

Abstract
In this study, we treated PC12 cells with 0–20 μM amyloid-β peptide (25–35) for 24 hours to induce cytotoxicity, and found that 5–20 μM amyloid-β peptide (25–35) decreased PC12 cell viability, but adenosine triphosphate-sensitive potassium channel activator diazoxide suppressed the decrease in PC12 cell viability induced by amyloid-β peptide (25–35). Diazoxide protected PC12 cells against amyloid-β peptide (25–35)-induced increases in mitochondrial membrane potential and intracellular reactive oxygen species levels. These protective effects were reversed by the selective mitochondrial adenosine triphosphate-sensitive potassium channel blocker 5-hydroxydecanoate. An inducible nitric oxide synthase inhibitor, Nω-nitro-L-arginine, also protected PC12 cells from amyloid-β peptide (25–35)-induced increases in both mitochondrial membrane potential and intracellular reactive oxygen species levels. However, the H2O2-degrading enzyme catalase could not reverse the amyloid-β peptide (25–35)-induced increase in intracellular reactive oxygen species. A 24-hour exposure to amyloid-β peptide (25–35) did not result in apoptosis or necrosis, suggesting that the increases in both mitochondrial membrane potential and reactive oxygen species levels preceded cell death. The data suggest that amyloid-β peptide (25–35) cytotoxicity is associated with adenosine triphosphate-sensitive potassium channels and nitric oxide. Regulation of adenosine triphosphate-sensitive potassium channels suppresses PC12 cell cytotoxicity induced by amyloid-β peptide (25–35).
Keywords: neural regeneration, neurodegenerative diseases, amyloid-β peptide (25–35), PC12 cell, adenosine triphosphate-sensitive potassium channel, inducible nitric oxide synthase, mitochondrial membrane potential, reactive oxygen species, grant-supported paper, photographs-containing paper, neuroregeneration
Research Highlights
(1) Amyloid-β peptide (25–35) increases mitochondrial membrane potential and intracellular reactive oxygen species levels, resulting in PC12 cell damage.
(2) Diazoxide increases the viability of PC12 cells damaged by amyloid-β peptide (25–35).
(3) An inducible nitric oxide synthase inhibitor, Nω-nitro-L-arginine, protects PC12 cells from amyloid-β peptide (25–35)-induced neurotoxic effects.
(4) Diazoxide and Nω-nitro-L-arginine protect PC12 cells from amyloid-β peptide (25–35)-induced increases in both mitochondrial membrane potential and intracellular reactive oxygen species levels.
Abbreviations
Aβ, amyloid-β peptide; KATP, adenosine triphosphate-sensitive potassium; ATP, adenosine triphosphate; DCF, 2’,7’-dichlorofluorescein; JC-1,5,5’,6,6’-tetrachloro-1,1’,3,3’- tetraethylbenzimidazolcarbocyanine iodide; DCFH-DA, 5,6-carboxy-2’,7’-dichlorofluorescin-diacetate
INTRODUCTION
The role of the amyloid β peptide (Aβ) during neurodegeneration has become the focus of studies of the pathogenesis of Alzheimer's disease[1,2]. Recent discoveries have shown that Aβ causes synaptic degeneration and cell apoptosis[3,4]. The Aβ25-35 fragment has also been shown to be cytotoxic[5]. Furthermore, Aβ might trigger mitochondrial dysfunction accompanied by the generation of free radicals, which in turn might impair ion-motive adenosine triphosphatase activity, resulting in membrane depolarization. Supporting this hypothesis, there is ample evidence for a central role of mitochondrial dysfunction in the pathogenesis of Alzheimer's disease[5,6,7,8].
Adenosine triphosphate (ATP)-sensitive potassium (KATP) channels link cell metabolism to membrane potential[9]. It has been shown that Aβ can induce KATP channel dysfunction[10]. Modulation of the KATP channels expressed in neurons may exert a neuroprotective effect. Diazoxide, an activator of mitochondrial KATP (mitoKATP) channels, is becoming a promising protective agent. Potassium channel activators have been shown to protect cardiac myocytes against ischemic injury[11]. Diazoxide has also been shown to protect cultured neurons against toxicity induced by neurotoxins including glutamate, Aβ, and hydrogen peroxide (H2O2)[12,13]. Additionally, the production of nitric oxide, which plays an important role in the regulation of mitochondrial function, is believed to be closely related to KATP channel activity[14]. The formation of nitric oxide is catalyzed by inducible nitric oxide synthase[15]. In Alzheimer's disease brain cortex, inducible nitric oxide synthase expression levels are markedly increased[16]. Excess nitric oxide could impair neuronal function[17]. Therefore, regulation of inducible nitric oxide synthase activity may produce a neuroprotective effect. Despite the abundance of data, the precise mechanisms of these beneficial effects are still obscure, especially in the brain.
