

Abstract
Gap junction blocking agents can inhibit spontaneous discharge frequency in cells. We established a rat model of posttraumatic epilepsy induced using ferric ions. Rats were intraperitoneally injected with carbenoxolone, 20 mg/kg, prior to and 30 minutes after model establishment, once a day for 14 consecutive days. Immunohistochemistry showed glial cell proliferation around a cortical focus and significantly increased connexin expression in posttraumatic epilepsy. However, carbenoxolone pretreatment or treatment significantly reduced connexin expression in the cortex, inhibited glial fibrillary acidic protein expression and ameliorated seizure degree in rats. These findings indicate that large amounts of glial cell proliferation and abnormal gap junction generation play a role in posttraumatic epilepsy, and that carbenoxolone may prevent and treat this disease.
Keywords: neural regeneration, brain injury, posttraumatic epilepsy, ferric ion, gap junction, carbenoxolone, glial fibrillary acidic protein, connexin 43, seizure, brain injury, grant-supported paper, photographs-containing paper, neuroregeneration
Research Highlights
(1) Glial cell proliferation around a cortical focus and significantly increased connexin expression were found in rats with posttraumatic epilepsy.
(2) Large amounts of glial cell proliferation and abnormal gap junction generation have a role in posttraumatic epilepsy.
(3) Carbenoxolone pretreatment or treatment could significantly reduce connexin expression, inhibit glial cell proliferation around a cortical focus and attenuate seizure degree of posttraumatic epilepsy.
INTRODUCTION
Posttraumatic epilepsy is a form of epilepsy that results from abnormal neuronal discharge caused by physical trauma to the brain[1,2]. The incidence of posttraumatic epilepsy ranges from 4.4–53% in different traumatic conditions[3]. It is estimated that 5 000–30 000 new posttraumatic epilepsy cases per year are caused by brain trauma[4]. Posttraumatic epilepsy further aggravates pathological injury of brain tissues and neurobiochemical changes, increases risk of death, and results in huge mental and employment pressure in addition to economic burden[5,6]. As an effective prevention of posttraumatic epilepsy has not been discovered, most posttraumatic epilepsy patients may develop refractory epilepsy. However, why posttraumatic epilepsy occurs post brain trauma remains poorly understood. Furthermore, why posttraumatic epilepsy occurs after a certain degree of recovery following brain injury, and discovering the triggering mechanism of neuronal discharge[7,8] are still questions that require answering. Given these gaps in our knowledge regarding this condition, increasing attention has been paid to posttraumatic epilepsy[9,10,11,12].
In the central nervous system, neuronal synaptic transmission includes chemical and electrical synapses. Gap junctions are the structural basis of the electrical synapse, transferring information via this structure. Gap junctions are the only known direct channel for cell substances and information exchange and may play a role between information communication between glial cells and neurons[13]. Meister et al[13] found that the interval time of action potential transfer between two adjacent ganglion cells is almost zero, and separated by 1 to 2 minutes of silence. This direct transfer of gap junction has been regarded as an important reason for synchronous activity of neurons[13]. Gap junctions play an important role in the regulation of growth, differentiation and physiological function of nerve cells; therefore, increasing studies have focused on the correlation between gap junction-generated electrical synapses between neurons and epilepsy pathogenesis[14,15]. Connexin is the basic structural and functional protein of intercellular gap junction channels. Connexin belongs to a large family of membrane proteins coded by a multigene family and specifically expressed in all tissues in the body[16]. Connexin 43 is mostly expressed in the brain of adult mammals[17], and is thought to have important physiological functions including regulating cell proliferation and differentiation, maintaining environmental homeostasis in cells, and protecting organism development and hemocyte formation[18].
