilia organization reflects polarity in the growth plate and implies loss of polarity and mosaicism in osteochondroma. Lab Invest. 2010;90:1091–101. doi: 10.1038/labinvest.2010.81. [DOI] [PubMed] [Google Scholar]
- 35.Zelzer E, McLean W, Ng YS, Fukai N, Reginato AM, Lovejoy S, D'Amore PA, Olsen BR. Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development. 2002;129:1893–904. doi: 10.1242/dev.129.8.1893. [DOI] [PubMed] [Google Scholar]
- 36.Duong LT, Rodan GA. Regulation of osteoclast formation and function. Rev Endocr Metab Disord. 2001;2:95–104. doi: 10.1023/a:1010063225902. [DOI] [PubMed] [Google Scholar]
- 37.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–37. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 38.Pitsillides AA, Beier F. Cartilage biology in osteoarthritis--lessons from developmental biology. Nat Rev Rheumatol. 2011;7:654–63. doi: 10.1038/nrrheum.2011.129. [DOI] [PubMed] [Google Scholar]
- 39.Iwamoto M, Yagami K, Shapiro IM, Leboy PS, Adams SL, Pacifici M. Retinoic acid is a major regulator of chondrocyte maturation and matrix mineralization. Microsc Res Tech. 1994;28:483–91. doi: 10.1002/jemt.1070280604. [DOI] [PubMed] [Google Scholar]
- 40.St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–86. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q, Green RP, Zhao G, Ornitz DM. Differential regulation of endochondral bone growth and joint development by FGFR1 and FGFR3 tyrosine kinase domains. Development. 2001;128:3867–76. doi: 10.1242/dev.128.19.3867. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi T, Soegiarto DW, Yang Y, Lanske B, Schipani E, McMahon AP, Kronenberg HM. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J Clin Invest. 2005;115:1734–42. doi: 10.1172/JCI24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Handler M, Yurchenco PD, Iozzo RV. Developmental expression of perlecan during murine embryogenesis. Dev Dyn. 1997;210:130–45. doi: 10.1002/(SICI)1097-0177(199710)210:2<130::AID-AJA6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 44.Cantley L, Saunders C, Guttenberg M, Candela ME, Ohta Y, Yasuhara R, Kondo N, Sgariglia F, Asai S, Zhang X, Qin L, Hecht JT, Chen D, Yamamoto M, Toyosawa S, Dormans JP, Esko JD, Yamaguchi Y, Iwamoto M, Pacifici M, Enomoto-Iwamoto M. Loss of beta-catenin induces multifocal periosteal chondroma-like masses in mice. Am J Pathol. 2013;182:917–27. doi: 10.1016/j.ajpath.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuasa T, Kondo N, Yasuhara R, Shimono K, Mackem S, Pacifici M, Iwamoto M, Enomoto-Iwamoto M. Transient activation of Wnt/{beta}-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol. 2009;175:1993–2003. doi: 10.2353/ajpath.2009.081173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook A, Raskind W, Blanton SH, Pauli RM, Gregg RG, Francomano CA, Puffenberger E, Conrad EU, Schmale G, Schellenberg G, et al. Genetic heterogeneity in families with hereditary multiple exostoses. Am J Hum Genet. 1993;53:71–9. [PMC free article] [PubMed] [Google Scholar]
- 47.Wu YQ, Heutink P, de Vries BB, Sandkuijl LA, van den Ouweland AM, Niermeijer MF, Galjaard H, Reyniers E, Willems PJ, Halley DJ. Assignment of a second locus for multiple exostoses to the pericentromeric region of chromosome 11. Hum Mol Genet. 1994;3:167–71. doi: 10.1093/hmg/3.1.167. [DOI] [PubMed] [Google Scholar]
- 48.Zuntini M, Pedrini E, Parra A, Sgariglia F, Gentile FV, Pandolfi M, Alberghini M, Sangiorgi L. Genetic models of osteochondroma onset and neoplastic progression: evidence for mechanisms alternative to EXT genes inactivation. Oncogene. 2010;29:3827–34. doi: 10.1038/onc.2010.135. [DOI] [PubMed] [Google Scholar]
- 49.Reijnders CM, Waaijer CJ, Hamilton A, Buddingh EP, Dijkstra SP, Ham J, Bakker E, Szuhai K, Karperien M, Hogendoorn PC, Stringer SE, Bovee JV. No haploinsufficiency but loss of heterozygosity for EXT in multiple osteochondromas. Am J Pathol. 2010;177:1946–57. doi: 10.2353/ajpath.2010.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trebicz-Geffen M, Robinson D, Evron Z, Glaser T, Fridkin M, Kollander Y, Vlodavsky I, Ilan N, Law KF, Cheah KS, Chan D, Werner H, Nevo Z. The molecular and cellular basis of exostosis formation in hereditary multiple exostoses. Int J Exp Pathol. 2008;89:321–31. doi: 10.1111/j.1365-2613.2008.00589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zuntini M, Salvatore M, Pedrini E, Parra A, Sgariglia F, Magrelli A, Taruscio D, Sangiorgi L. MicroRNA profiling of multiple osteochondromas: identification of disease-specific and normal cartilage signatures. Clin Genet. 2010;78:507–16. doi: 10.1111/j.1399-0004.2010.01490.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.