The present study was designed to determine whether the cytotoxic Aβ25-35 fragment could change mitochondrial membrane potential and intracellular reactive oxygen species levels, and whether a KATP channel activator, diazoxide, and an inducible nitric oxide synthase inhibitor, Nω-nitro-L-arginine, could protect cells against Aβ25-35 cytotoxicity.
RESULTS
Diazoxide counteracted the effected of Aβ25-35 on PC12 cell viability
Cultured PC12 cells were treated with several concentrations of Aβ25-35 and cellular viability was measured by assessing MTT reduction. At 5, 10 and 20 µM, Aβ25-35 significantly lowered cellular viability (P < 0.05 or P < 0.01). However, when cultured cells were pretreated with diazoxide (1 mM) for 1 hour and then exposed to Aβ25-35 for 24 hours, their viability was significantly greater than that of cells exposed to Aβ25-35 alone (P < 0.05 or P < 0.01). The protective effect of diazoxide was abolished by 5-hydroxydecanoate (500 μM), a KATP channel blocker (P < 0.05 or P < 0.01). The control reverse Aβ35-25 had no effect on cellular viability (Figure 1).
Figure 1.
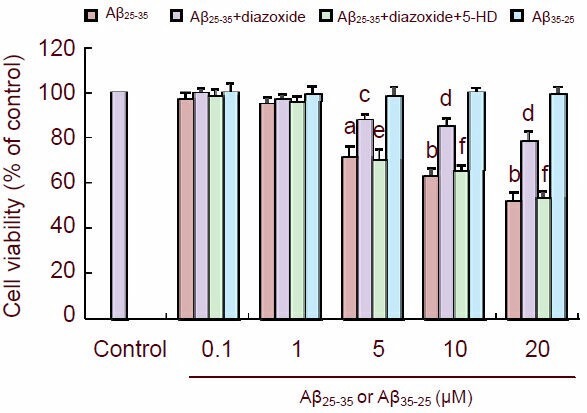
Diazoxide (a potassium channel activator) protected the cultured PC12 cells against amyloid-β peptide (25–35) (Aβ25-35) toxicity.
Cultured cells were treated with the indicated concentration of Aβ25-35 or Aβ35-25 for 24 hours. Cell viability (A570 nm) was detected by MTT reduction assay.
The data are expressed as mean ± SEM from five independent experiments. aP < 0.05, bP < 0.01, vs. control; cP < 0.05, dP < 0.01, vs. Aβ25-35; eP < 0.05, fP < 0.01, vs. Aβ25-35 + diazoxide (one-way analysis of variance, followed by two-tailed Student's t-test).
5-HD: 5-hydroxydecanoate.
Diazoxide and Nω-nitro-L-arginine suppressed the increase in mitochondrial membrane potential induced by Aβ25-35 in PC12 cells
5,5’,6,6’-tetrachloro-1,1’,3,3’- tetraethylbenzimidazolcarbocyanine iodide (JC-1) produces two fluorescence emission peaks that reflect the existence of two physical forms of the dye. The monomer, which is the predominant form at low mitochondrial membrane potential, emits green fluorescence (emission maximum at 527 nm), and the so-called “J-aggregate”, which is the predominant species at high mitochondrial membrane potential, emits orange-red fluorescence (emission maximum at 590 nm). Earlier observations suggested that the fluorescence intensity of the monomer is insensitive to changes in mitochondrial membrane potential[18]. Therefore, it was concluded that the lower the JC-1 590/527 nm fluorescence ratio is, the higher the mitochondrial membrane potential is (Figure 2A).
Figure 2.
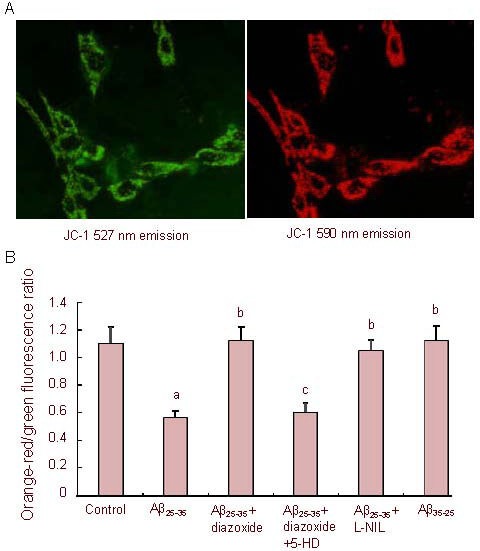
Effects of amyloid-β peptide (25–35) (Aβ25-35) on mitochondrial membrane potential in PC12 cells.