Carbenoxolone is a gap junction blocking agent and can pass the blood-brain barrier. The mode of action of carbenoxolone is to phosphorylate connexin through activating protein kinases and G proteins in several minutes. This action leads to channel blocking and galvanic couple reduction, thereby blocking gap junctions, while not affecting neuronal properties[19]. Gap junctions are a new focus in epilepsy studies and their role in brain injury and glial cell apoptosis remain controversial[20,21,22]. Thus, we established a rat model of posttraumatic epilepsy induced by ferric ion to investigate the role of gap junctions in posttraumatic epilepsy progression.
RESULTS
Quantitative analysis of experimental animals
A total of 40 Sprague-Dawley rats were equally and randomly assigned to the normal, model, sham-surgery, carbenoxolone pretreatment and carbenoxolone treatment groups. Posttraumatic epilepsy was induced by ferric ion (FeCl3) injection into the model, carbenoxolone pretreatment and carbenoxolone treatment groups, while the sham-surgery group was injected with normal saline. In addition, carbenoxolone pretreatment and carbenoxolone treatment groups were intraperitoneally injected with carbenoxolone 30 minutes prior to and following model establishment, respectively. All 40 rats were included in the final analysis.
Influence of ferric ion injection on gross morphology of rat brain
In the model group, sensorimotor cortex in the frontal lobe was significantly shrunken at the injection site in the left hemisphere. Brown yellow cortex, 1–3 mm, was observed in the cortical region from hemosiderin pigmentation (Figure 1A). An injection pinhole was observed in the sham-surgery group, with no obvious pathological changes (Figure 1B).
Figure 1.
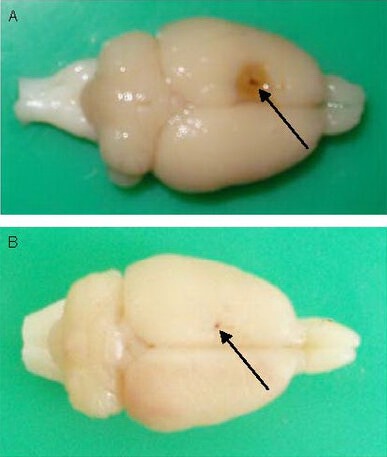
Gross morphology of rat brain after ferric ion injection. Arrows: Pin hole.
(A) Sensorimotor cortex in the frontal lobe was significantly shrunken at the injection site of the left hemisphere, and the cortex turned brown yellow following FeCl3 injection.
(B) Only an injection pin hole was observed in rats injected with normal saline.
Carbenoxolone ameliorated seizure degree
No epileptic behaviors were found in normal and sham-surgery groups, but various types of epileptic seizure occurred following model establishment. Both carbenoxolone pretreatment and carbenoxolone treatment significantly ameliorated epileptic seizure scores compared with the model group (P < 0.05); moreover, carbenoxolone pretreatment (within 5 days) exhibited better effects compared with carbenoxolone treatment (P < 0.05; Table 1).
Table 1.
Mean highest degree of Racine scores of daily seizure in epileptic rats

Carbenoxolone reduced cortical glial fibrillary acidic protein and connexin 43 expression in epileptic rats
Immunohistochemistry staining was conducted to detect glial fibrillary acidic protein and connexin 43 expression. In normal and sham-surgery groups, a few glial fibrillary acidic protein-positive cells were observed. Cells were lightly stained, with small cell bodies and thin processes, while connexin 43-positive cells were observed occasionally, with light staining. In the model group, the number of glial fibrillary acidic protein-positive cells was significantly increased (P < 0.01). Cells were darkly stained, with large cell bodies and abundant processes, in particular in the injection hemisphere. The number of connexin 43-positive cells was significantly increased compared with the normal group (P < 0.01), and cells were darkly stained. In addition, the number of glial fibrillary acidic protein- and connexin 43-positive cells was significantly reduced in carbenoxolone pretreatment and carbenoxolone treatment groups compared with the model group (P < 0.05). The cortical connexin 43-positive cells in the carbenoxolone treatment group were scattered and lightly stained, and was similar to the sham-surgery group (Table 2, Figures 2, 3).
Table 2.