Cells stained with 5,5’,6,6’-tetrachloro-1,1’,3,3’- tetraethylbenzimidazolcarbocyanine iodide (JC-1) (A, × 400). Green fluorescence (emission maximum at 527 nm) and orange-red fluorescence (emission maximum at 590 nm) are shown.
Cells were exposed to Aβ25-35 (5 µM) or Aβ35-25 (5 µM) in the presence or absence of 1 mM diazoxide (a potassium channel activator) or 1 mM diazoxide plus 500 μM 5-hydroxydecanoate (5-HD, a KATP channel blocker) or 500 μM Nω-nitro-L-arginine (L-NIL, a nitric oxide synthase inhibitor) for 24 hours, followed by staining with JC-1 and laser scan confocal microscopy semi-quantitative analysis of the JC-1 590/527 nm fluorescence ratio (B) (the lower the JC-1 590/527 nm fluorescence ratio is, the higher the mitochondrial membrane potential is).
The data are expressed as mean ± SEM from five independent experiments. aP < 0.01, vs. control; bP < 0.01, vs. Aβ25-35; cP < 0.01, vs. Aβ25-35 + diazoxide (one-way analysis of variance, followed by two-tailed Student's t-test).
Aβ25-35 (5 µM) treatment for 24 hours caused a marked increase in mitochondrial membrane potential (P < 0.01). However, addition of diazoxide rescued the membrane potential. 5-hydroxydecanoate abolished the protective effect of diazoxide, reverting the membrane potential to that of the Aβ25-35-treated cells, suggesting that diazoxide was responsible for the observed maintenance of mitochondrial membrane potential during Aβ25-35 challenge.
When cultured PC12 cells were co-treated with Aβ25-35 and inducible nitric oxide synthase inhibitor Nω-nitro-L-arginine for 24 hours, the JC-1 590/527 nm fluorescence ratio significantly increased compared with cells exposed to Aβ25-35 alone (P < 0.01), suggesting that the Aβ25-35-induced increase in mitochondrial membrane potential was prevented by Nω-nitro-L-arginine. The reverse control peptide Aβ35-25 had no effect on mitochondrial membrane potential (Figure 2B).
Diazoxide and Nω-nitro-L-arginine suppressed the increase in reactive oxygen species levels induced by Aβ25-35 in PC12 cells
In control PC12 cells, flow cytometry indicated an average 2’,7’-dichlorofluorescein (DCF) fluorescence intensity (527 nm) of 600.6 ± 85.2; whereas in the Aβ25-35 (5 µM) group, the average DCF fluorescence intensity was 1 285.5 ± 110.2. This showed that reactive oxygen species levels were significantly higher in Aβ25-35 -treated cells than in controls (P < 0.05), indicating oxidative stress. The Aβ25-35-induced increase in DCF fluorescence was counteracted by the addition of Nω-nitro-L-arginine (average DCF fluorescence intensity 700.6 ± 101.7), suggesting that nitric oxide was responsible for the Aβ25-35-induced production of intracellular reactive oxygen species.
The Aβ25-35-induced increase in DCF fluorescence was also counteracted by 1 mM diazoxide (average DCF fluorescence intensity 750.5 ± 99.3), suggesting that diazoxide could protect cells from oxidative stress induced by Aβ25-35 treatment. The protective effect of diazoxide was abolished by 5-hydroxydecanoate (500 μM).
We also determined whether H2O2 production was necessary for the Aβ25-35-induced increase in reactive oxygen species levels. Incubating cells with H2O2 increased DCF fluorescence compared with control cells (P < 0.05). However, incubation with the H2O2-degrading enzyme catalase (100 mg/mL) did not reverse the Aβ25-35-induced increase in DCF fluorescence intensity (P > 0.05), suggesting intracellular H2O2 levels did not play an important role in the Aβ25-35-induced increase in reactive oxygen species levels (Figure 3).
Figure 3.
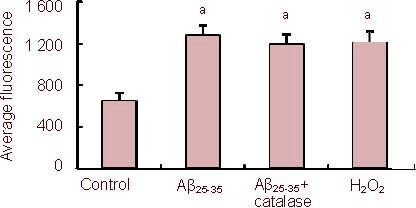
Effects of amyloid-β peptide (25–35) (Aβ25-35) on reactive oxygen levels in PC12 cells using 5,6-carboxy-2’, 7’-dichlorofluorescin-diacetate (DCFH-DA) fluorescence.
Cells were exposed to Aβ25-35 (5 µM) in the presence or absence of 100 mg/mL catalase for 24 hours, followed by staining with DCFH-DA and flow cytometry.
Data are expressed as mean ± SEM from five independent experiments (527 nm fluorescence). aP < 0.05, vs. control (one-way analysis of variance, followed by two-tailed Student's t-test).
Effect of Aβ25-35 peptide on apoptosis and necrosis in PC12 cells
To study whether the Aβ25-35-induced increases in mitochondrial membrane potential and reactive oxygen species levels were associated with apoptosis and necrosis, cells were exposed to Aβ25-35 (5 µM) for 24 hours and then examined with annexin-V-FITC/propidium iodide flow cytometry. The percentage of apoptotic and necrotic PC12 cells exposed to Aβ25-35 was 6.4 ± 1.8% (Figure 4). There was no difference in cell death in Aβ25-35-treated PC12 cells compared with the control.
Figure 4.
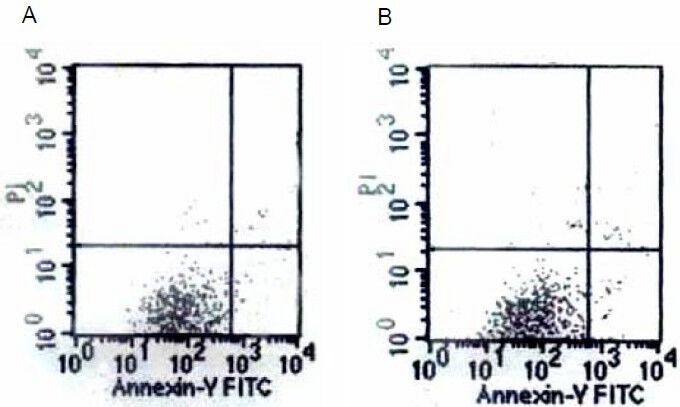
Graphs of the annexin-V-FITC flow cytometry results of control (A) and amyloid-β peptide (25–35)-treated (B) cells.
There was no difference in cell death.
DISCUSSION
Previous studies confirmed that H2O2 induces neuronal mitochondrial depolarization, and that simultaneous application of diazoxide and H2O2 completely inhibits H2O2-induced mitochondrial membrane depolarization[11,12,13]. This inhibitory action of diazoxide is antagonized by the addition of 5-hydroxydecanoate (a mitoKATP channel blocker), suggesting that opening of the mitoKATP channel was responsible for the maintenance of mitochondrial membrane potential during H2O2 challenge[19]. The mitoKATP channel plays an important role in the regulation of mitochondrial membrane potential[20].
This study determined that diazoxide protects cells from the neurotoxic effects of the Aβ25-35 fragment implicated in the molecular pathogenesis of Alzheimer's disease. An early indicator of toxicity is the inhibition of cellular MTT reduction to MTT formazan, a widely used assay for measuring cell viability[21,22]. We used this assay to investigate the toxicity of Aβ25-35 in PC12 cells. Our study indicated that Aβ25-35 induces a decrease in PC12 cell viability in a concentration- dependent manner, and this decrease can be significantly counteracted by diazoxide. This neuroprotective effect is attenuated by 5-hydroxydecanoate. These data indicate that the neuroprotective effect of diazoxide against Aβ25-35-induced toxicity in PC12 cells is mediated through the mitoKATP channel.
Few studies have focused on the role of mitochondrial membrane integrity in the neuroprotection evoked by diazoxide. Measuring mitochondrial activity with the membrane potential-sensitive probe JC-1 revealed that exposure of PC12 cells to Aβ25-35 for 24 hours significantly increases mitochondrial membrane potential and strongly decreases the proportion of active mitochondria, and this effect can be significantly reversed by diazoxide. Potassium channel activators for KATP channels protected cardiac myocytes from ischemic injury[23] and neurons from oxidative injury induced by glutamate[12]. A possible mechanism for this protective effect is that diazoxide might activate K+ channels, decrease mitochondrial membrane potential and maintain normal physiological mitochondrial functions, protecting cells from amyloid β25-35 peptide-induced intracellular oxidative stress.