Number of glial fibrillary acidic protein (GFAP)- and connexin 43-positive cells (cells per 400-fold field of view) in rat cortex
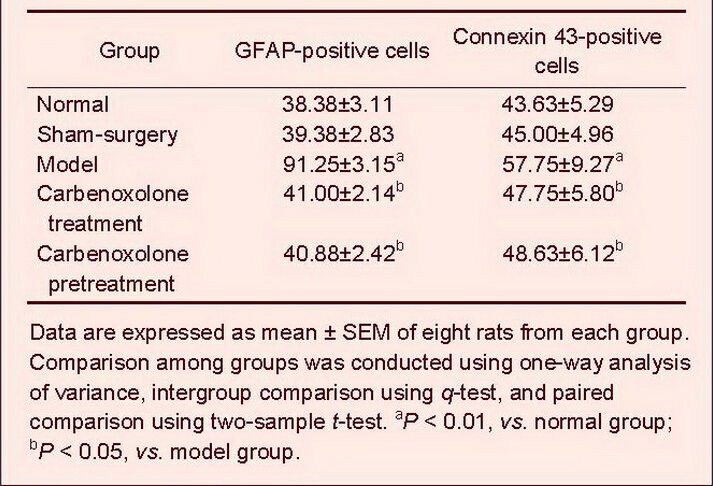
Figure 2.
Cortical glial fibrillary acidic protein expression (immunohistochemical staining, × 200).
The nuclei of glial fibrillary acidic protein-positive cells stained lavender with hematoxylin, while cytoplasm stained brown with diaminobenzidine. Arrows: Glial fibrillary acidic protein-positive cells.
Glial fibrillary acidic protein expression was low in the cortex of normal (A) and sham-surgery groups (B); the glial fibrillary acidic protein expression was increased in the model group (C), but decreased in the carbenoxolone pretreatment (D) and carbenoxolone treatment groups (E) compared with the model group.
Figure 3.
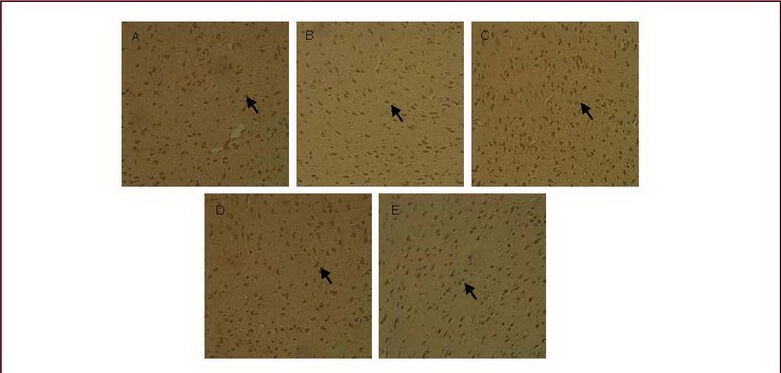
Cortical connexin 43 expression (immunohistochemical staining, × 200).
The nuclei of connexin 43-positive cells stained lavender with hematoxylin, while cytoplasm stained brown with diaminobenzidine. Arrows: Connexin 43-positive cells.
Connexin 43 expression was low in the cortex of normal (A) and sham-surgery groups (B); connexin 43 expression was increased in the model group (C), but decreased in the carbenoxolone pretreatment (D) and carbenoxolone treatment groups (E) compared with the model group.
DISCUSSION
When an electric circuit has a certain electrical loading and intensity of current, there is a junction point, which may trigger discharge because of bad contact induced by electrolysis or oxidation, resulting in damage or dysfunction of an electrical appliance in the electric circuit. Discharge interrupts or better connects the junction point, leading to transient or permanent disappearance of discharge at the junction point. To prevent this discharge, the junction point should be welded firmly to recover the function of the electrical circuit or completely separate the junction point to prevent further damage.