Since oxidative stress is believed to be another important contributor to the neurodegenerative process in Alzheimer's disease[24], we measured reactive oxygen species levels in PC12 cells treated with Aβ25-35 by flow cytometric analysis using the peroxide-sensitive fluorescence probe 5,6-carboxy-2’,7’-dichlorofluorescin- diacetate (DCFH-DA). Quantitative analysis revealed that, in cells exposed to Aβ25-35 for 24 hours, intracellular reactive oxygen species levels are significantly higher than in control cells. Diazoxide significantly inhibits the increase in reactive oxygen species levels induced by Aβ25-35 via interaction with mitochondria. Previous reports showed that mitochondrial deficits lead to oxidative stress[25,26]. Thus, a possible mechanism of cellular damage by Aβ25-35 involves an increase in reactive oxygen species and intracellular oxidative stress caused by impairment of the normal physiological function of KATP channels in the mitochondria. Both intracellular reactive oxygen species and impaired mitochondria form a vicious cycle such that cells are subjected to increasing oxidative stress. Ozcan et al[27] found that potassium channel activators decreased the production of reactive oxygen species, such as superoxide and hydrogen peroxide. Therefore, we believe that diazoxide reverses the cytotoxic effect of Aβ25-35 by maintaining the conductance of K+ channels and decreasing reactive oxygen species production.
Physiologically, normal plasma concentrations of nitric oxide play a critical role in signal transduction, memory and synaptic plasticity in the central nervous system, but excess nitric oxide synthesized by inducible nitric oxide synthase has been implicated in neuronal cytotoxicity[17]. In the present study, Aβ25-35 induced an increase in mitochondrial membrane potential, and the associated increase in intracellular reactive oxygen species levels was counteracted by the inducible nitric oxide synthase inhibitor Nω-nitro-L-arginine, suggesting that nitric oxide played a significant role in regulating intracellular reactive oxygen species production and the increase in mitochondrial membrane potential. Supporting our data, previous research showed that synthetic Aβ elicited a marked and sustained induction of inducible nitric oxide synthase activity and the formation of nitric oxide metabolites in primary cultures of mixed rat neuronal and glial cells[28,29]. The generation of reactive oxygen species in response to Aβ25-35 treatment was mimicked by H2O2, which is converted to the highly toxic hydroxyl radical by the Fenton reaction. However, the H2O2-degrading enzyme catalase (a reactive oxygen species inhibitor) could not reverse the Aβ25-35-induced increase in intracellular reactive oxygen species, suggesting that intracellular H2O2 does not play an important role in the Aβ25-35-induced increases in reactive oxygen species levels. However, since the Aβ25-35-induced elevation of intracellular reactive oxygen species levels was counteracted by an inducible nitric oxide synthase inhibitor, we suggest that nitric oxide plays an important role in this increase. Aβ25-35 activated inducible nitric oxide synthase activity and increased the formation of nitric oxide, resulting in increased intracellular reactive oxygen species levels and changes in mitochondrial membrane potential. In addition, increased intracellular reactive oxygen species levels might be responsible for nitric oxide-induced intracellular oxidative stress and cytotoxicity.
Flow cytometry found no difference in apoptosis and necrosis between the control and Aβ25-35-treated cells, suggesting that cell death was preceded by the increase in mitochondrial membrane potential and in reactive oxygen species levels. These results are of significant importance for the understanding of Alzheimer's disease pathogenesis.
In summary, this study demonstrated for the first time that diazoxide and the inducible nitric oxide synthase inhibitor Nω-nitro-L-arginine exert their neuroprotective effects through inhibition of the increase of mitochondrial membrane potential and the associated increase in intracellular reactive oxygen species levels induced by Aβ25-35 application. A possible mechanism is that diazoxide might activate K+ channels, decrease the mitochondrial membrane potential and maintain normal physiological mitochondrial functions. The effect of Nω-nitro-L-arginine indicates that nitric oxide plays an important role in regulating the production of reactive oxygen species and increasing mitochondrial membrane potential in response to Aβ25-35. Increased mitochondrial membrane potential and intracellular reactive oxygen species levels might promote each other, eventually leading to cell death. Further studies are required to explore in more detail the mechanisms of the observed effects, which may offer a novel therapeutic strategy for neurodegenerative disorders such as Alzheimer's disease.
MATERIALS AND METHODS
Design
A comparative in vitro cell culture experiment.
Time and setting
The experiments were performed at a laboratory in the Shanghai Jiao Tong University School of Medicine from December 2010 to June 2011.
Materials
Aβ25-35 and Aβ35-25 purchased from Sigma (St. Louis, MO, USA) were dissolved in sterile distilled water at 500 μM and incubated at 37°C for 4 days.
Methods
PC12 cell culture and differentiation
Rat PC12 pheochromocytoma cells (Chinese Academy of Sciences, Shanghai, China) were cultured at 37°C in Dulbecco's modified Eagle's medium (Gibco, Carlsbad, CA, USA) supplemented with 5% horse serum (Gibco), 10% fetal bovine serum (Gibco), 2 mM L-glutamine (Sigma), 100 IU/mL penicillin (Sigma), and 100 μg/mL streptomycin (Sigma) in a humidified atmosphere containing 5% CO2 in air. Neuronal-like differentiation of PC12 cells was induced by 7-day treatment with neural growth factor (100 ng/mL) (Gibco) (Figure 5).