In the central nervous system, bioelectricity constitutes an electric circuit in neurons. Among the electric circuits, electrical synapses based on gap junctions are the junction points. In recent years, gap junctions have become a focus in studies of epilepsy, but most emphasize synchronization of gap junctions and central nervous system paradoxical discharge[13,15,23] and attempt to develop a novel pathway for treating epilepsy using gap junction blocking agents. Based on discharge phenomenon in the macroscopic world, we hypothesized that as the junction point of biological electrical circuits in the nervous system, gap junctions altered in structure and function in response to abnormal physical, chemical and biological factors. They may also block bioelectricity conduction because of bad contact.
When electrical loading and current intensity at two sides of a gap junction reach a certain amount, paradoxical discharge occurs, leading to dysfunction of corresponding regions in the brain[24]. This hypothesis explains some clinical doubts, for example, the focus of symptomatic epilepsy is always distributed in cerebral gray matter containing gap junctions[25]. Furthermore, there are certain associations between epileptic seizure patterns and gap junctions and different connexin expression in different age stages[26]. In fact, attacks of posttraumatic epilepsy at several months or years post trauma are associated with abnormal gap junctions during brain repair[27]. If this hypothesis is true, we can develop novel therapies for epilepsy by finding substances to repair poorly connected gap junctions and prevent seizures in populations with high risk of epilepsy, such as brain trauma.
The present study successfully established a rat model of ferric ion-induced posttraumatic epilepsy and recorded behavior manifestations in epileptic seizures. Moreover, immunohistochemistry was used to assess cortical glial fibrillary acidic protein and connexin 43 expression. The differences in the above indexes were compared prior to and following carbenoxolone treatment. We selected the ferric ion-induced posttraumatic epilepsy model, cortical glial fibrillary acidic protein and connexin 43 as observation indexes because after brain trauma, glial cell scars form and new neuronal networks are established. Gap junctions are the major connection pathway between glial cells and neurons, so structural and functional changes in gap junctions may play a key role in posttraumatic epilepsy occurrence[28,29]. Cortical gap junctions are abundant, and the cerebral cortex is an important site for posttraumatic epilepsy and following epileptic seizure formation and transmission. Glial fibrillary acidic protein is a specific marker of astrocytes, and its positive expression reflects increased activity of these cells. Connexin 43 is most expressed in the central nervous system of adult mammals, especially in astrocytes[30].
In the present study, FeCl3 was injected into the sensorimotor cortex to trigger neuronal burst discharge, inducing epileptic seizures. Moreover, the course of the disease was clearly divided into the acute, rest and chronic stages, similar to the human posttraumatic epilepsy process. Immunohistochemistry showed that at 14 days after model establishment, glial fibrillary acidic protein and connexin 43 expression was significantly increased compared with normal and sham-surgery groups. This indicates that with glial cell proliferation post brain injury, glial fibrillary acidic protein-positive cells were increased, connexin 43 expression was increased, and posttraumatic epilepsy gradually formed which is consistent with previous results[31]. These findings further confirm that gap junctions are involved in posttraumatic epilepsy occurrence and progression[32].
The present study treated rats with the gap junction blocking agent, carbenoxolone, to further investigate the role of gap junctions in posttraumatic epilepsy occurrence and progression. Carbenoxolone has been clinically used to treat gastric ulcers and exhibits strong effects in blocking gap junctions[33]. Carbenoxolone treatment prior to model establishment significantly reduced epileptic seizure degree within 5 days following modeling, and the effective time of carbenoxolone treatment group was significantly later than carbenoxolone pretreatment group. Effects of carbenoxolone treatment following model establishment were similar to results seen by Medina-Ceja et al[34], further confirming that gap junctions are a promising target in epilepsy treatment. Carbenoxolone pretreatment was seen to be more valuable and may inhibit abnormal gap junction formation. This information provides a new pathway for preventing posttraumatic epilepsy post brain injury. Our results confirm, to a certain degree, the hypothesis that structural and functional changes in gap junctions are the source of abnormal neuronal discharge resulting from “bad contact” in neuronal electrical conduction.
MATERIALS AND METHODS
Design
A randomized, controlled, animal study.
Time and setting
Experiments were conducted in the Institute of Nautical Medicine, Nantong University, China from January 2009 to December 2010.
Materials
Animals
A total of 40 healthy adult male Sprague-Dawley rats, aged 6–8 weeks, weighing 220–250 g, were provided by the Laboratory Animal Center of Nantong University [license No. SCXK (Su) 2008-0010]. They were housed at 25 ± 2°C with 40–60% humidity in natural day/night cycle and allowed free access to food and water. Animal procedures were conducted in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology of China[35].
Drugs
Carbenoxolone, with the chemical formule of C34H48O7Na2, purity ≥ 98%, was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Methods
Posttraumatic epilepsy model establishment
A posttraumatic epilepsy model was established by injection of FeCl3 as previously described[36] after some modifications. The rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (350 mg/kg) and placed in a stereotaxic instrument (Stoelting, Wood Dale, IL, USA). Following routine skin disinfection, a 3.0 cm scalp incision was made along the vertex median line. According to the Rat Brain in Stereotaxic Coordinates[37], a hole of 2.0 mm diameter was drilled at 2.0 mm posterior to coronal suture, 2.0 mm lateral to sagittal suture of the left cranium (Figure 4), and a micro-injection pump (WPI, Sarasota, FL, USA) was used to slowly inject 10 μL FeCl3 solution (0.1 M; Shanghai Jinshan Medicine Chemical Industry Factory, Shanghai, China) to a depth of 2.5 mm, 1 μL/min. The needle was maintained for 10 minutes to prevent FeCl3 leakage. The skin was closed, and rats were removed from the instrument after routine disinfection. The sham-surgery group was injected with 10 μL normal saline into the sensorimotor area of the left cortical frontal lobe. Rat behaviors were observed during and 120 minutes after injection, and 9:00 a.m. to 11:00 a.m. every day thereafter. Behaviors of rats were evaluated according to Racine scale of epileptic seizure[38]: grade 0, no behaviors of epileptic seizure; grade 1, face clonus, chewing, yawning; grade 2, nodding, face clonus, neck muscle convulsion in addition to behaviors of grade 1; grade 3, forelimb or hindlimb convulsion in addition to behaviors of grade 2; grade 4, hindlimb standing, sudden standing in addition to behaviors of grade 3; grade 5, rhythmic convulsion of four limbs, hindlimb stiffness and dorsal flexure or convulsion in addition to behaviors of grade 4. According to the Racine scale, rats at grade 4 or higher were selected as models.
Figure 4.
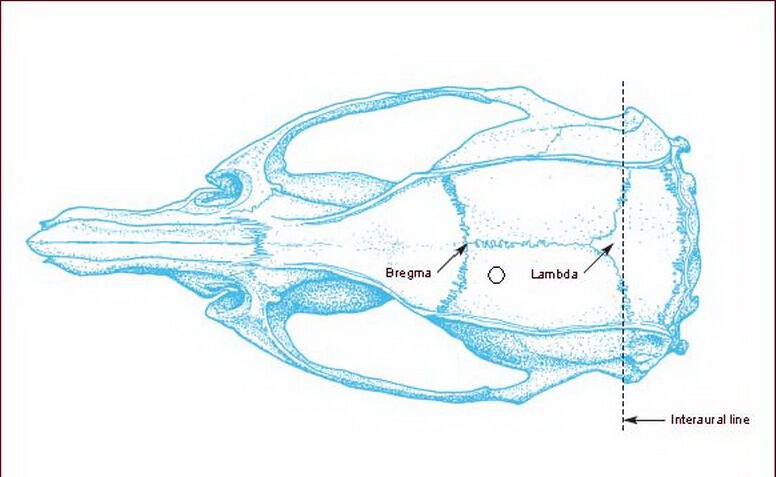
FeCl3 injection site in the cranium[37].
The hollowed dot represents the injection site at the left cranium.