Figure 5.
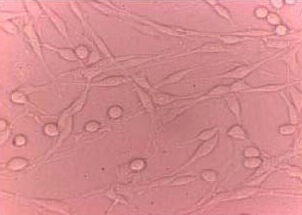
Identification of neuronal-like differentiation of PC12 cells (Axioplan 2 microscope, × 400).
PC12 cells with neuronal-like projections appeared after differentiation with nerve growth factor.
Grouping and intervention
Aggregated Aβ25-35 (0.1, 1, 5, 10, 20 µM) or Aβ35-25 (0.1, 1, 5, 10, 20 µM) were applied to the differentiated cells (at a density of 6 × 104/cm2) for 24 hours. Diazoxide (Sigma) was prepared as a stock solution of 50 mg/mL in dimethyl sulfoxide (final concentration 0.08%) and added to the media for a final concentration of 1 mM. Cells were preincubated for 1 hour with 1 mM diazoxide and then co-treated with Aβ25-35 and diazoxide for 24 hours. The Nω-nitro-L-arginine (Roche, Basel, Switzerland) was dissolved in 0.2 mM PBS (pH 7.4) and added to the media for a final concentration of 500 µM. Cells were co-incubated with Nω-nitro-L-arginine and Aβ25-35 for 24 hours.
Cellular viability as detected by MTT
The viability of cultured cells exposed to various drugs for 24 hours was evaluated using the MTT colorimetric assay[30]. A total of 10 μL MTT reagent (Sigma) was added to plated cells for 2 hours until a purple precipitate was visible. A total of 100 μL detergent reagent was added at room temperature in the dark for 2 hours. MTT reduction was quantified at 570 nm using a microplate reader (Bio-Tek Instruments Inc., Winooski, VT, USA). The ratio of MTT reduction in samples treated with various drugs to the MTT reduction in control samples was used to determine cellular viability.
Determination of the mitochondrial membrane potential using JC-1 fluorescence
Mitochondrial membrane potential was quantified using the ratiometric probe JC-1[18]. In apoptotic cells, in which the mitochondrial membrane potential had collapsed, monomeric JC-1 remained in the cytosol and appeared green. Following exposure to 5 μM Aβ25-35 or Aβ35-25 in the presence or absence of 1 mM diazoxide or 1 mM diazoxide plus 500 μM 5-hydroxydecanoate (Sigma) or 500 μM Nω-nitro-L- arginine for 24 hours, cells were incubated in culture medium-free serum containing 10 µg/mL JC-1 (solubilized in N, N-dimethylformamide, 1% v/v; Molecular Probes Europe, Leiden, Netherlands) for 30 minutes at 37°C under 5% CO2, and then washed with 0.1 M PBS twice. Laser confocal microscopy (Leica, Wetzlar, Germany) semi-quantitative analysis of JC-1 fluorescence was used to determine the average intensity of green (527 nm) fluorescent monomers and J-aggregates with orange-red (590 nm) fluorescence. The ratio of fluorescence at 590 nm/fluorescence at 527 nm was used to monitor changes in mitochondrial membrane potential.
Measurement of reactive oxygen species by DCFH-DA fluorescence
The production of reactive oxygen species was estimated by flow cytometry using the oxidation-sensitive fluorescent probe DCFH-DA[31], a cell-permeable dye that, once inside the cell, is cleaved by intracellular esterase into its non-fluorescent form DCFH. This form, which is no longer membrane permeable, may be further oxidized by H2O2 or OH- to its fluorescent form, DCF. Following exposure to 5 μM Aβ25-35 or Aβ35-25 in the presence or absence of 1 mM diazoxide or 1 mM diazoxide plus 500 μM 5-hydroxydecanoate or 500 μM Nω-nitro-L-arginine or 100 mg/mL catalase (Sigma) or 1 µM H2O2 (Sigma) for 24 hours, the cultured cells were incubated in serum-free culture medium containing 10 µM DCFH-DA (Molecular Probes Europe) for 30 minutes at 37°C and were then washed with 0.2 M PBS once. Cells were then trypsinized and washed, followed by flow cytometer detection using a FACS Calibur cytometry instrument (BD Biosciences, San Jose, CA, USA). Fluorescence emission from DCF (green) was detected at a wavelength of 527 nm.
Detection of apoptosis and necrosis using flow cytometry
After exposure to 5 μM Aβ25-35 or Aβ35-25 for 24 hours, free-floating cells pooled with cells detached by mild trypsinization were incubated in 100 mL annexin-V- FLUOS solution (Roche) containing 2 mL Annexin-V reagent (Roche) and 2 mL propidium iodide reagent (Roche) for 10–15 minutes at 20°C and then cooled so that the reaction was terminated. FITC-fluorescence was measured with a FACS Calibur flow cytometer at an excitation wavelength of 488 nm.