Carbenoxolone pretreatment and treatment
The carbenoxolone pretreatment group was intraperitoneally injected with 20 mg/kg carbenoxolone 30 minutes prior to model establishment. The carbenoxolone treatment group was injected intraperitoneally with 20 mg/kg carbenoxolone 30 minutes following model establishment, once a day for 14 consecutive days[39].
Immunohistochemistry for cortical glial fibrillary acidic protein and connexin 43 expression
At 14 days after model establishment, the rats were anesthetized, followed by thoracotomy to expose the heart. The rats were perfused with normal saline and 4% paraformaldehyde phosphate buffer through the left ventricle. The rats were sacrificed and the brain was harvested, postfixed with 4% paraformaldehyde PBS for 24 hours and dehydrated with phosphate buffer containing 20% and 30% sucrose. The cross-section of 3.20–3.80 mm posterior to Bregma was harvested for serial sections on ice, 6 μm thick. Immunohistochemistry was conducted using the streptavidin peroxidase conjugated method[40]. Briefly, the brain sections were rinsed with 0.01 M PBS, placed in peroxidase blocking solution at room temperature for 10 minutes, washed and incubated with mouse anti-rat glial fibrillary acidic protein monoclonal antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit anti-connexin 43 polyclonal antibody (1:100; Bioss, Beijing, China) at 4°C for 1 day. The antibodies were removed, and the sections were washed, incubated with secondary antibodies, ready-to-use goat anti-mouse/rabbit IgG (1:50; Maixin, Fuzhou, China) at room temperature for 15 minutes. The secondary antibody was removed, and sections were washed, visualized using diaminobenzidine (Maixin), stained with hematoxylin (Sinopharm Chemical Reagent Co., Shanghai, China) for 30 seconds, washed with tap water, dehydrated, mounted, and observed using a light microscope (Nikon, Tokyo, Japan). The negative control was treated with immunohistochemical staining primary antibody dilution buffer (Beyotime, Nantong, China) rather than primary antibody. Six serial sections from each rat were selected, and the number of glial fibrillary acidic protein- and connexin 43-positive cells was quantified under a 400 × magnification light microscope. The mean value was calculated as the number of positive cells in each rat.
Statistical analysis
Data were analyzed using Stata version 10.0 (Stata, College Station, TX, USA) and expressed as mean ± SEM. Comparison among groups was conducted using one-way analysis of variance, and intergroup comparison using q-test and paired comparison using two-sample t-test. A value of P < 0.05 was considered statistically significant.
Acknowledgments:
We thank the staff at the Institute of Nautical Medicine, Nantong University, China, for technical support.
Footnotes
Weiguan Chen, Master, Attending physician, Lecturer.
Funding: This study was supported by the Social Development Program of Nantong, No. S2009035.
Conflicts of interest: None declared.
Ethical approval: This study received permission from the Animal Ethics Committee of Affiliated Hospital, Nantong University, China.