Statistical analysis
All data are expressed as mean ± SEM. Statistical analyses between controls and samples treated with various drugs were done by SPSS 10.0 software (SPSS, Chicago, IL, USA) using one-way analysis of variance, followed by two-tailed Student's t-test or multiple comparison test where appropriate. A value of P < 0.05 was considered statistically significant for all analyses.
Footnotes
Min Kong, Ph.D., Attending physician.
Funding: This study was financially supported by the Project Sponsored by Yantai Science and Technology Bureau, China, No. 2010232.
Conflicts of interest: None declared.
(Edited by Liu KD, Zhang XL/Qiu Y/Song LP)
REFERENCES
- [1].Lehéricy S, Hirsch EC, Cervera-Piérot P, et al. Heterogeneity and selectivity of the degeneration of cholinergic neurons in the basal forebrain of patients with Alzheimer's disease. J Comp Neurol. 1993;330(1):15–31. doi: 10.1002/cne.903300103. [DOI] [PubMed] [Google Scholar]
- [2].Barnham KJ, Cappai R, Beyreuther K, et al. Delineating common molecular mechanisms in Alzheimer's and prion diseases. Trends Biochem Sci. 2006;31(8):465–472. doi: 10.1016/j.tibs.2006.06.006. [DOI] [PubMed] [Google Scholar]
- [3].Abad MA, Enguita M, DeGregorio-Rocasolano N, et al. Neuronal pentraxin 1 contributes to the neuronal damage evoked by amyloid-beta and is overexpressed in dystrophic neurites in Alzheimer's brain. J Neurosci. 2006;26(49):12735–12747. doi: 10.1523/JNEUROSCI.0575-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tickler AK, Wade JD, Separovic F. The role of Abeta peptides in Alzheimer's disease. Protein Pept Lett. 2005;12(6):513–519. doi: 10.2174/0929866054395905. [DOI] [PubMed] [Google Scholar]
- [5].Kaminsky YG, Marlatt MW, Smith MA, et al. Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: evidence for Abeta(25-35) Exp Neurol. 2010;221(1):26–37. doi: 10.1016/j.expneurol.2009.09.005. [DOI] [PubMed] [Google Scholar]
- [6].Gao X, Tang XC. Huperzine A attenuates mitochondrial dysfunction in beta-amyloid-treated PC12 cells by reducing oxygen free radicals accumulation and improving mitochondrial energy metabolism. J Neurosci Res. 2006;83(6):1048–1057. doi: 10.1002/jnr.20791. [DOI] [PubMed] [Google Scholar]
- [7].Silva DF, Esteves AR, Oliveira CR, et al. Mitochondria: the common upstream driver of amyloid-β and tau pathology in Alzheimer's disease. Curr Alzheimer Res. 2011;8(5):563–572. doi: 10.2174/156720511796391872. [DOI] [PubMed] [Google Scholar]
- [8].Maruszak A, Żekanowski C. Mitochondrial dysfunction and Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(2):320–330. doi: 10.1016/j.pnpbp.2010.07.004. [DOI] [PubMed] [Google Scholar]
- [9].Zawar C, Plant TD, Schirra C, et al. Cell-type specific expression of ATP-sensitive potassium channels in the rat hippocampus. J Physiol. 1999;514(Pt 2):327–341. doi: 10.1111/j.1469-7793.1999.315ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ma G, Fu Q, Zhang Y, et al. Effects of Abeta1-42 on the subunits of KATP expression in cultured primary rat basal forebrain neurons. Neurochem Res. 2008;33(7):1419–1424. doi: 10.1007/s11064-008-9603-2. [DOI] [PubMed] [Google Scholar]
- [11].Ala-Rämi A, Ylitalo KV, Hassinen IE. Ischaemic preconditioning and a mitochondrial KATP channel opener both produce cardioprotection accompanied by F1F0- ATPase inhibition in early ischaemia. Basic Res Cardiol. 2003;98(4):250–258. doi: 10.1007/s00395-003-0413-z. [DOI] [PubMed] [Google Scholar]
- [12].Goodman Y, Mattson MP. K+ channel openers protect hippocampal neurons against oxidative injury and amyloid b-peptide toxicity. Brain Res. 1996;706(2):328–332. doi: 10.1016/0006-8993(95)01367-9. [DOI] [PubMed] [Google Scholar]
- [13].Hu LF, Wang S, Shi XR, et al. ATP-sensitive potassium channel opener iptakalim protected against the cytotoxicity of MPP+ on SH-SY5Y cells by decreasing extracellular glutamate level. J Neurochem. 2005;94(6):1570–1579. doi: 10.1111/j.1471-4159.2005.03306.x. [DOI] [PubMed] [Google Scholar]