(Edited by Shen GY, Su YH/Su LL/Song LP)
REFERENCES
- [1].Kharatishvili I, Pitkänen A. Posttraumatic epilepsy. Curr Opin Neurol. 2010;23(2):183–188. doi: 10.1097/WCO.0b013e32833749e4. [DOI] [PubMed] [Google Scholar]
- [2].Agrawal A, Timothy J, Pandit L, et al. Post-traumatic epilepsy: an overview. Clin Neurol Neurosurg. 2006;108(5):433–439. doi: 10.1016/j.clineuro.2005.09.001. [DOI] [PubMed] [Google Scholar]
- [3].Frey LC. Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia. 2003;44(Suppl 10):11–17. doi: 10.1046/j.1528-1157.44.s10.4.x. [DOI] [PubMed] [Google Scholar]
- [4].Diaz-Arrastia R, Gong Y, Fair S, et al. Increased risk of late posttraumatic seizures associated with inheritance of APOE epsilon4 allele. Arch Neurol. 2003;60(6):818–822. doi: 10.1001/archneur.60.6.818. [DOI] [PubMed] [Google Scholar]
- [5].Medvdovsky M, Ifergane G, Wirguin I, et al. Traumatic intracranial hemorrhage in patients with seizures: descriptive characteristics. Epilepsy Behav. 2006;8(2):429–433. doi: 10.1016/j.yebeh.2005.12.010. [DOI] [PubMed] [Google Scholar]
- [6].Li H, McDonald W, Parada I, et al. Targets for preventing epilepsy following cortical injury. Neurosci Lett. 2011;497(3):172–176. doi: 10.1016/j.neulet.2011.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jensen FE. Introduction. Posttraumatic epilepsy: treatable epileptogenesis. Epilepsia. 2009;50(Suppl 2):1–3. doi: 10.1111/j.1528-1167.2008.02003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dichter MA. Posttraumatic epilepsy: the challenge of translating discoveries in the laboratory to pathways to a cure. Epilepsia. 2009;50(Suppl 2):41–45. doi: 10.1111/j.1528-1167.2008.02009.x. [DOI] [PubMed] [Google Scholar]
- [9].Pagni CA, Zenga F. Posttraumatic epilepsy with special emphasis on prophylaxis and prevention. Acta Neurochir Suppl. 2005;93:27–34. doi: 10.1007/3-211-27577-0_3. [DOI] [PubMed] [Google Scholar]
- [10].Hunt RF, Scheff SW, Smith BN. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol. 2009;215(2):243–252. doi: 10.1016/j.expneurol.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10–13. doi: 10.1111/j.1528-1167.2008.02005.x. [DOI] [PubMed] [Google Scholar]
- [12].Diaz-Arrastia R, Agostini MA, Madden CJ, et al. Posttraumatic epilepsy: the endophenotypes of a human model of epileptogenesis. Epilepsia. 2009;50(Suppl 2):14–20. doi: 10.1111/j.1528-1167.2008.02006.x. [DOI] [PubMed] [Google Scholar]
- [13].Meister M, Wong RO, Baylor DA, et al. Synchronous bursts of action potentials in ganglion cells of the developing mammalian retina. Science. 1991;252(5008):939–943. doi: 10.1126/science.2035024. [DOI] [PubMed] [Google Scholar]
- [14].Jin MM, Chen Z. Role of gap junctions in epilepsy. Neurosci Bull. 2011;27(6):389–406. doi: 10.1007/s12264-011-1944-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Steinhäuser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia. 2012;60(8):1192–1202. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- [16].Söhl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res. 2004;62(2):228–232. doi: 10.1016/j.cardiores.2003.11.013. [DOI] [PubMed] [Google Scholar]
- [17].Dermietzel R, Traub O, Hwang TK, et al. Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci U S A. 1989;86(24):10148–10152. doi: 10.1073/pnas.86.24.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nagy JI, Rash JE. Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev. 2000;32(1):29–44. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- [19].Gigout S, Louvel J, Kawasaki H, et al. Effects of gap junction blockers on human neocortical synchronization. Neurobiol Dis. 2006;22(3):496–508. doi: 10.1016/j.nbd.2005.12.011. [DOI] [PubMed] [Google Scholar]
- [20].Prince DA, Parada I, Scalise K, et al. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia. 2009;50(Suppl 2):30–40. doi: 10.1111/j.1528-1167.2008.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Volman V, Perc M, Bazhenov M. Gap junctions and epileptic seizures--two sides of the same coin? PLoS One. 2011;6(5):e20572. doi: 10.1371/journal.pone.0020572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Amiri M, Bahrami F, Janahmadi M. On the role of astrocytes in epilepsy: a functional modeling approach. Neurosci Res. 2012;72(2):172–180. doi: 10.1016/j.neures.2011.11.006. [DOI] [PubMed] [Google Scholar]
- [23].Timofeev I, Bazhenov M, Avramescu S, et al. Posttraumatic epilepsy: the roles of synaptic plasticity. Neuroscientist. 2010;16(1):19–27. doi: 10.1177/1073858409333545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cunningham MO, Roopun A, Schofield IS, et al. Glissandi: transient fast electrocorticographic oscillations of steadily increasing frequency, explained by temporally increasing gap junction conductance. Epilepsia. 2012;53(7):1205–1214. doi: 10.1111/j.1528-1167.2012.03530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lowenstein DH. Epilepsy after head injury: an overview. Epilepsia. 2009;50(Suppl 2):4–9. doi: 10.1111/j.1528-1167.2008.02004.x. [DOI] [PubMed] [Google Scholar]
- [26].Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5(12):1055–1063. doi: 10.1016/S1474-4422(06)70626-3. [DOI] [PubMed] [Google Scholar]
- [27].Frantseva MV, Kokarovtseva L, Naus CG, et al. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002;22(3):644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pitkänen A, Immonen RJ, Gröhn OH, et al. From traumatic brain injury to posttraumatic epilepsy: what animal models tell us about the process and treatment options. Epilepsia. 2009;50(Suppl 2):21–29. doi: 10.1111/j.1528-1167.2008.02007.x. [DOI] [PubMed] [Google Scholar]
- [29].Ueda Y, Kitamoto A, Willmore LJ, et al. Hippocampal gene network analysis in an experimental model of posttraumatic epilepsy. Neurochem Res. 2011;36(7):1323–1328. doi: 10.1007/s11064-010-0386-x. [DOI] [PubMed] [Google Scholar]
- [30].Rouach N, Avignone E, Même W, et al. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell. 2002;94(7-8):457–475. doi: 10.1016/s0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- [31].Samoilova M, Wentlandt K, Adamchik Y, et al. Connexin 43 mimetic peptides inhibit spontaneous epileptiform activity in organotypic hippocampal slice cultures. Exp Neurol. 2008;210(2):762–775. doi: 10.1016/j.expneurol.2008.01.005. [DOI] [PubMed] [Google Scholar]
- [32].Traub RD, Draguhn A, Whittington MA, et al. Axonal gap junctions between principal neurons: a novel source of network oscillations, and perhaps epileptogenesis. Rev Neurosci. 2002;13(1):1–30. doi: 10.1515/revneuro.2002.13.1.1. [DOI] [PubMed] [Google Scholar]
- [33].Wallraff A, Köhling R, Heinemann U, et al. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26(20):5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Medina-Ceja L, Cordero-Romero A, Morales-Villagrán A. Antiepileptic effect of carbenoxolone on seizures induced by 4-aminopyridine: a study in the rat hippocampus and entorhinal cortex. Brain Res. 2008;1187:74–81. doi: 10.1016/j.brainres.2007.10.040. [DOI] [PubMed] [Google Scholar]
- [35].The Ministry of Science and Technology of the People's Republic of China. Guidance suggestions for the care and use of laboratory animals 2006-09-30 [Google Scholar]
- [36].Willmore LJ, Sypert GW, Munson JV, et al. Chronic focal epileptiform discharges induced by injection of iron into rat and cat cortex. Science. 1978;200(4349):1501–1503. doi: 10.1126/science.96527. [DOI] [PubMed] [Google Scholar]
- [37].Paxinos G, Watson C. 5th ed. Oxford(UK): Elsevier Academic Press; 2005. The Rat Brain in Stereotaxic Coordinates. [Google Scholar]
- [38].Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- [39].Gareri P, Condorelli D, Belluardo N, et al. Anticonvulsant effects of carbenoxolone in genetically epilepsy prone rats (GEPRs) Neuropharmacology. 2004;47(8):1205–1216. doi: 10.1016/j.neuropharm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- [40].Ramos-Vara JA. Technical aspects of immunohistochemistry. Vet Pathol. 2005;42(4):405–426. doi: 10.1354/vp.42-4-405. [DOI] [PubMed] [Google Scholar]