- [14].Lacza Z, Pankotai E, Busija DW. Mitochondrial nitric oxide synthase: current concepts and controversies. Front Biosci. 2009;14:4436–4443. doi: 10.2741/3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Blanco S, Molina FJ, Castro L, et al. Study of the nitric oxide system in the rat cerebellum during aging. BMC Neurosci. 2010;11:78. doi: 10.1186/1471-2202-11-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Haas J, Storch-Hagenlocher B, Biessmann A, et al. Inducible nitric oxide synthase and argininosuccinate synthetase: co-induction in brain tissue of patients with Alzheimer's dementia and following stimulation with beta- amyloid 1-42 in vitro. Neurosci Lett. 2002;322(2):121–125. doi: 10.1016/s0304-3940(02)00095-2. [DOI] [PubMed] [Google Scholar]
- [17].Abramson SB, Amin AR, Clancy RM, et al. The role of nitric oxide in tissue destruction. Best Pract Res Clin Rheumatol. 2001;15(5):831–845. doi: 10.1053/berh.2001.0196. [DOI] [PubMed] [Google Scholar]
- [18].Mathur A, Hong Y, Kemp BK, et al. Evaluation of fluorescent dyes for the detection of mitochondrial membrane potential changes in cultured cardiomyocytes. Cardiovasc Res. 2000;46(1):126–138. doi: 10.1016/s0008-6363(00)00002-x. [DOI] [PubMed] [Google Scholar]
- [19].Teshima Y, Akao M, Li RA, et al. Mitochondrial ATP-sensitive potassium channel activation protects cerebellar granule neurons from apoptosis induced by oxidative stress. Stroke. 2003;34(7):1796–1802. doi: 10.1161/01.STR.0000077017.60947.AE. [DOI] [PubMed] [Google Scholar]
- [20].Bednarczyk P. Potassium channels in brain mitochondria. Acta Biochim Pol. 2009;56(3):385–392. [PubMed] [Google Scholar]
- [21].Shearman MS, Ragan CI, Iversen LL. Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of beta-amyloid-mediated cell death. Proc Natl Acad Sci U S A. 1994;91(4):1470–1474. doi: 10.1073/pnas.91.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu Y, Schubert D. Cytotoxic amyloid peptides inhibit cellular 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis. J Neurochem. 1997;69(6):2285–2293. doi: 10.1046/j.1471-4159.1997.69062285.x. [DOI] [PubMed] [Google Scholar]
- [23].Han JS, Wang HS, Yan DM, et al. Myocardial ischemic and diazoxide preconditioning both increase PGC-1alpha and reduce mitochondrial damage. Acta Cardiol. 2010;65(6):639–644. doi: 10.1080/ac.65.6.2059860. [DOI] [PubMed] [Google Scholar]
- [24].Dumont M, Beal MF. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med. 2011;51(5):1014–1026. doi: 10.1016/j.freeradbiomed.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pagani L, Eckert A. Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis 2011. 2011 doi: 10.4061/2011/925050. 925050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer's disease. J Alzheimers Dis. 2010;20(Suppl 2):S569–578. doi: 10.3233/JAD-2010-100357. [DOI] [PubMed] [Google Scholar]
- [27].Ozcan C, Bienengraeber M, Dzeja PP, et al. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282(2):H531–539. doi: 10.1152/ajpheart.00552.2001. [DOI] [PubMed] [Google Scholar]
- [28].Ishii K, Muelhauser F, Liebl U, et al. Subacute NO generation induced by Alzheimer's beta-amyloid in the living brain: reversal by inhibition of the inducible NO synthase. FASEB J. 2000;14(11):1485–1489. doi: 10.1096/fj.14.11.1485. [DOI] [PubMed] [Google Scholar]
- [29].Tsai SJ, Liu WH, Yin MC. Trans fatty acids enhanced beta-amyloid induced oxidative stress in nerve growth factor differentiated PC12 cells. Neurochem Res. 2012;37(4):786–794. doi: 10.1007/s11064-011-0673-1. [DOI] [PubMed] [Google Scholar]
- [30].Ahmadian S, Barar J, Saei AA, et al. Cellular toxicity of nanogenomedicine in MCF-7 cell line: MTT assay. J Vis Exp. 2009;26:pii:1191. doi: 10.3791/1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol. 2010;594:57–72. doi: 10.1007/978-1-60761-411-1_4. [DOI] [PubMed] [Google Scholar]